Discovery of a potent and highly β1 specific proteasome inhibitor from a focused library of urea-containing peptide vinyl sulfones and peptide epoxyketones†
Wouter A.
van der Linden
a,
Lianne I.
Willems
a,
Tamer B.
Shabaneh
b,
Nan
Li
a,
Mark
Ruben
a,
Bogdan I.
Florea
a,
Gijs A.
van der Marel
a,
Markus
Kaiser
c,
Alexei F.
Kisselev
*b and
Herman S.
Overkleeft
*a
aGorlaeus Laboratories, Einsteinweg 55, 2300 CC, Leiden, The Netherlands. E-mail: h.s.overkleeft@chem.leidenuniv.nl; Fax: +31-71-5274307; Tel: +31-71-5274342
bNorris Cotton Cancer Center, Department of Pharmacology and Toxicology, Dartmouth Medical School, Dartmouth College, Lebanon, NH 03756, USA. E-mail: alexei.f.kisselev@dartmouth.edu
cZMB/Faculty of Biology, Building S03 S02 A55, Universität Duisburg-Essen, Campus Essen, 45117 Essen, Germany. E-mail: Markus.Kaiser@uni-due.de; Tel: +49-201-183-4980
First published on 13th October 2011
Abstract
Syringolins, a class of natural products, potently and selectively inhibit the proteasome and show promising antitumour activity. To gain insight in the mode of action of syringolins, the ureido structural element present in syringolins is incorporated in oligopeptide vinyl sulfones and peptide epoxyketones yielding a focused library of potent new proteasome inhibitors. The distance of the ureido linkage with respect to the electrophilic trap strongly influences subunit selectivity within the proteasome. Compounds 13 and 15 are β5 selective and their potency exceeds that of syringolin A. In contrast, 5 may well be the most potent β1 selective compound active in living cells reported to date.
1 Introduction
Syringolins form a class of small molecule natural products that are secreted by strains of the plant pathogen Pseudomonas syringae pv. syringae (Pss) when these are invading a plant.1Syringolin A and B (Fig. 1) are of particular interest as these molecules act upon the plant proteasome to compromise plant defense mechanisms against invading pathogens.2,3 The eukaryotic 20S proteasome contains three catalytically active β subunits, β1, β2 and β5, which display caspase-like, trypsin-like and chymotrypsin-like activity, respectively.4 The plant 20S proteasome is homologous to the mammalian proteasome and syringolins are potent and selective inhibitors of the human 20S proteasome as well.5 Small molecule proteasome inhibitors show promising antitumour activity and bortezomib is used in the clinic against multiple myeloma, whereas several other proteasome inhibitors are in clinical trials.6–10 Biological evaluation of syringolins on tumour cells has shown inhibition of cell proliferation and induction of apoptosis.11,12 This sparked the interest of several organic chemists and lately, the total syntheses of several syringolins have been published.13–16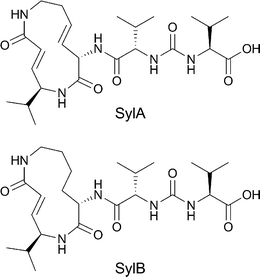 | ||
| Fig. 1 Structure of syringolin A and B. | ||
With the total synthesis of syringolin A and B completed, the opportunity opened up to perform structure–activity relationships of syringolins and their analogues. Syringolins contain a 12-membered lactam core structure the nature of which differs among the members of this family of compounds. Syringolin A (SylA) has two (E)-configured double bonds resulting in a strained system, and syringolin B has only one ring unsaturation. The α,β unsaturated amide in the ring system is responsible for covalently and irreversibly inactivating the catalytic Thr1 Oγ of the 20S proteasomeβ subunits viaMichael addition.5 SylA inhibits β2 and β5 potently and β1 at higher concentration.17 The electrophilic lactam is quite rigid in structure and probably determines to a large extent the proteasome specificity. Attached to the lactam core structure is a peptoid fragment, which differs among the syringolins. SylA and SylB contain a valine-urea-valine moiety at their pseudo N-terminus. An ureido linkage induces chain reversal in a peptide, and for this reason has been installed in protease inhibitors.18–20 It is also found in several natural peptide-based compounds with antibiotic activity.21–23 However, the syringolins are the only examples of proteasome inhibitors that contain this moiety. Some research has already been conducted (Fig. 2) on the exocyclic part of syringolins, including a D-AA scan of the two valines, revealing that the naturally occurring configuration yields the most potent compounds.16 Addition of aliphatic15 or hydrophilic16 tails yielded inhibitors with increased potency with respect to SylA. Addition of a fluorophore was tolerated, yielding a proteasome probe.24 Hybrids between SylA and glidobactin A were designed that appear to possess improved potency compared to SylA.17,25
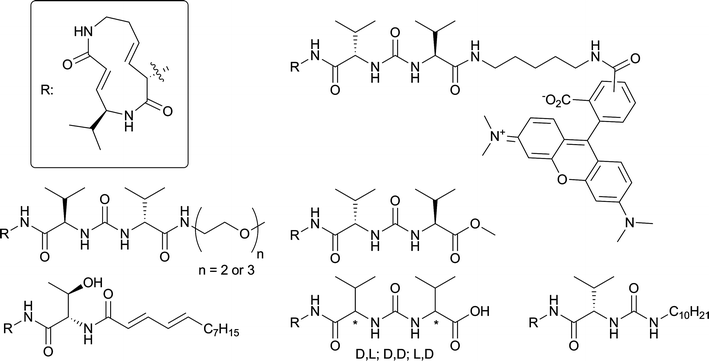 | ||
| Fig. 2 Literature SylA modifications. | ||
The proteasome subunit selectivity pattern of an inhibitor is important for its antitumour activity. Inhibition of β5 is essential, but coinhibition of β1 or β2 is usually needed to be cytotoxic in cell lines derived from various cancers.26–28 The structural elements in syringolins that are responsible for proteasome subunit selectivity are therefore useful in the design of new proteasome inhibitors. To determine the effect of the valine-urea-valine motif in the biological profile of syringolins we incorporated this moiety in a more conventional linear peptide based proteasome inhibitor design. In this design, we replaced the unsaturated lactam Michael acceptor with leucine vinyl sulfone, another Michael acceptor commonly used to arrive at potent proteasome inhibitors. A leucine epoxyketone is used as well since this warhead often yields more potent and selective inhibitors than the vinyl sulfone.29
2 Results and discussion
The first set of target compounds for this study are displayed in Fig. 3. Direct attachment of the leucine-derived electrophilic trap to the valine-urea-valine moiety yields two tripeptide compounds (2 and 6). The carboxylic acid functionality is masked as a tert-butyl ester as its synthetic precursor (1 and 5). A benzylamide cap is also used (3 and 7), since aromaticity at the N-terminus of a short proteasome inhibitor could be beneficial for its potency.30,31 The decyl chain, improving syringolin A activity,15 is also used in this library (4 and 8). When the tripeptide structure is compared to SylA, the distance between the electrophile of the inhibitor and the urea group is shorter in the tripeptide than in SylA. Therefore, one more amino acid is incorporated between the urea and electrophile, yielding tetrapeptide inhibitors (9–16).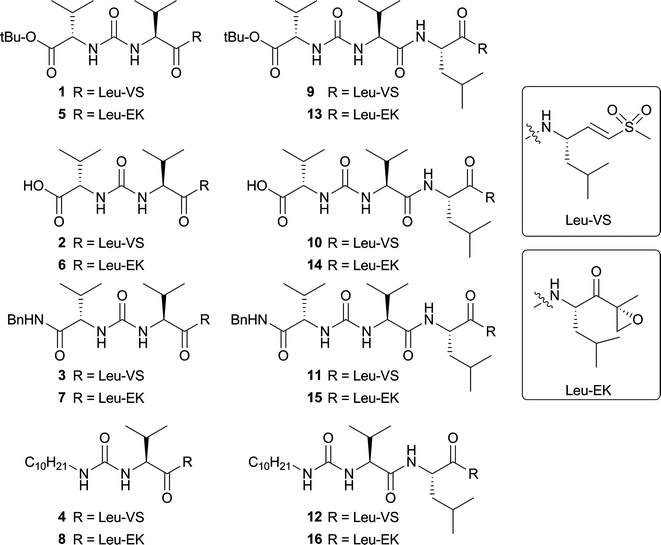 | ||
| Fig. 3 Panel of synthesized potential proteasome inhibitors. | ||
The synthesis of the tripeptide vinyl sulfones (VS) commenced with coupling the TFA·H-LeuVS32 to BocValOH to yield Boc-protected dipeptide 17 from which the Boc protecting group was removed to yield dipeptide 18 (Fig. 4). This dipeptide was reacted with valine tert-butyl ester isocyanate, valine benzylamide isocyanate or decyl isocyanate to yield three inhibitors 1, 3 and 4. Peptide 1 was deprotected with TFA to yield 2.
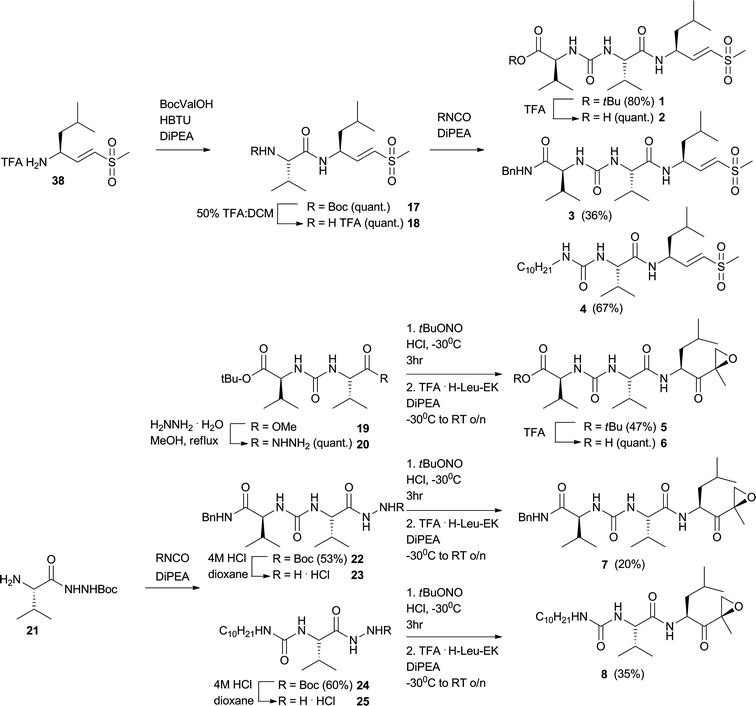 | ||
| Fig. 4 Synthesis of tripeptide vinyl sulfones and epoxyketones. | ||
In an attempt to react epoxyketone (EK) TFA·H-Val-Leu-EK with valine tert-butyl ester isocyanate, no formation of compound 5 was detected. Rather, TFA·H-Val-Leu-EK cyclizes in this basic reaction medium. As an alternative approach to obtain compound 5, the methyl ester in compound 1915 was transformed to the hydrazide (20) which was then in situ transformed to the acyl azide and coupled to TFA·HLeu-EK to arrive at 5, which was subsequently deprotected to gain 6 (Fig. 4). Compounds 7 and 8 were synthesized by a modified approach. Reacting 2133 with valine benzylamide isocyanate or decyl isocyanate yielded 22 and 24, which were then Boc-deprotected to yield hydrazides 23 and 25 which were transformed to the acyl azidein situ and coupled to TFA·H-Leu-EK to obtain 7 and 8.
The synthesis of tetrapeptide vinyl sulfones and epoxyketones followed a general strategy (Fig. 5). Methyl ester 2634 was transformed to the corresponding hydrazide 27 by hydrazine hydrate in methanol. This compound was in situ transformed to its acyl azide and then coupled to TFA·H-Leu-VS or TFA·H-Leu-EK to arrive at 28 and 30. 28 was deprotected with TFA in DCM to give 29, which in the next step was reacted with the three isocyanates mentioned in the section above to yield tetrapeptides 9, 11 and 12. 9 was deprotected with TFA to yield 10. The same strategy, employing 30, led to 13–16.
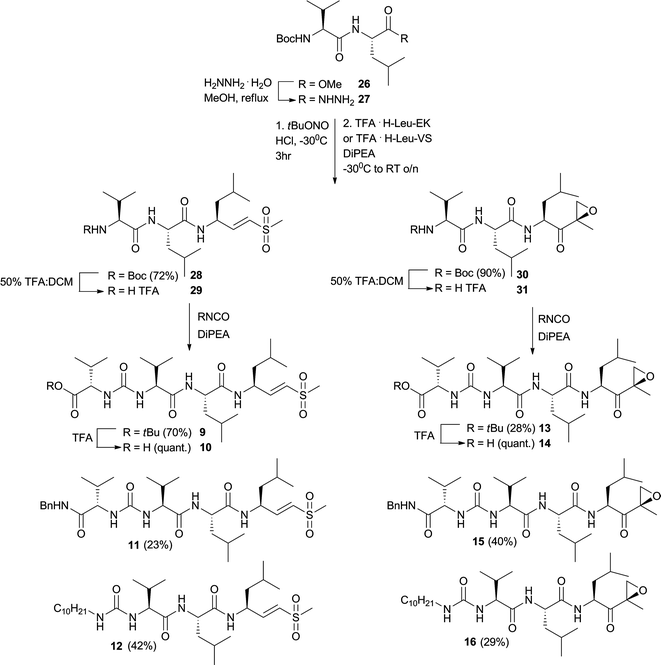 | ||
| Fig. 5 Synthesis of tetrapeptide vinyl sulfones and epoxyketones. | ||
In a first assessment of inhibitor potency the sixteen compounds were subjected to a competition assay versus Bodipy-TMR-epoxomicin (MVB003).35 First, extracts from HEK293T cells were incubated with a broad concentration range of inhibitor for one hour, after which remaining proteasome activity was labeled with MVB003. After SDS-PAGE separation of the proteome, the wet gel slabs were scanned on a Typhoon fluorescence scanner. Results are displayed in Fig. 6. Proteasome subunits were assigned based on earlier work.35
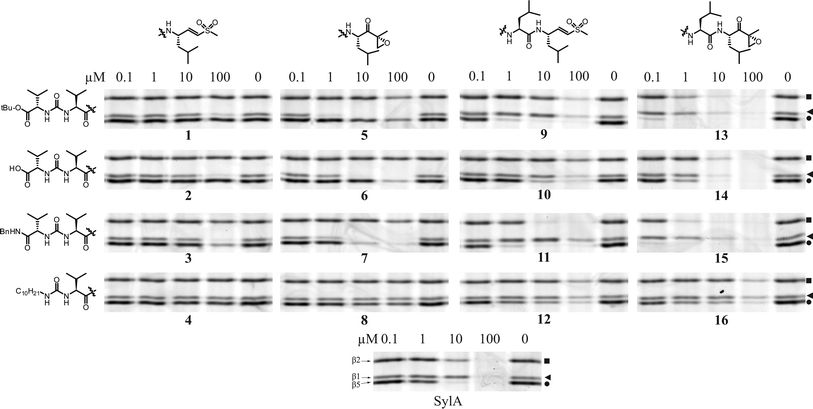 | ||
| Fig. 6 Competition assay in HEK293T lysate. Lysates (15 μg) were incubated with indicated end concentration of inhibitor for 1 h at 37 °C. Residual proteasome activity was labeled by MVB003 (0.5 μM end concentration) for 1 h at 37 °C). Upper band β2 (■), middle band β1 (◀), lower band β5 (●). | ||
One first obvious conclusion from this broad concentration scan is that the syringolin A inspired ureido peptide moiety, coupled to leucine epoxyketone or vinyl sulfone, yields active proteasome inhibitors. Generally, the inhibitors containing a vinyl sulfone are less potent than their epoxyketone counterparts, a phenomenon observed earlier.27,29 A second obvious trend is that tripeptide inhibitors show some selectivity for β1. The most distinct selectivity for β1 is displayed by 5 in HEK lysate (Fig. 6). The tetrapeptide inhibitors, on the other hand, appear to preferentially target β5. Compounds containing the long aliphatic tail (4, 8, 12, 16) are far less potent than their counterparts lacking this moiety. This is in sharp contrast to SylA, where the addition of this tail increased potency dramatically.15
To gain better insight in subunit selectivity of the most potent inhibitors, the competition assay was repeated in a more detailed concentration series (Fig. 7). Tripeptide vinyl sulfones 2–4 as well as 8 and 12 were omitted because the inhibitory effect of these compounds falls mostly outside the more diluted concentration range. In HEK lysate (Fig. 7), several library members indeed are subunit selective; 9 inhibits β5 by more than 90% at about 1 μM, at which concentration β1 and β2 appear untouched. Compound 13 fully inhibits β5 at 100 nM, before inhibiting β1 and β2. Compound 5 is very selective for the β1 subunit. Compared to NC001, a very selective tetrapeptide epoxyketone, compound 5 is only slightly less potent in HEK lysate.26,36 The benzyl amides in this library are generally less selective than their tert-butyl counterparts; 11 coinhibits β2 before complete β5 inhibition is reached. Benzyl amide 7 coinhibits β5 much earlier than the selective tert-butyl ester 5. The difference in selectivity between 13 and 15, however, is less pronounced.
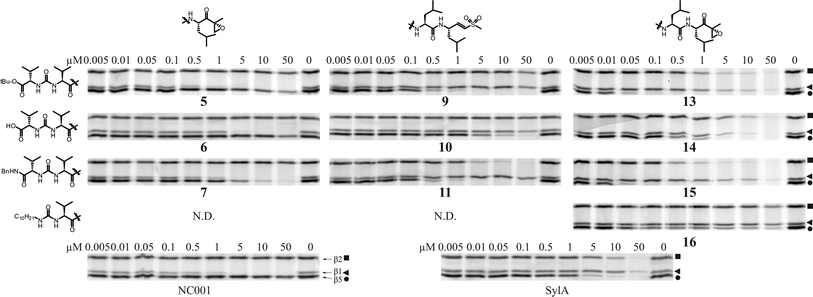 | ||
| Fig. 7 Competition assay in HEK293T lysate. Lysates (15 μg) were incubated with indicated end concentration of inhibitor for 1 h at 37 °C. Residual proteasome activity was labeled by MVB003 (0.5 μM end concentration) for 1 h at 37 °C. N.D. not determined. Upper band β2 (■), middle band β1 (◀), lower band β5 (●). | ||
The parent compound, SylA, inhibits β5 and β2 in the low micromolar range and only inhibits β1 at higher concentration (Fig. 7). In this respect, the activities of the tetrapeptide inhibitors of this library in general more resemble that of SylA than the activities of the tripeptides do. Interestingly, there are several compounds in this library that display more potent proteasome inhibition than the parent compound, SylA. Compounds 13 and 15 are the most potent inhibitors in this study; compared to SylA in HEK lysate (Fig. 7), these two compounds inhibit β5 at about 100-fold lower concentration. The most potent and selective compounds were also analysed for their inhibitory capacities against purified rabbit 26S proteasome with subunit specific fluorogenic peptides. To better compare these results with the competition assay gels from Fig. 7, the gel bands were quantified and plotted against inhibitor concentrations. From these plots, the IC50 values were calculated and the results are summarised in Table 1. The IC50 values found with the fluorogenic peptide assay are higher than those found in the competition assay, but one should be careful when comparing IC50 values obtained with two very different experimental setups. The subunit selectivity trends found with the fluorogenic peptide assay, however, in most cases agree with the results from the competition assay. In both settings, 5 is a selective β1 proteasome inhibitor, while 15 is a selective β5 inhibitor. In the fluorogenic substrate assay, compound 13 shows onset of inhibition of the β1 and β2 sites when β5 is targeted, while in the competition assay this compound appears more selective. We have no explanation for this discrepancy yet. The differences could be caused by the presence of other proteasome forms in cell lysate (i.e. PA200 and PA28 activated proteasomes) or post-translational modifications that affect active site specificity and that are either species or tissue specific and may be lost during preparation of proteasomes from muscle.
| Quantified competition assay gels | Fluorogenic peptide assay | |||||
|---|---|---|---|---|---|---|
| β5 | β1 | β2 | β5 | β1 | β2 | |
| Compound | IC50 (μM) | IC50 (μM) | ||||
| 5 | ∼50 | 0.46 | >50 | >30 | >30 | |
| 6 | >50 | 1.4 | >50 | |||
| 7 | 2.9 | 0.52 | >50 | >10 | 1.2 | >10 |
| 9 | 0.23 | >50 | >50 | 1.0 | >30 | >30 |
| 10 | 4.2 | >50 | >50 | |||
| 11 | 0.17 | >50 | 2.1 | 0.64 | 18 | 10 |
| 13 | 0.016 | 1.3 | 0.69 | 0.3 | >0.3 | >0.3 |
| 14 | 0.48 | 2.8 | 5.9 | |||
| 15 | 0.008 | 2.4 | 0.28 | 0.013 | 0.24 | >0.3 |
| 16 | 0.5 | >50 | >50 | |||
| SylA | 1.3 | 25 | 6.6 | |||
| NC001 | >50 | 0.33 | >50 | |||
Compounds 5, 13 and 15 were evaluated on their proteasomal inhibition capacities in living HEK cells to assess cell permeability of these compounds and compared to literature subunit selective proteasome inhibitors NC001 and NC005-VS.26,36 Living HEK cells were incubated with the inhibitors for 4 h. after which remaining proteasomal activity was labeled by cell permeable probe MVB003. The results are summarised in Table 2. Interestingly, 5 shows fivefold higher potency in living HEK cells than NC001, while NC001 is slightly more potent in cell extract. This difference could be ascribed to better cell permeability of 5, possibly due to its smaller size than NC001. Compound 5 is the most potent and selective β1 inhibitor to date. Compounds 13 and 15 do inhibit the proteasome in living HEK cells and therefore are cell permeable, but their β5 selectivity is much lower than observed in HEK extracts and NC005-VS is much more β5 selective in cells than 13 and 15.
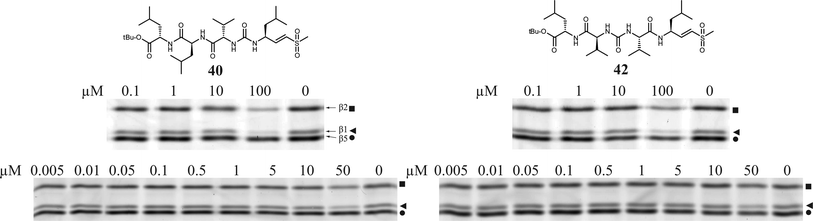
Having evaluated a small library of proteasome inhibitors containing tri- and tetrapeptides with the valine-urea-valine motif, a dramatic shift in selectivity from β1 to β5 is observed when the ureido linkage is moved one place in the molecule. We chose a tetrapeptide vinyl sulfone scaffold in which the ureido-linkage is ‘shifted’ through the molecule to arrive at two more potential proteasome inhibitors with the urea after the first or second amino acid in the peptide instead of the third (Fig. 8). The Fmoc group in 34 and 3237 was removed by DBU.38Dipeptide 35 was coupled to Fmoc-Val-OH and again deprotected with DBU to arrive at tripeptide 37. TFA·H-Leu-VS was converted to its isocyanate by phosgene and DiPEA in DCM15 and subsequently reacted with amine 33 which resulted in tetrapeptide 40. Amine 33 was converted to the isocyanate by phosgene and sat. aq. NaHCO339 and reacted with amine 18 which resulted in tetrapeptide 42. Compounds 40 and 42 were subjected to the competition assay in HEK lysate versusMVB003 (Fig. 9). Only at high concentrations (100 μM), these two compounds show limited proteasome inhibition. Apparently, the place of the ureido-linkage in the peptide inhibitor determines its activity and selectivity for proteasome subunits. The lack of activity could also be the result of ‘inversed’ amino acid side chain configuration caused by chain reversal due to the ureido linkage. Substitution for D-amino acids at P2-4 for 40 or P3 and P4 for 42 might restore activity of these scaffolds.
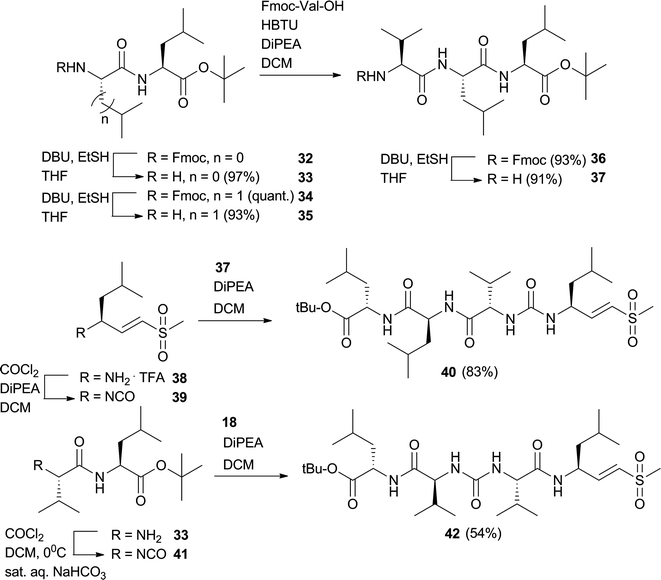 | ||
| Fig. 8 Synthesis of two potential tetrapeptide proteasome inhibitors with ureido-linkage after P1 or P2. | ||
 | ||
| Fig. 9 Competition assay in HEK lysate (15 μg protein). Lysates were incubated with indicated end concentration of inhibitor for 1 h at 37 °C. Residual proteasome activity was labeled by MVB003 (0.5 μM end concentration) for 1 h at 37 °C. Upper band β2 (■), middle band β1 (◀), lower band β5 (●). | ||
3 Conclusions
Incorporating the exocyclic valine-urea-valine motif, which is found in syringolin A, into a vinyl sulfone or epoxyketone oligopeptide results in a new set of potent proteasome inhibitors. In general, epoxyketone containing inhibitors are more potent than their vinyl sulfone counterparts, but this increase in potency is often accompanied by a decrease of subunit selectivity, a phenomenon observed earlier.27,29,36 The position of the ureido linkage with respect to the electrophilic trap has a profound effect on proteasome subunit selectivity. A short distance, as in compounds 1–4 and 5–8 results in preference for the β1 subunit of the proteasome. Tripeptide epoxyketone 5 is a potent and very selective inhibitor of the β1 subunit. In living cells, this compound is fivefold more potent than NC001 and is the most potent β1 selective compound known to date. Further optimisation of this compound, for example by changing the P2 valine for a norleucine residue or changing the tert-butyl for a smaller methyl group, might make this molecule even more selective for β1. When an extra amino acid is incorporated in between the ureido linkage and the warhead, as in inhibitors 9–12 and 13–16, a preference for the β5 subunit is observed, with tetrapeptide epoxyketones 13 and 15 being quite selective inhibitors of the β5 site. When the ureido linkage is incorporated at different places in the peptide, two more compounds are obtained, 40 and 42, but both are hardly active as a proteasome inhibitors. Tetrapeptide epoxyketones 13 and 15 are the most potent compounds in this library, more potent than the parent compound SylA. The subunit selectivity pattern of 15 shows similarities to that of SylA, but displays higher potency, indicating that the antitumour activity of this compound should be assessed. The activities of the tetrapeptide inhibitors in this study resemble those of compounds Z-L3-EK and Z-L3-VS in terms of selectivity. Z-L3-EK and Z-L3-VS have a slight preference for β5 while the tetrapeptides in this study appear to possess more β5 selectivity.29 Translating the findings of this study back to the case of syringolin A meets difficulties. One could envision that the molecular basis of selectivity of 5, 13 and 15 could be unraveled with the crystal structures of these compounds in complex with 20S proteasomes.5,40,414 Experimental
4.1 Synthesis
All reagents were commercial grade and were used as received unless indicated otherwise. Toluene (Tol.) (purum), ethyl acetate (EtOAc) (puriss.), and light petroleum ether (PetEt) (puriss.) were obtained from Riedel-de Haën or Biosolve and were distilled prior to use. Dichloromethane (DCM), dimethylformamide (DMF), and dioxane were stored on 4 Å molecular sieves. Tetrahydrofuran (THF) was distilled from LiAlH4 prior to use. Reactions were conducted under an argon atmosphere. Reactions were monitored by TLC analysis by using DC-fertigfolien (Schleicher & Schuell, F1500, LS254) with detection by UV absorption (254 nm), spraying with 20% H2SO4 in ethanol or (NH4)6Mo7O24C4H2O (25 g L−1) and (NH4)4Ce(SO4)4C2H2O (10 g L−1) in 10% sulfuric acid followed by charring at ∼150 °C or by spraying with an aqueous solution of KMnO4 (7%) and KOH (2%). Column chromatography was performed on Screening devices (0.040–0.063 nm). LC/MS analysis was performed on a LCQ AdvantageMax (Thermo Finnigan) equipped with an Gemini C18 column (Phenomenex). HRMS were recorded on a LTQ Orbitrap (Thermo Finnigan). 1H- and 13C-APT-NMR spectra were recorded on a Bruker DPX-300 (300/75 MHz) or Bruker AV-400 (400/100 MHz) equipped with a pulsed field gradient accessory. Chemical shifts are given in ppm (δ) relative to tetramethylsilane as an internal standard. Coupling constants are given in Hz. All presented 13C-APT spectra are proton decoupled. TFA.H-Leu-EK2 and TFA.H-Leu-VS32 were obtained by literature procedures. NC001 and NC005-mvs were obtained as described.26,36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM:sat. aq. NaHCO3 at 0 °C. The layers were allowed to separate and phosgene (1.2 equiv., 20 wt-% in Tol.) was added to the DCM layer and stirring was continued for 10 min after which the mixture was extracted with DCM (3×). The organic layers were combined and dried over Na2SO4 and concentrated. The resulting crude isocyanate was used without further purification.
:EA 3:1 and cooled to −30° C. tBuONO (1.1 equiv.) and HCl (4 M in dioxane, 2.8 equiv.) were added and the resulting mixture stirred for 3 h. TFA·H-Leu-EK or TFA·H-Leu-VS (1.1 equiv.) in DMF was added followed by DiPEA (5 equiv.) and the mixture was allowed to warm to RT overnight. The mixture was diluted with EA and washed with H2O (3×). The organic layer was dried over Na2SO4 and concentrated before flash column chromatography.
:PE→60% EA:PE) to yield the title compound (587 mg, 1.5 mmol, quant.). 1H-NMR NMR (400 MHz, CDCl3): δ 6.88 (d, J = 6.6 Hz, 1H), 6.79 (dd, J1 = 15.1, J2 = 5.3 Hz, 1H), 6.52 (d, J = 15.1 Hz, 1H), 5.33 (d, J = 7.5 Hz, 1H), 4.87–4.62 (m, 1H), 3.86 (dd, J1 = 8.3, J2 = 7.1 Hz, 1H), 2.89 (s, 3H), 2.15–2.00 (m, 1H), 1.70–1.56 (m, 1H), 1.40 (s, 9H), 0.96–0.81 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 171.63, 155.89, 147.63, 129.16, 79.80, 60.25, 47.65, 42.62, 42.48, 30.17, 28.15, 24.50, 22.63, 21.65, 19.27, 17.85.
:1 DCM:TFA for 30 min before coevaporation with toluene (3×) yielded the title compound, which was immediately used in the next reaction without further purification.
:PE→70% EA:PE) yielded the title compound (119 mg, 243 μmol, 80%). LCMS: Rt 10.28 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42 (d, J = 7.98 Hz, 1H), 6.80 (dd, J1 = 15.06, J2 = 5.76 Hz, 1H), 6.70 (d, J = 15.16 Hz, 1H), 6.34 (d, J = 7.99 Hz, 1H), 6.00 (d, J = 8.97 Hz, 1H), 4.76–4.67 (m, 1H), 4.24 (dd, J1 = 8.82, J2 = 5.29 Hz, 1H), 4.03 (t, J = 8.40, 1H), 2.93 (s, 3H), 2.13–1.94 (m, 2H), 1.73–1.58 (m, 1H), 1.47 (s, 9H), 1.46–1.38 (m, 2H), 0.97–0.84 (m, 18H). 13C NMR (100 MHz, CDCl3): δ (ppm) 219.35, 172.97, 172.34, 158.28, 147.04, 129.88, 81.59, 60.16, 58.69, 48.02, 42.66, 42.53, 31.37, 30.76, 28.04, 24.60, 22.56, 22.08, 19.47, 18.99, 18.56, 17.98. HRMS calcd. for [C23H44N3O6S1]+ 490.29453, found 490.29445.
:DCM yielded the title compound (43 mg, 81 μmol, 36%). LCMS: Rt 7.53 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3/CD3OD 1/1): δ (ppm) 7.36–7.21 (m, 5H), 6.81 (dd, J1 = 15.13, J2 = 5.17 Hz, 1H), 6.59 (dd, J1 = 15.12, J2 = 1.49 Hz, 1H), 4.75–4.65 (m, 1H), 4.41 (q, J = 14.94, 2H), 4.02 (d, J = 6.69 Hz, 1H), 3.96 (d, J = 6.64 Hz, 1H), 2.96 (s, 3H), 2.06 (m, 2H), 1.74–1.61 (m, 1H), 1.59–1.37 (m, 2H), 1.02–0.84 (m, 18H).). 13C NMR (100 MHz, CDCl3/CD3OD): δ (ppm) 172.64, 147.33, 128.73, 128.00, 127.02, 126.78, 59.24, 58.95, 47.35, 42.71, 41.92, 41.84, 30.66, 30.25, 24.22, 22.08, 20.90, 18.70, 18.60, 17.14, 17.09. HRMS calcd. for [C26H43N4O5S1]+ 523.29487, found 523.29478.
:PE→80% EA:PE) yielded the title compound (85 mg, 179 μmol, 67%). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.28 (d, J = 7.54 Hz, 1H), 6.88 (dd, J1 = 15.10, J2 = 6.04 Hz, 1H), 6.68 (d, J = 14.98 Hz, 1H), 6.55–6.43 (m, 1H), 5.91–5.83 (m, 1H), 4.77–4.64 (m, 1H), 4.12–4.04 (m, 1H), 3.24–3.09 (m, 1H), 3.05–2.95 (m, 1H), 2.93 (s, 1H), 2.02–1.85 (m, 1H), 1.72–1.58 (m, 1H), 1.54–1.35 (m, 5H), 1.33–1.21 (m, 16H), 1.02–0.81 (m, 12H). 13C NMR (100 MHz, CDCl3): δ (ppm) 173.49, 158.70, 147.46, 129.73, 60.05, 47.94, 42.73, 42.69, 40.42, 31.87, 31.35, 30.47, 29.64, 29.60, 29.52, 29.32, 27.06, 24.66, 22.75, 22.64, 22.25, 19.52, 18.82, 14.06. HRMS calcd. for [C24H48N3O4S1]+ 474.33600, found 474.33595.
:DCM, 2×) yielded the title compound (22 mg, 47 μmol, 47%). LCMS: Rt 8.93 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ (ppm) 6.49 (d, J = 7.55 Hz, 1H), 5.51 (d, J = 8.52 Hz, 1H), 5.39 (d, J = 8.82 Hz, 1H), 4.60 (ddd, J1 = 10.60, J2 = 7.72, J3 = 3.23 Hz, 1H), 4.26 (dd, J1 = 8.78, J2 = 4.62 Hz, 1H), 4.08 (dd, J1 = 8.56, J2 = 7.07 Hz, 1H), 3.36 (d, J = 4.98 Hz, 1H), 2.88 (d, J = 4.97 Hz, 1H), 2.15–1.93 (m, 2H), 1.76–1.59 (m, 2H), 1.46 (s, 9H), 1.51 (s, 3H), 1.58–1.51 (m, 1H), 0.95–0.82 (m, 18H). 13 C NMR (100 MHz, CDCl3): δ (ppm) 208.44, 172.72, 172.24, 157.72, 81.63, 59.27, 59.11, 58.47, 52.48, 50.41, 39.97, 31.49, 31.18, 29.71, 28.08, 25.13, 23.36, 21.33, 19.27, 18.98, 18.10, 17.65, 16.78. HRMS calcd. for [C24H44N3O6]+ 470.32246, found 470.32233.
:DCM) yielded the title compound (230 mg, 496 μmol, 53%). LCMS: Rt 7.08 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min), (ESI-MS (m/z): 464.00 (M + H)+). 1H NMR (400 MHz, CD3OD, CDCl3): δ 7.89 (t, J = 5.7 Hz, 1H), 7.37–7.18 (m, 5H), 4.43–4.29 (m, 2H), 4.02 (d, J = 6.5 Hz, 1H), 3.96 (d, J = 6.7 Hz, 1H), 2.19–1.91 (m, 2H), 1.44 (s, 9H), 1.00–0.83 (m, 12H). 13C NMR (100 MHz, CD3OD, CDCl3): δ 172.70, 158.11, 137.59, 127.78, 126.82, 126.52, 58.93, 57.01, 42.46, 30.72, 30.39, 27.16, 18.37, 18.27, 16.97.
:DCM) yielded the title compound (10 mg, 20 μmol, 20%). LCMS: Rt 8.41 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H-NMR (400 MHz, CDCl3:CD3OD): δ 7.43–7.18 (m, 5H), 4.54 (dd, J1 = 10.6, J2 = 3.1 Hz, 1H), 4.37 (dd, J1 = 36.1, J2 = 14.9 Hz, 2H), 4.04 (dd, J1 = 6.7, J2 = 4.9 Hz, 2H), 3.29 (d, J = 5.0 Hz, 1H), 2.91 (d, J = 5.1 Hz, 1H), 2.09–1.93 (m, 2H), 1.80–1.64 (m, 1H), 1.48 (s, 3H), 1.57–1.21 (m, 1H), 0.99–0.85 (m, 18H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 208.39, 173.42, 173.29, 158.71, 138.24, 128.21, 127.28, 126.93, 59.28, 58.85, 58.71, 51.96, 50.41, 42.84, 39.06, 31.08, 31.05, 24.94, 22.69, 20.49, 18.74, 18.71, 17.30, 16.05. HRMS calcd. for [C27H43N4O5]+ 503.32280, found 503.32271.
:PE) yielded the title compound (250 mg, 600 μmol, 60%). LCMS: Rt 10.23 min (linear gradient 10 →90% ACN + 0.1% TFA, 15 min), (ESI-MS (m/z): 415.00 (M + H)+). 1H NMR (400 MHz, CD3OD, CDCl3): δ 6.03 (d, J = 8.8 Hz, 1H), 5.98 (t, J = 5.2 Hz, 1H), 5.56 (t, J = 5.1 Hz, 1H), 4.07 (s, 1H), 3.22–2.99 (m, 2H), 2.09–2.00 (m, 1H), 1.47 (s, 9H), 1.34–1.20 (m, 16H), 0.98 (dd, J1 = 14.5, J2 = 6.8 Hz, 6H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD, CDCl3): δ 172.75, 158.61, 57.04, 39.53, 31.38, 30.71, 29.71, 29.61, 29.10, 29.06, 28.89, 28.80, 27.43, 26.40, 22.12, 18.52, 17.27, 13.36.
:DCM and 10% EA:Tol. → 30% EA:Tol.) yielded the title compound (24 mg, 53 μmol, 35%). LCMS: Rt 11.18 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ 6.84 (d, J = 7.4 Hz, 1H), 5.56 (d, J = 8.5 Hz, 1H), 5.03 (t, J = 5.6 Hz, 1H), 4.71–4.43 (m, 1H), 4.10 (t, J = 7.8 Hz, 1H), 3.35 (d, J = 5.0 Hz, 1H), 3.25–3.00 (m, 2H), 2.89 (d, J = 5.0 Hz, 1H), 2.10–1.85 (m, 1H), 1.52 (s, 3H), 1.73–1.36 (m, 3H), 1.40–1.16 (m, 16H), 0.97–0.80 (m, 15H). 13C NMR (100 MHz, CDCl3): δ 208.37, 173.26, 158.22, 59.27, 59.15, 52.41, 50.56, 40.75, 40.57, 39.63, 31.88, 31.40, 30.28, 30.20, 29.68, 29.57, 29.40, 29.34, 29.31, 26.95, 25.16, 23.33, 22.66, 21.29, 19.19, 18.23, 16.78, 14.07. HRMS calcd. for [C25H48N3O4]+ 454.36393, found 454.36392.
:Tol.) yielded the title compound (362 mg, 719 μmol, 72%). 1H NMR (400 MHz, CDCl3): δ 7.39–7.22 (m, 1H), 6.97 (d, J = 6.8 Hz, 1H), 6.85 (dd, J1 = 15.1, J2 = 5.3 Hz, 1H), 6.61 (d, J = 15.1 Hz, 1H), 5.45 (d, J = 6.6 Hz, 1H), 4.85–4.69 (m, 1H), 4.57–4.42 (m, 1H), 3.92 (t, J = 6.7 Hz, 1H), 2.94 (s, 3H), 2.14 (dq, J1 = 13.5, J2 = 6.8 Hz, 1H), 1.76–1.39 (m, 6H), 1.45 (s, 9H), 0.97–0.91 (m, 18H). 13C NMR (100 MHz, CDCl3): δ 172.02, 171.57, 156.30, 147.44, 129.36, 80.22, 60.69, 52.16, 47.72, 42.76, 42.71, 40.41, 30.09, 28.25, 24.87, 24.61, 22.83, 22.58, 21.91, 19.23, 17.94.
:Tol. → 60% EA:Tol.) yielded the title compound (437 mg, 903 μmol, 90%). 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 7.1 Hz, 1H), 7.44 (d, J = 6.9 Hz, 1H), 5.95 (d, J = 9.0 Hz, 1H), 4.70–4.45 (m, 2H), 4.05 (t, J = 8.5 Hz, 1H), 3.45 (d, J = 4.2 Hz, 1H), 2.87 (d, J = 5.0 Hz, 1H), 2.09–1.94 (m, 1H), 1.75–1.30 (m, 6H), 1.51 (s, 3H), 1.45 (s, 9H), 0.98–0.82 (m, 18H). 13C NMR (100 MHz, CDCl3): δ 208.57, 172.02, 155.86, 79.18, 59.67, 59.13, 52.28, 51.33, 50.09, 41.00, 39.16, 30.99, 28.32, 24.83, 24.39, 23.21, 22.75, 22.32, 19.02, 18.38, 16.66.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (40 μmol, 244 μmol, 2.4 equiv.) was added and this mixture was added to a solution of valine tert-butyl ester isocyanate (prepared from HCl·H-Val-OtBu (23 mg, 110 μmol, 1.1 equiv.) using general procedure A). The resulting mixture was stirred for 30 min before being washed with 1 M HCl and sat. aq. NaHCO3 and dried over MgSO4. Column chromatography (DCM → 1% MeOH:DCM) afforded the title compound (42 mg, 70 μmol, 70%). LCMS: Rt 8.80 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3/CD3OD 1/1): δ (ppm) 6.81 (dd, J1 = 15.13, J2 = 4.91 Hz, 1H), 6.62 (dd, J1 = 15.14, J2 = 1.36 Hz, 1H), 4.74–4.63 (m, 1H), 4.39 (t, J = 7.37, 1H), 4.04 (d, J = 5.13 Hz, 1H), 3.95 (d, J = 5.78 Hz, 1H), 2.97 (s, 3H), 2.16–2.03 (m, 2H), 1.73–1.57 (m, 6H), 1.47 (s, 1H), 1.04–0.88 (m, 24H). 13C NMR (100 MHz, CDCl3/CD3OD): δ (ppm) 173.00, 172.27, 171.43, 158.72, 146.50, 128.55, 80.62, 59.40, 58.16, 51.38, 47.07, 41.18, 41.06, 39.34, 29.89, 29.78, 26.57, 23.94, 23.84, 21.59, 21.55, 20.09, 19.95, 17.97, 17.80, 16.43, 16.37. HRMS calcd. for [C29H55N4O7S]+ 603.37860, found 603.37866.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (37 μl, 222 μmol, 2.2 equiv.) was added and this mixture was added to a solution of valine benzylamide isocyanate (prepared from HCl·H-Val-NHBn (35 mg, 144 μmol, 1.4 equiv.) using general procedure B). The resulting mixture was stirred overnight. Upon quenching with H2O the title compound precipitated as a white solid (15 mg, 23 μmol, 23%). LCMS: Rt 7.88 min (linear gradient 10 →90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3:CD3OD): δ 7.37–7.15 (m, 5H), 6.83 (dd, J1 = 15.1, J2 = 5.3 Hz, 6H), 6.59 (dd, J1 = 15.1, J2 = 1.2 Hz, 6H), 4.58–4.24 (m, 4H), 4.08 (dd, J1 = 15.4, J2 = 7.1 Hz, 2H), 2.93 (s, 3H), 2.20–1.88 (m, 2H), 1.80–1.53 (m, 4H), 1.54–1.13 (m, 2H), 1.19–0.59 (m, 24H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 173.14, 172.46, 172.09, 158.43, 147.20, 137.61, 128.53, 127.92, 126.89, 126.84, 126.65, 59.16, 58.76, 51.60, 47.28, 42.51, 41.78, 41.68, 39.81, 30.97, 30.57, 24.29, 24.06, 22.07, 21.97, 21.01, 20.83, 18.53, 18.47, 17.40, 17.32. HRMS calcd. for [C32H54N5O6S]+ 636.37893, found 636.37909.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (36 mmol, 220 μmol, 2.2 equiv.) was added and this mixture was added to a solution of decyl isocyanate (prepared from decylamine (24 mg, 160 μmol, 1.6 equiv.) using general procedure A). The resulting mixture was stirred for 3 h before being washed with 1 M HCl (3×) and H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 2% MeOH:DCM) afforded the title compound (25 mg, 43 μmol, 43%). LCMS: Rt 10.91 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min).1H NMR (400 MHz, CD3OD): δ (ppm) 6.82 (dd, J1 = 15.2, J2 = 4.9 Hz, 1H), 6.66 (dd, J1 = 15.2, J2 = 1.5 Hz, 1H), 4.74–4.66 (m, 1H), 4.42 (dd, J1 = 9.6, J2 = 5.3 Hz, 1H), 3.97 (d, J = 5.8 Hz, 1H), 3.23–3.06 (m, 2H), 3.00 (s, 3H), 2.16–2.02 (m, 1H), 1.80–1.57 (m, 5H), 1.54–1.43 (m, 3H), 1.39–1.27 (m, 14H), 1.08–0.87 (m, 21H). 13C NMR (100 MHz, CD3OD): δ 175.43, 174.52, 161.13, 148.39, 130.83, 61.47, 53.45, 43.22, 42.81, 41.27, 41.06, 33.07, 31.87, 31.30, 30.76, 30.70, 30.52, 30.46, 28.02, 26.01, 25.86, 23.74, 23.47, 23.42, 21.90, 21.69, 19.85, 18.30, 14.45. HRMS calcd. for [C30H59N4O5S]+ 587.42007, found 587.42017.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (31 μl, 187 μmol, 2.2 equiv.) was added and this mixture was added to a solution of valine tert-butyl ester isocyanate (prepared from HCl·H-Val-OtBu (27 mg, 128 μmol, 1.6 equiv.) using general procedure A). The resulting mixture was stirred overnight. The reaction mixture was washed with 1 M HCl (3×), H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 2% MeOH:DCM yielded the title compound (14 mg, 24 μmol, 28%). LCMS: Rt 9.84 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.02 (s, 1H) 6.49 (d, J = 7.5 Hz, 1H), 6.19 (s, 1H), 4.74–4.60 (m, 1H), 4.53 (dd, J1 = 14.7, J2 = 7.4 Hz, 1H), 4.38 (dd, J1 = 9.2, J2 = 4.9 Hz, 1H), 4.25 (t, J = 8.3 Hz, 1H), 3.41 (d, J = 4.8 Hz, 1H), 2.87 (d, J = 4.8 Hz, 1H), 2.09–1.95 (m, 1H), 1.90–1.30 (m, 7H), 1.52 (s, 3H), 1.49 (s, 1H), 1.01–0.74 (m, 24H). 13C NMR (100 MHz, CDCl3): δ (ppm) 173.37, 172.83, 171.92, 157.73, 81.41, 59.13, 58.85, 58.11, 52.56, 52.01, 50.32, 40.96, 39.12, 32.42, 31.96, 28.11, 24.92, 24.46, 23.37, 23.09, 22.17, 21.09, 19.25, 18.99, 18.74, 18.07, 16.89. HRMS calcd. for [C30H55N4O7]+ 583.40653 found 583.40668.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (37 μl, 222 μmol, 2.2 equiv.) was added and this mixture was added to a solution of valine benzylamide isocyanate (prepared from HCl·H-Val-NHBn (34 mg, 144 μmol, 1.4 equiv.) using general procedure B). The resulting mixture was stirred for 3 h. The reaction mixture was washed with H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 4% MeOH:DCM) followed by crystallisation (DCM:PE) yielded the title compound (25 mg, 40 μmol, 40%). LCMS: Rt 8.51 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CD3OD): δ 7.35–7.18 (m, 5H), 6.57 (dd, J1 = 8.9, J2 = 6.1 Hz, 1H), 4.58–4.49 (m, 2H), 4.44 (d, J = 15.0 Hz, 1H), 4.33 (d, J = 14.9 Hz, 1H), 4.18–4.04 (m, 2H), 3.25 (d, J = 5.0 Hz, 1H), 2.89 (d, J = 5.0 Hz, 1H), 2.05–1.88 (m, 2H), 1.77–1.24 (m, 6H), 1.48 (s, 3H), 0.99–0.83 (m, 24H). 13C NMR (100 MHz, CD3OD, CDCl3): δ 207.71, 172.54, 172.42, 172.22, 157.96, 137.61, 127.50, 126.59, 126.20, 58.39, 58.15, 58.08, 51.24, 50.56, 49.71, 42.09, 39.95, 38.11, 30.74, 30.69, 24.17, 23.68, 22.01, 21.45, 20.82, 19.69, 18.06, 16.87, 16.79, 15.34. HRMS calcd. for [C33H54N5O6]+ 616.40686 found 616.40698.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (37 μl, 222 μmol, 2.2 equiv.) was added and this mixture was added to a solution of decyl isocyanate (prepared from decylamine (24 μl, 121 μmol, 1.2 equiv.) using general procedure A). The resulting mixture was stirred overnight. The reaction mixture was washed with 1 M HCl (3×), H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 2% MeOH:DCM yielded the title compound (16 mg, 29 μmol, 29%). LCMS: Rt 11.30 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CD3OD, CDCl3): δ (ppm) 4.55–4.51 (m, 1H), 4.43 (dd, J1 = 8.9, J2 = 5.5 Hz, 1H), 4.03 (d, J = 6.5 Hz, 1H), 3.29 (d, J = 5.0 Hz, 1H), 3.20–2.99 (m, 2H), 2.90 (d, J = 5.0 Hz, 1H), 2.11–1.92 (m, 1H), 1.77–1.32 (m, 6H), 1.49 (s, 3H), 1.39–1.10 (m, 16H), 1.05–0.78 (m, 21H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 208.04, 172.86, 172.33, 158.66, 58.57, 58.53, 51.76, 50.85, 49.99, 40.15, 39.50, 38.69, 31.34, 30.60, 29.65, 29.60, 29.04, 29.00, 28.84, 28.75, 26.34, 24.55, 24.04, 22.54, 22.07, 22.00, 21.11, 20.28, 18.50, 17.02, 15.97, 13.25. HRMS calcd. for [C25H47N4O7S]+ 547.31600 found 547.31610.
:PE:MeOH 50:50:0 → 100:0:0 → 95:0:5) yielded the title compound (524 mg, 1.83 mmol, 97%). 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.5 Hz, 1H), 4.57–4.42 (m, 1H), 3.27 (d, J = 4.0 Hz, 1H), 2.35–2.19 (m, 1H), 2.05 (s, 2H), 1.80–1.49 (m, 3H), 1.46 (s, 9H), 0.99 (d, J = 7.0 Hz, 3H), 0.97–0.89 (m, 6H), 0.85 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 173.83, 171.91, 81.09, 59.76, 50.58, 41.23, 30.57, 27.57, 24.56, 22.48, 21.51, 19.23, 15.87.
:Tol. 0 → 16%) afforded the title compound (1.09 g, 2.04 mmol, quant.). 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 7.5 Hz, 2H), 7.56 (t, J = 7.0 Hz, 2H), 7.40–7.29 (m, 2H), 7.23 (t, J = 7.0 Hz, 2H), 7.13 (d, J = 7.6 Hz, 1H), 7.02 (d, J = 7.9 Hz, 1H), 5.99 (d, J = 8.7 Hz, 1H), 4.49 (dd, J1 = 13.8, J2 = 7.9 Hz, 1H), 4.43–4.33 (m, 2H), 4.32–4.24 (m, 1H), 4.20–4.13 (m, 1H), 1.80–1.46 (m, 6H), 1.43 (s, 9H), 0.98–0.83 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 172.11, 171.62, 156.05, 143.65, 143.50, 140.97, 127.38, 126.79, 124.89, 119.64, 81.33, 66.80, 53.11, 51.27, 46.84, 41.53, 41.18, 27.69, 24.60, 24.38, 22.77, 22.44, 21.88.
:PE:MeOH 50:50:0 → 100:0:0 → 95:0:5) yielded the title compound (586 mg, 1.95 mmol, 93%). 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.6 Hz, 1H), 4.46 (dt, J1 = 8.7, J2 = 5.2 Hz, 1H), 3.42 (dd, J1 = 9.6, J2 = 4.2 Hz, 1H), 1.87 (s, 2H), 1.81–1.29 (m, 6H), 1.46 (s, 9H), 1.07–0.81 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 175.06, 171.77, 80.92, 53.03, 50.39, 43.69, 41.17, 27.46, 24.42, 24.28, 22.93, 22.39, 21.53, 20.95.
:Tol. 0 → 30%) yielded the title compound (1.15 g, 1.85 mmol, 95%). 1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 7.5 Hz, 2H), 7.64–7.52 (m, 2H), 7.37 (dd, J1 = 9.5, J2 = 5.1 Hz, 2H), 7.30–7.22 (m, 2H), 7.15 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 8.2 Hz, 1H), 6.11 (d, J = 9.1 Hz, 1H), 4.66 (dd, J1 = 15.1, J2 = 7.5 Hz, 1H), 4.53 (dd, J1 = 13.5, J2 = 8.5 Hz, 1H), 4.44 (dd, J1 = 10.1, J2 = 7.3 Hz, 1H), 4.30–4.22 (m, 1H), 4.19 (t, J = 7.2 Hz, 1H), 4.12 (t, J = 8.2 Hz, 1H), 2.15–1.99 (m, 1H), 1.77–1.46 (m, 6H), 1.43 (s, 9H), 0.94 (t, J = 6.4 Hz, 6H), 0.86 (dd, J1 = 10.1, J2 = 6.2 Hz, 12H). 13C NMR (100 MHz, CDCl3): δ 171.88, 171.45, 171.35, 156.47, 143.82, 141.20, 127.60, 126.99, 125.21, 125.13, 119.86, 81.70, 67.07, 60.31, 51.58, 51.29, 47.09, 41.53, 41.31, 31.46, 27.91, 24.74, 24.60, 22.83, 22.52, 21.96, 19.05, 18.21.
:PE:MeOH 50:50:0 → 100:0:0 → 95:0:5) to yield the title compound (672 mg, 1.68 mmol, 91%) 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 8.8 Hz, 3H), 7.40 (d, J = 7.7 Hz, 3H), 4.74–4.58 (m, 3H), 4.40 (dd, J = 13.9, 7.9 Hz, 3H), 3.23 (d, J = 4.1 Hz, 3H), 2.36–2.18 (m, 3H), 1.74–1.49 (m, 6H), 1.46 (s, 9H), 0.98 (d, J = 6.9 Hz, 3H), 0.95–0.87 (m, 12H), 0.84 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 174.15, 171.75, 171.41, 80.93, 59.87, 51.25, 50.67, 41.00, 40.87, 30.63, 27.60, 24.46, 24.33, 22.64, 22.31, 21.86, 21.83, 19.31, 15.94.
:1 TFA:DCM for 30 min. before being coevaporated with toluene (3×). The residue was dissolved in DCM and treated with the isocyanate of Val-Leu-OtBu 41 (obtained from H-Val-Leu-OtBu 33 (54 mg, 188 μmol, 1.15 equiv.) employing general procedure B) and DiPEA (60 μl, 361 μmol, 2.2 equiv.). The mixture was stirred for 2 h before being washed with 1 M HCl (2×) and dried over MgSO4. Column chromatography (MeOH:DCM 1 → 3%) yielded the title compound (54 mg, 90 μmol, 54%). LCMS: Rt 9.02 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CD3OD): δ 6.81 (dd, J1 = 15.2, J2 = 5.4 Hz, 1H), 6.68 (d, J = 15.2 Hz, 1H), 6.57–6.39 (m, 1H), 4.83–4.66 (m, 1H), 4.36 (t, J = 7.5 Hz, 1H), 4.15 (t, J = 6.5 Hz, 1H), 4.05 (t, J = 6.4 Hz, 1H), 2.98 (s, 3H), 2.04 (dq, J = 13.0, 6.5 Hz, 2H), 1.71 (td, J1 = 13.3, J2 = 6.6 Hz, 2H), 1.67–1.53 (m, 4H), 1.47 (s, 9H), 1.15–0.82 (m, 24H). 13C NMR (100 MHz, CD3OD): δ 174.66, 174.59, 173.20, 148.49, 130.97, 82.56, 60.91, 60.84, 60.00, 52.90, 49.12, 43.34, 42.84, 41.66, 32.63, 32.30, 28.27, 25.91, 25.83, 23.37, 23.29, 22.10, 22.06, 20.00, 18.54, 18.49. HRMS: calcd. for [C29H55N4O7S]+ 603.37860 found 603.37881
:DCM 0 → 3%) yielded the title compound (113 mg, 183 μmol, 83%). LCMS: Rt 9.40 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3, CD3OD): δ 7.93 (t, J = 7.9 Hz, 2H), 6.84 (dd, J1 = 15.1, J2 = 5.1 Hz, 1H), 6.53 (d, J = 15.1 Hz, 1H), 6.31 (d, J = 8.0 Hz, 1H), 6.02 (d, J = 8.8 Hz, 1H), 4.55–4.42 (m, 2H), 4.35 (dd, J1 = 14.5, J2 = 8.1 Hz, 1H), 4.13–4.03 (m, 1H), 2.98 (s, 3H), 2.09–1.95 (m, 1H), 1.85–1.50 (m, 9H), 1.47 (s, 9H), 1.10–0.77 (m, 24H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 172.55, 172.15, 171.43, 157.68, 148.96, 127.92, 81.13, 51.14, 50.99, 48.16, 42.58, 41.69, 40.28, 40.11, 30.76, 26.96, 24.12, 23.96, 21.90, 20.96, 20.74, 18.39, 16.92. HRMS: calcd. for [C30H57N4O7S]+ 617.39425, found 617.39466.
:1 DCM:sat. aq. NaHCO3 at 0 °C. The layers were allowed to separate and phosgene (1.2 equiv., 20 wt-% in Tol.) was added to the DCM layer and stirring was continued for 10 min after which the mixture was extracted with DCM (3×). The organic layers were combined and dried over Na2SO4 and concentrated. The resulting crude isocyanate was used without further purification.
:EA 3:1 and cooled to −30° C. tBuONO (1.1 equiv.) and HCl (4 M in dioxane, 2.8 equiv.) were added and the resulting mixture stirred for 3 h. TFA·H-Leu-EK or TFA·H-Leu-VS (1.1 equiv.) in DMF was added followed by DiPEA (5 equiv.) and the mixture was allowed to warm to RT overnight. The mixture was diluted with EA and washed with H2O (3×). The organic layer was dried over Na2SO4 and concentrated before flash column chromatography.
:PE→60% EA:PE) to yield the title compound (587 mg, 1.5 mmol, quant.). 1H-NMR NMR (400 MHz, CDCl3): δ 6.88 (d, J = 6.6 Hz, 1H), 6.79 (dd, J1 = 15.1, J2 = 5.3 Hz, 1H), 6.52 (d, J = 15.1 Hz, 1H), 5.33 (d, J = 7.5 Hz, 1H), 4.87–4.62 (m, 1H), 3.86 (dd, J1 = 8.3, J2 = 7.1 Hz, 1H), 2.89 (s, 3H), 2.15–2.00 (m, 1H), 1.70–1.56 (m, 1H), 1.40 (s, 9H), 0.96–0.81 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 171.63, 155.89, 147.63, 129.16, 79.80, 60.25, 47.65, 42.62, 42.48, 30.17, 28.15, 24.50, 22.63, 21.65, 19.27, 17.85.
:1 DCM:TFA for 30 min before coevaporation with toluene (3×) yielded the title compound, which was immediately used in the next reaction without further purification.
:PE→70% EA:PE) yielded the title compound (119 mg, 243 μmol, 80%). LCMS: Rt 10.28 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42 (d, J = 7.98 Hz, 1H), 6.80 (dd, J1 = 15.06, J2 = 5.76 Hz, 1H), 6.70 (d, J = 15.16 Hz, 1H), 6.34 (d, J = 7.99 Hz, 1H), 6.00 (d, J = 8.97 Hz, 1H), 4.76–4.67 (m, 1H), 4.24 (dd, J1 = 8.82, J2 = 5.29 Hz, 1H), 4.03 (t, J = 8.40, 1H), 2.93 (s, 3H), 2.13–1.94 (m, 2H), 1.73–1.58 (m, 1H), 1.47 (s, 9H), 1.46–1.38 (m, 2H), 0.97–0.84 (m, 18H). 13C NMR (100 MHz, CDCl3): δ (ppm) 219.35, 172.97, 172.34, 158.28, 147.04, 129.88, 81.59, 60.16, 58.69, 48.02, 42.66, 42.53, 31.37, 30.76, 28.04, 24.60, 22.56, 22.08, 19.47, 18.99, 18.56, 17.98. HRMS calcd. for [C23H44N3O6S1]+ 490.29453, found 490.29445.
:DCM yielded the title compound (43 mg, 81 μmol, 36%). LCMS: Rt 7.53 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3/CD3OD 1/1): δ (ppm) 7.36–7.21 (m, 5H), 6.81 (dd, J1 = 15.13, J2 = 5.17 Hz, 1H), 6.59 (dd, J1 = 15.12, J2 = 1.49 Hz, 1H), 4.75–4.65 (m, 1H), 4.41 (q, J = 14.94, 2H), 4.02 (d, J = 6.69 Hz, 1H), 3.96 (d, J = 6.64 Hz, 1H), 2.96 (s, 3H), 2.06 (m, 2H), 1.74–1.61 (m, 1H), 1.59–1.37 (m, 2H), 1.02–0.84 (m, 18H).). 13C NMR (100 MHz, CDCl3/CD3OD): δ (ppm) 172.64, 147.33, 128.73, 128.00, 127.02, 126.78, 59.24, 58.95, 47.35, 42.71, 41.92, 41.84, 30.66, 30.25, 24.22, 22.08, 20.90, 18.70, 18.60, 17.14, 17.09. HRMS calcd. for [C26H43N4O5S1]+ 523.29487, found 523.29478.
:PE→80% EA:PE) yielded the title compound (85 mg, 179 μmol, 67%). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.28 (d, J = 7.54 Hz, 1H), 6.88 (dd, J1 = 15.10, J2 = 6.04 Hz, 1H), 6.68 (d, J = 14.98 Hz, 1H), 6.55–6.43 (m, 1H), 5.91–5.83 (m, 1H), 4.77–4.64 (m, 1H), 4.12–4.04 (m, 1H), 3.24–3.09 (m, 1H), 3.05–2.95 (m, 1H), 2.93 (s, 1H), 2.02–1.85 (m, 1H), 1.72–1.58 (m, 1H), 1.54–1.35 (m, 5H), 1.33–1.21 (m, 16H), 1.02–0.81 (m, 12H). 13C NMR (100 MHz, CDCl3): δ (ppm) 173.49, 158.70, 147.46, 129.73, 60.05, 47.94, 42.73, 42.69, 40.42, 31.87, 31.35, 30.47, 29.64, 29.60, 29.52, 29.32, 27.06, 24.66, 22.75, 22.64, 22.25, 19.52, 18.82, 14.06. HRMS calcd. for [C24H48N3O4S1]+ 474.33600, found 474.33595.
:DCM, 2×) yielded the title compound (22 mg, 47 μmol, 47%). LCMS: Rt 8.93 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ (ppm) 6.49 (d, J = 7.55 Hz, 1H), 5.51 (d, J = 8.52 Hz, 1H), 5.39 (d, J = 8.82 Hz, 1H), 4.60 (ddd, J1 = 10.60, J2 = 7.72, J3 = 3.23 Hz, 1H), 4.26 (dd, J1 = 8.78, J2 = 4.62 Hz, 1H), 4.08 (dd, J1 = 8.56, J2 = 7.07 Hz, 1H), 3.36 (d, J = 4.98 Hz, 1H), 2.88 (d, J = 4.97 Hz, 1H), 2.15–1.93 (m, 2H), 1.76–1.59 (m, 2H), 1.46 (s, 9H), 1.51 (s, 3H), 1.58–1.51 (m, 1H), 0.95–0.82 (m, 18H). 13 C NMR (100 MHz, CDCl3): δ (ppm) 208.44, 172.72, 172.24, 157.72, 81.63, 59.27, 59.11, 58.47, 52.48, 50.41, 39.97, 31.49, 31.18, 29.71, 28.08, 25.13, 23.36, 21.33, 19.27, 18.98, 18.10, 17.65, 16.78. HRMS calcd. for [C24H44N3O6]+ 470.32246, found 470.32233.
:DCM) yielded the title compound (230 mg, 496 μmol, 53%). LCMS: Rt 7.08 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min), (ESI-MS (m/z): 464.00 (M + H)+). 1H NMR (400 MHz, CD3OD, CDCl3): δ 7.89 (t, J = 5.7 Hz, 1H), 7.37–7.18 (m, 5H), 4.43–4.29 (m, 2H), 4.02 (d, J = 6.5 Hz, 1H), 3.96 (d, J = 6.7 Hz, 1H), 2.19–1.91 (m, 2H), 1.44 (s, 9H), 1.00–0.83 (m, 12H). 13C NMR (100 MHz, CD3OD, CDCl3): δ 172.70, 158.11, 137.59, 127.78, 126.82, 126.52, 58.93, 57.01, 42.46, 30.72, 30.39, 27.16, 18.37, 18.27, 16.97.
:DCM) yielded the title compound (10 mg, 20 μmol, 20%). LCMS: Rt 8.41 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H-NMR (400 MHz, CDCl3:CD3OD): δ 7.43–7.18 (m, 5H), 4.54 (dd, J1 = 10.6, J2 = 3.1 Hz, 1H), 4.37 (dd, J1 = 36.1, J2 = 14.9 Hz, 2H), 4.04 (dd, J1 = 6.7, J2 = 4.9 Hz, 2H), 3.29 (d, J = 5.0 Hz, 1H), 2.91 (d, J = 5.1 Hz, 1H), 2.09–1.93 (m, 2H), 1.80–1.64 (m, 1H), 1.48 (s, 3H), 1.57–1.21 (m, 1H), 0.99–0.85 (m, 18H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 208.39, 173.42, 173.29, 158.71, 138.24, 128.21, 127.28, 126.93, 59.28, 58.85, 58.71, 51.96, 50.41, 42.84, 39.06, 31.08, 31.05, 24.94, 22.69, 20.49, 18.74, 18.71, 17.30, 16.05. HRMS calcd. for [C27H43N4O5]+ 503.32280, found 503.32271.
:PE) yielded the title compound (250 mg, 600 μmol, 60%). LCMS: Rt 10.23 min (linear gradient 10 →90% ACN + 0.1% TFA, 15 min), (ESI-MS (m/z): 415.00 (M + H)+). 1H NMR (400 MHz, CD3OD, CDCl3): δ 6.03 (d, J = 8.8 Hz, 1H), 5.98 (t, J = 5.2 Hz, 1H), 5.56 (t, J = 5.1 Hz, 1H), 4.07 (s, 1H), 3.22–2.99 (m, 2H), 2.09–2.00 (m, 1H), 1.47 (s, 9H), 1.34–1.20 (m, 16H), 0.98 (dd, J1 = 14.5, J2 = 6.8 Hz, 6H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CD3OD, CDCl3): δ 172.75, 158.61, 57.04, 39.53, 31.38, 30.71, 29.71, 29.61, 29.10, 29.06, 28.89, 28.80, 27.43, 26.40, 22.12, 18.52, 17.27, 13.36.
:DCM and 10% EA:Tol. → 30% EA:Tol.) yielded the title compound (24 mg, 53 μmol, 35%). LCMS: Rt 11.18 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ 6.84 (d, J = 7.4 Hz, 1H), 5.56 (d, J = 8.5 Hz, 1H), 5.03 (t, J = 5.6 Hz, 1H), 4.71–4.43 (m, 1H), 4.10 (t, J = 7.8 Hz, 1H), 3.35 (d, J = 5.0 Hz, 1H), 3.25–3.00 (m, 2H), 2.89 (d, J = 5.0 Hz, 1H), 2.10–1.85 (m, 1H), 1.52 (s, 3H), 1.73–1.36 (m, 3H), 1.40–1.16 (m, 16H), 0.97–0.80 (m, 15H). 13C NMR (100 MHz, CDCl3): δ 208.37, 173.26, 158.22, 59.27, 59.15, 52.41, 50.56, 40.75, 40.57, 39.63, 31.88, 31.40, 30.28, 30.20, 29.68, 29.57, 29.40, 29.34, 29.31, 26.95, 25.16, 23.33, 22.66, 21.29, 19.19, 18.23, 16.78, 14.07. HRMS calcd. for [C25H48N3O4]+ 454.36393, found 454.36392.
:Tol.) yielded the title compound (362 mg, 719 μmol, 72%). 1H NMR (400 MHz, CDCl3): δ 7.39–7.22 (m, 1H), 6.97 (d, J = 6.8 Hz, 1H), 6.85 (dd, J1 = 15.1, J2 = 5.3 Hz, 1H), 6.61 (d, J = 15.1 Hz, 1H), 5.45 (d, J = 6.6 Hz, 1H), 4.85–4.69 (m, 1H), 4.57–4.42 (m, 1H), 3.92 (t, J = 6.7 Hz, 1H), 2.94 (s, 3H), 2.14 (dq, J1 = 13.5, J2 = 6.8 Hz, 1H), 1.76–1.39 (m, 6H), 1.45 (s, 9H), 0.97–0.91 (m, 18H). 13C NMR (100 MHz, CDCl3): δ 172.02, 171.57, 156.30, 147.44, 129.36, 80.22, 60.69, 52.16, 47.72, 42.76, 42.71, 40.41, 30.09, 28.25, 24.87, 24.61, 22.83, 22.58, 21.91, 19.23, 17.94.
:Tol. → 60% EA:Tol.) yielded the title compound (437 mg, 903 μmol, 90%). 1H NMR (400 MHz, CDCl3): δ 7.59 (d, J = 7.1 Hz, 1H), 7.44 (d, J = 6.9 Hz, 1H), 5.95 (d, J = 9.0 Hz, 1H), 4.70–4.45 (m, 2H), 4.05 (t, J = 8.5 Hz, 1H), 3.45 (d, J = 4.2 Hz, 1H), 2.87 (d, J = 5.0 Hz, 1H), 2.09–1.94 (m, 1H), 1.75–1.30 (m, 6H), 1.51 (s, 3H), 1.45 (s, 9H), 0.98–0.82 (m, 18H). 13C NMR (100 MHz, CDCl3): δ 208.57, 172.02, 155.86, 79.18, 59.67, 59.13, 52.28, 51.33, 50.09, 41.00, 39.16, 30.99, 28.32, 24.83, 24.39, 23.21, 22.75, 22.32, 19.02, 18.38, 16.66.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (40 μmol, 244 μmol, 2.4 equiv.) was added and this mixture was added to a solution of valine tert-butyl ester isocyanate (prepared from HCl·H-Val-OtBu (23 mg, 110 μmol, 1.1 equiv.) using general procedure A). The resulting mixture was stirred for 30 min before being washed with 1 M HCl and sat. aq. NaHCO3 and dried over MgSO4. Column chromatography (DCM → 1% MeOH:DCM) afforded the title compound (42 mg, 70 μmol, 70%). LCMS: Rt 8.80 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3/CD3OD 1/1): δ (ppm) 6.81 (dd, J1 = 15.13, J2 = 4.91 Hz, 1H), 6.62 (dd, J1 = 15.14, J2 = 1.36 Hz, 1H), 4.74–4.63 (m, 1H), 4.39 (t, J = 7.37, 1H), 4.04 (d, J = 5.13 Hz, 1H), 3.95 (d, J = 5.78 Hz, 1H), 2.97 (s, 3H), 2.16–2.03 (m, 2H), 1.73–1.57 (m, 6H), 1.47 (s, 1H), 1.04–0.88 (m, 24H). 13C NMR (100 MHz, CDCl3/CD3OD): δ (ppm) 173.00, 172.27, 171.43, 158.72, 146.50, 128.55, 80.62, 59.40, 58.16, 51.38, 47.07, 41.18, 41.06, 39.34, 29.89, 29.78, 26.57, 23.94, 23.84, 21.59, 21.55, 20.09, 19.95, 17.97, 17.80, 16.43, 16.37. HRMS calcd. for [C29H55N4O7S]+ 603.37860, found 603.37866.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (37 μl, 222 μmol, 2.2 equiv.) was added and this mixture was added to a solution of valine benzylamide isocyanate (prepared from HCl·H-Val-NHBn (35 mg, 144 μmol, 1.4 equiv.) using general procedure B). The resulting mixture was stirred overnight. Upon quenching with H2O the title compound precipitated as a white solid (15 mg, 23 μmol, 23%). LCMS: Rt 7.88 min (linear gradient 10 →90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3:CD3OD): δ 7.37–7.15 (m, 5H), 6.83 (dd, J1 = 15.1, J2 = 5.3 Hz, 6H), 6.59 (dd, J1 = 15.1, J2 = 1.2 Hz, 6H), 4.58–4.24 (m, 4H), 4.08 (dd, J1 = 15.4, J2 = 7.1 Hz, 2H), 2.93 (s, 3H), 2.20–1.88 (m, 2H), 1.80–1.53 (m, 4H), 1.54–1.13 (m, 2H), 1.19–0.59 (m, 24H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 173.14, 172.46, 172.09, 158.43, 147.20, 137.61, 128.53, 127.92, 126.89, 126.84, 126.65, 59.16, 58.76, 51.60, 47.28, 42.51, 41.78, 41.68, 39.81, 30.97, 30.57, 24.29, 24.06, 22.07, 21.97, 21.01, 20.83, 18.53, 18.47, 17.40, 17.32. HRMS calcd. for [C32H54N5O6S]+ 636.37893, found 636.37909.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (36 mmol, 220 μmol, 2.2 equiv.) was added and this mixture was added to a solution of decyl isocyanate (prepared from decylamine (24 mg, 160 μmol, 1.6 equiv.) using general procedure A). The resulting mixture was stirred for 3 h before being washed with 1 M HCl (3×) and H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 2% MeOH:DCM) afforded the title compound (25 mg, 43 μmol, 43%). LCMS: Rt 10.91 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min).1H NMR (400 MHz, CD3OD): δ (ppm) 6.82 (dd, J1 = 15.2, J2 = 4.9 Hz, 1H), 6.66 (dd, J1 = 15.2, J2 = 1.5 Hz, 1H), 4.74–4.66 (m, 1H), 4.42 (dd, J1 = 9.6, J2 = 5.3 Hz, 1H), 3.97 (d, J = 5.8 Hz, 1H), 3.23–3.06 (m, 2H), 3.00 (s, 3H), 2.16–2.02 (m, 1H), 1.80–1.57 (m, 5H), 1.54–1.43 (m, 3H), 1.39–1.27 (m, 14H), 1.08–0.87 (m, 21H). 13C NMR (100 MHz, CD3OD): δ 175.43, 174.52, 161.13, 148.39, 130.83, 61.47, 53.45, 43.22, 42.81, 41.27, 41.06, 33.07, 31.87, 31.30, 30.76, 30.70, 30.52, 30.46, 28.02, 26.01, 25.86, 23.74, 23.47, 23.42, 21.90, 21.69, 19.85, 18.30, 14.45. HRMS calcd. for [C30H59N4O5S]+ 587.42007, found 587.42017.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (31 μl, 187 μmol, 2.2 equiv.) was added and this mixture was added to a solution of valine tert-butyl ester isocyanate (prepared from HCl·H-Val-OtBu (27 mg, 128 μmol, 1.6 equiv.) using general procedure A). The resulting mixture was stirred overnight. The reaction mixture was washed with 1 M HCl (3×), H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 2% MeOH:DCM yielded the title compound (14 mg, 24 μmol, 28%). LCMS: Rt 9.84 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.02 (s, 1H) 6.49 (d, J = 7.5 Hz, 1H), 6.19 (s, 1H), 4.74–4.60 (m, 1H), 4.53 (dd, J1 = 14.7, J2 = 7.4 Hz, 1H), 4.38 (dd, J1 = 9.2, J2 = 4.9 Hz, 1H), 4.25 (t, J = 8.3 Hz, 1H), 3.41 (d, J = 4.8 Hz, 1H), 2.87 (d, J = 4.8 Hz, 1H), 2.09–1.95 (m, 1H), 1.90–1.30 (m, 7H), 1.52 (s, 3H), 1.49 (s, 1H), 1.01–0.74 (m, 24H). 13C NMR (100 MHz, CDCl3): δ (ppm) 173.37, 172.83, 171.92, 157.73, 81.41, 59.13, 58.85, 58.11, 52.56, 52.01, 50.32, 40.96, 39.12, 32.42, 31.96, 28.11, 24.92, 24.46, 23.37, 23.09, 22.17, 21.09, 19.25, 18.99, 18.74, 18.07, 16.89. HRMS calcd. for [C30H55N4O7]+ 583.40653 found 583.40668.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (37 μl, 222 μmol, 2.2 equiv.) was added and this mixture was added to a solution of valine benzylamide isocyanate (prepared from HCl·H-Val-NHBn (34 mg, 144 μmol, 1.4 equiv.) using general procedure B). The resulting mixture was stirred for 3 h. The reaction mixture was washed with H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 4% MeOH:DCM) followed by crystallisation (DCM:PE) yielded the title compound (25 mg, 40 μmol, 40%). LCMS: Rt 8.51 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CD3OD): δ 7.35–7.18 (m, 5H), 6.57 (dd, J1 = 8.9, J2 = 6.1 Hz, 1H), 4.58–4.49 (m, 2H), 4.44 (d, J = 15.0 Hz, 1H), 4.33 (d, J = 14.9 Hz, 1H), 4.18–4.04 (m, 2H), 3.25 (d, J = 5.0 Hz, 1H), 2.89 (d, J = 5.0 Hz, 1H), 2.05–1.88 (m, 2H), 1.77–1.24 (m, 6H), 1.48 (s, 3H), 0.99–0.83 (m, 24H). 13C NMR (100 MHz, CD3OD, CDCl3): δ 207.71, 172.54, 172.42, 172.22, 157.96, 137.61, 127.50, 126.59, 126.20, 58.39, 58.15, 58.08, 51.24, 50.56, 49.71, 42.09, 39.95, 38.11, 30.74, 30.69, 24.17, 23.68, 22.01, 21.45, 20.82, 19.69, 18.06, 16.87, 16.79, 15.34. HRMS calcd. for [C33H54N5O6]+ 616.40686 found 616.40698.
:1 TFA:DCM for 30 min, coevaporated with toluene (3×). The residue was dissolved in DCM and DiPEA (37 μl, 222 μmol, 2.2 equiv.) was added and this mixture was added to a solution of decyl isocyanate (prepared from decylamine (24 μl, 121 μmol, 1.2 equiv.) using general procedure A). The resulting mixture was stirred overnight. The reaction mixture was washed with 1 M HCl (3×), H2O (3×) and dried over Na2SO4. Column chromatography (DCM → 2% MeOH:DCM yielded the title compound (16 mg, 29 μmol, 29%). LCMS: Rt 11.30 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CD3OD, CDCl3): δ (ppm) 4.55–4.51 (m, 1H), 4.43 (dd, J1 = 8.9, J2 = 5.5 Hz, 1H), 4.03 (d, J = 6.5 Hz, 1H), 3.29 (d, J = 5.0 Hz, 1H), 3.20–2.99 (m, 2H), 2.90 (d, J = 5.0 Hz, 1H), 2.11–1.92 (m, 1H), 1.77–1.32 (m, 6H), 1.49 (s, 3H), 1.39–1.10 (m, 16H), 1.05–0.78 (m, 21H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 208.04, 172.86, 172.33, 158.66, 58.57, 58.53, 51.76, 50.85, 49.99, 40.15, 39.50, 38.69, 31.34, 30.60, 29.65, 29.60, 29.04, 29.00, 28.84, 28.75, 26.34, 24.55, 24.04, 22.54, 22.07, 22.00, 21.11, 20.28, 18.50, 17.02, 15.97, 13.25. HRMS calcd. for [C25H47N4O7S]+ 547.31600 found 547.31610.
:PE:MeOH 50:50:0 → 100:0:0 → 95:0:5) yielded the title compound (524 mg, 1.83 mmol, 97%). 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.5 Hz, 1H), 4.57–4.42 (m, 1H), 3.27 (d, J = 4.0 Hz, 1H), 2.35–2.19 (m, 1H), 2.05 (s, 2H), 1.80–1.49 (m, 3H), 1.46 (s, 9H), 0.99 (d, J = 7.0 Hz, 3H), 0.97–0.89 (m, 6H), 0.85 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 173.83, 171.91, 81.09, 59.76, 50.58, 41.23, 30.57, 27.57, 24.56, 22.48, 21.51, 19.23, 15.87.
:Tol. 0 → 16%) afforded the title compound (1.09 g, 2.04 mmol, quant.). 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 7.5 Hz, 2H), 7.56 (t, J = 7.0 Hz, 2H), 7.40–7.29 (m, 2H), 7.23 (t, J = 7.0 Hz, 2H), 7.13 (d, J = 7.6 Hz, 1H), 7.02 (d, J = 7.9 Hz, 1H), 5.99 (d, J = 8.7 Hz, 1H), 4.49 (dd, J1 = 13.8, J2 = 7.9 Hz, 1H), 4.43–4.33 (m, 2H), 4.32–4.24 (m, 1H), 4.20–4.13 (m, 1H), 1.80–1.46 (m, 6H), 1.43 (s, 9H), 0.98–0.83 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 172.11, 171.62, 156.05, 143.65, 143.50, 140.97, 127.38, 126.79, 124.89, 119.64, 81.33, 66.80, 53.11, 51.27, 46.84, 41.53, 41.18, 27.69, 24.60, 24.38, 22.77, 22.44, 21.88.
:PE:MeOH 50:50:0 → 100:0:0 → 95:0:5) yielded the title compound (586 mg, 1.95 mmol, 93%). 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 8.6 Hz, 1H), 4.46 (dt, J1 = 8.7, J2 = 5.2 Hz, 1H), 3.42 (dd, J1 = 9.6, J2 = 4.2 Hz, 1H), 1.87 (s, 2H), 1.81–1.29 (m, 6H), 1.46 (s, 9H), 1.07–0.81 (m, 12H). 13C NMR (100 MHz, CDCl3): δ 175.06, 171.77, 80.92, 53.03, 50.39, 43.69, 41.17, 27.46, 24.42, 24.28, 22.93, 22.39, 21.53, 20.95.
:Tol. 0 → 30%) yielded the title compound (1.15 g, 1.85 mmol, 95%). 1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 7.5 Hz, 2H), 7.64–7.52 (m, 2H), 7.37 (dd, J1 = 9.5, J2 = 5.1 Hz, 2H), 7.30–7.22 (m, 2H), 7.15 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 8.2 Hz, 1H), 6.11 (d, J = 9.1 Hz, 1H), 4.66 (dd, J1 = 15.1, J2 = 7.5 Hz, 1H), 4.53 (dd, J1 = 13.5, J2 = 8.5 Hz, 1H), 4.44 (dd, J1 = 10.1, J2 = 7.3 Hz, 1H), 4.30–4.22 (m, 1H), 4.19 (t, J = 7.2 Hz, 1H), 4.12 (t, J = 8.2 Hz, 1H), 2.15–1.99 (m, 1H), 1.77–1.46 (m, 6H), 1.43 (s, 9H), 0.94 (t, J = 6.4 Hz, 6H), 0.86 (dd, J1 = 10.1, J2 = 6.2 Hz, 12H). 13C NMR (100 MHz, CDCl3): δ 171.88, 171.45, 171.35, 156.47, 143.82, 141.20, 127.60, 126.99, 125.21, 125.13, 119.86, 81.70, 67.07, 60.31, 51.58, 51.29, 47.09, 41.53, 41.31, 31.46, 27.91, 24.74, 24.60, 22.83, 22.52, 21.96, 19.05, 18.21.
:PE:MeOH 50:50:0 → 100:0:0 → 95:0:5) to yield the title compound (672 mg, 1.68 mmol, 91%) 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 8.8 Hz, 3H), 7.40 (d, J = 7.7 Hz, 3H), 4.74–4.58 (m, 3H), 4.40 (dd, J = 13.9, 7.9 Hz, 3H), 3.23 (d, J = 4.1 Hz, 3H), 2.36–2.18 (m, 3H), 1.74–1.49 (m, 6H), 1.46 (s, 9H), 0.98 (d, J = 6.9 Hz, 3H), 0.95–0.87 (m, 12H), 0.84 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 174.15, 171.75, 171.41, 80.93, 59.87, 51.25, 50.67, 41.00, 40.87, 30.63, 27.60, 24.46, 24.33, 22.64, 22.31, 21.86, 21.83, 19.31, 15.94.
:1 TFA:DCM for 30 min. before being coevaporated with toluene (3×). The residue was dissolved in DCM and treated with the isocyanate of Val-Leu-OtBu 41 (obtained from H-Val-Leu-OtBu 33 (54 mg, 188 μmol, 1.15 equiv.) employing general procedure B) and DiPEA (60 μl, 361 μmol, 2.2 equiv.). The mixture was stirred for 2 h before being washed with 1 M HCl (2×) and dried over MgSO4. Column chromatography (MeOH:DCM 1 → 3%) yielded the title compound (54 mg, 90 μmol, 54%). LCMS: Rt 9.02 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CD3OD): δ 6.81 (dd, J1 = 15.2, J2 = 5.4 Hz, 1H), 6.68 (d, J = 15.2 Hz, 1H), 6.57–6.39 (m, 1H), 4.83–4.66 (m, 1H), 4.36 (t, J = 7.5 Hz, 1H), 4.15 (t, J = 6.5 Hz, 1H), 4.05 (t, J = 6.4 Hz, 1H), 2.98 (s, 3H), 2.04 (dq, J = 13.0, 6.5 Hz, 2H), 1.71 (td, J1 = 13.3, J2 = 6.6 Hz, 2H), 1.67–1.53 (m, 4H), 1.47 (s, 9H), 1.15–0.82 (m, 24H). 13C NMR (100 MHz, CD3OD): δ 174.66, 174.59, 173.20, 148.49, 130.97, 82.56, 60.91, 60.84, 60.00, 52.90, 49.12, 43.34, 42.84, 41.66, 32.63, 32.30, 28.27, 25.91, 25.83, 23.37, 23.29, 22.10, 22.06, 20.00, 18.54, 18.49. HRMS: calcd. for [C29H55N4O7S]+ 603.37860 found 603.37881
:DCM 0 → 3%) yielded the title compound (113 mg, 183 μmol, 83%). LCMS: Rt 9.40 min (linear gradient 10 → 90% ACN + 0.1% TFA, 15 min). 1H NMR (400 MHz, CDCl3, CD3OD): δ 7.93 (t, J = 7.9 Hz, 2H), 6.84 (dd, J1 = 15.1, J2 = 5.1 Hz, 1H), 6.53 (d, J = 15.1 Hz, 1H), 6.31 (d, J = 8.0 Hz, 1H), 6.02 (d, J = 8.8 Hz, 1H), 4.55–4.42 (m, 2H), 4.35 (dd, J1 = 14.5, J2 = 8.1 Hz, 1H), 4.13–4.03 (m, 1H), 2.98 (s, 3H), 2.09–1.95 (m, 1H), 1.85–1.50 (m, 9H), 1.47 (s, 9H), 1.10–0.77 (m, 24H). 13C NMR (100 MHz, CDCl3, CD3OD): δ 172.55, 172.15, 171.43, 157.68, 148.96, 127.92, 81.13, 51.14, 50.99, 48.16, 42.58, 41.69, 40.28, 40.11, 30.76, 26.96, 24.12, 23.96, 21.90, 20.96, 20.74, 18.39, 16.92. HRMS: calcd. for [C30H57N4O7S]+ 617.39425, found 617.39466.
4.2 Competition experiments in vitro
HEK293T cells were cultured on DMEM supplemented with 10% Fetal Calf Serum (FCS), 10 units ml−1penicillin and 10 μg ml−1streptomycin in a 7% CO2 humidified incubator at 37 °C. Cells were harvested, washed 2× with PBS and permeated in digitonin lysis buffer (4 × pellet volume, 50 mM Tris pH 7.5, 250 mM sucrose, 5 mM MgCl2, 5 mM DTT, 0.025% digitonin) for 15 min on ice and centrifuged at 16.100 rcf. for 20 min at 4 °C. The supernatant containing the cytosolic fraction was collected and the protein content was determined by Bradford assay (Biorad). 15 μg (HEK293T) total protein per experiment was exposed to the inhibitors (10 × solution in DMSO) for 1 h. at 37 °C prior to incubation with MVB003 (500 nM) for 1 h. at 37 °C. Reaction mixtures were boiled with Laemmli’s buffer containing β-mercaptoethanol for 3 min before being resolved on 12.5% SDS-PAGE. In-gel detection of fluorescently labeled proteins was performed in the wet gel slabs directly on the Typhoon Variable Mode Imager (Amersham Biosciences) using the Cy3/Tamra settings (λex 532, λem 560).4.3 Competition experiments in vivo
1 × 106HEK cells were seeded and grown overnight. 100 × stock solution of inhibitor was added to 1 ml medium and the cells were incubated for 4 h at 37 °C. MVB003 (100 × stock solution in DMSO, 5 μM end concentration) was added and incubated for 2 h at 37 °C. Cells were harvested, washed 2× with PBS and permeated in digitonin lysis buffer (4 × pellet volume, 50 mM Tris pH 7.5, 250 mM sucrose, 5 mM MgCl2, 5 mM DTT, 0.025% digitonin) for 15 min on ice and centrifuged at 16.100 rcf. for 20 min at 4 °C. The supernatant containing the cytosolic fraction was collected and the protein content was determined by Bradford assay (Biorad). 10 μg protein (dilution with lysis buffer) was boiled in Laemmli's sample buffer and resolved as described above. Coomassie Brilliant Blue staining of the gel verified protein loading.4.4 Fluorogenic substrate assay
26S proteasomes were purified from rabbit muscle as described.27 To determine inhibition of purified proteasomes, they were incubated with inhibitors for 30 min at 37 °C followed by assay of activity with fluorogenic substrates Suc-LLVY-amc (chymotrypsin-like site), Ac-RLR-amc (trypsin-like site), and Ac-nLPnLD-amc (caspase-like site).43Acknowledgements
This work was supported by the Netherlands Organisation for Scientific Research (NWO) and the Netherlands Genomics Initiative (NGI) by the U.S. National Cancer Institute (5RO1CA124634 and ARRA Supplement to this Award). Thanks to Hans van der Elst for HRMS analysis, Kees Erkelens and Fons Lefeber for assistance with NMR.References
- U. Wäspi, D. Blanc, T. Winkler, P. Rüedi and R. Dudler, Mol. Plant-Microbe Interact., 1998, 11, 727–733 CrossRef.
- I. Kolodziejek, J. C. Misas-Villamil, F. Kaschani, J. Clerc, C. Gu, D. Krahn, S. Niessen, M. Verdoes, L. I. Willems, H. S. Overkleeft, M. Kaiser and R. A. van der Hoorn, Plant Physiol., 2011, 155, 477–489 CrossRef CAS.
- B. Schellenberg, C. Ramel and R. Dudler, Mol. Plant-Microbe Interact., 2010, 23, 1287–93 CrossRef CAS.
- T. P. Dick, A. K. Nussbaum, M. Deeg, W. Heinemeyer, M. Groll, M. Schirle, W. Keilholz, S. Stevanović, D. H. Wolf, R. Huber, H.-G. Rammensee and H. Schild, J. Biol. Chem., 1998, 273, 25637–25646 CrossRef CAS.
- M. Groll, B. Schellenberg, A. S. Bachmann, C. R. Archer, R. Huber, T. K. Powell, S. Lindow,M. Kaiser and R. Dudler, Nature, 2008, 452, 755–758 CrossRef CAS.
- O. A. O'Connor, A. K. Stewart, M. Vallone, C. J. Molineaux, L. A. Kunkel, J. F. Gerecitano and R. Z. Orlowski, Clin. Cancer Res., 2009, 15, 7085–7091 CrossRef CAS.
- R. Piva, B. Ruggeri, M. Williams, G. Costa, I. Tamagno, D. Ferrero, V. Giai, M. Coscia, S. Peola, M. Massaia, G. Pezzoni, C. Allievi, N. Pescalli, M. Cassin, S. di Giovine, P. Nicoli, P. de Feudis, I. Strepponi, I. Roato, R. Ferracini, B. Bussolati, G. Camussi, S. Jones-Bolin, K. Hunter, H. Zhao, A. Neri, A. Palumbo, C. Berkers, H. Ovaa, A. Bernareggi and G. Inghirami, Blood, 2008, 111, 2765–2775 CrossRef CAS.
- D. Chauhan, L. Catley, G. Li, K. Podar, T. Hideshima, M. Velankar, C. Mitsiades, N. Mitsiades, H. Yasui, A. Letai, H. Ovaa, C. Berkers, B. Nicholson, T.-H. Chao, S. T. Neuteboom, P. Richardson, M. A. Palladino and K. C. Anderson, Cancer Cell, 2005, 8, 407–419 CrossRef CAS.
- H.-J. Zhou, M. A. Aujay, M. K. Bennett, M. Dajee, S. D. Demo, Y. Fang, M. N. Ho, J. Jiang, C. J. Kirk, G. J. Laidig, E. R. Lewis, Y. Lu, T. Muchamuel, F. Parlati, E. Ring, K. D. Shenk, J. Shields, P. J. Shwonek, T. Stanton, C. M. Sun, C. Sylvain, T. M. Woo and J. Yang, J. Med. Chem., 2009, 52, 3028–3038 CrossRef CAS.
- E. Kupperman, E. C. Lee, Y. Y. Cao, B. Bannerman, M. Fitzgerald, A. Berger, J. Yu, Y. Yang, P. Hales, F. Bruzzese, J. Liu, J. Blank, K. Garcia, C. Tsu, L. Dick, P. Fleming, L. Yu, M. Manfredi, M. Rolfe and J. Bolen, Cancer Res., 2010, 70, 1970–1980 CrossRef CAS.
- C. R. Archer, D.-L. T. Koomoa, E. M. Mitsunaga, J. Clerc, M. Shimizu, M. Kaiser, B. Schellenberg, R. Dudler and A. S. Bachmann, Biochem. Pharmacol., 2010, 80, 170–178 CrossRef CAS.
- C. S. Coleman, J. P. Rocetes, D. J. Park, C. J. Wallick, B. J. Warn-Cramer, K. Michel, R. Dudler and A. S. Bachmann, Cell Proliferation, 2006, 39, 599–609 CrossRef CAS.
- C. Dai and C. R. J. Stephenson, Org. Lett., 2010, 12, 3453–3455 CrossRef CAS.
- M. C. Pirrung, G. Biswas and T. R. Ibarra-Rivera, Org. Lett., 2010, 12, 2402–2405 CrossRef CAS.
- J. Clerc, M. Groll, D. J. Illich, A. S. Bachmann, R. Huber, B. Schellenberg, R. Dudler and M. Kaiser, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6507–6512 CrossRef CAS.
- J. Clerc, B. Schellenberg, M. Groll, A. S. Bachmann, R. Huber, R. Dudler and M. Kaiser, Eur. J. Org. Chem., 2010, 3991–4003 CrossRef CAS.
- J. Clerc, N. Li, D. Krahn, M. Groll, A. S. Bachmann, B. I. Florea, H. S. Overkleeft and M. Kaiser, Chem. Commun., 2011, 47, 385–387 RSC.
- A. C. Myers, J. A. Kowalski and M. A. Lipton, Bioorg. Med. Chem. Lett., 2004, 14, 5219–5222 CrossRef CAS.
- B. Barth, A. Myers and M. Lipton, J. Pept. Res., 2005, 65, 352–354 CrossRef CAS.
- M. L. Sanders and I. O. Donkor, Bioorg. Med. Chem. Lett., 2006, 16, 1965–1968 CrossRef CAS.
- M. Winn, R. J. M. Goss, K.-I. Kimura and T. D. H. Bugg, Nat. Prod. Rep., 2010, 27, 279–304 RSC.
- T. Walther, S. Renner, H. Waldmann and H.-D. Arndt, ChemBioChem, 2009, 10, 1153–1162 CrossRef CAS.
- M. Namikoshi and K. Rinehart, J. Ind. Microbiol., 1996, 17, 373–384 CrossRef CAS.
- J. Clerc, B. I. Florea, M. Kraus, M. Groll, R. Huber, A. S. Bachmann, R. Dudler, C. Driessen, H. S. Overkleeft and M. Kaiser, ChemBioChem, 2009, 10, 2638–2643 CrossRef CAS.
- A. Anshu, S. Thomas, P. Agarwal, T. R. Ibarra-Rivera, M. C. Pirrung and A. H. Schnthal, Biochem. Pharmacol., 2011, 82, 600–609 CrossRef CAS.
- M. Britton, M. M. Lucas, S. L. Downey, M. Screen, A. A. Pletnev, M. Verdoes, R. A. Tokhunts, O. Amir, A. L. Goddard, P. M. Pelphrey, D. L. Wright, H. S. Overkleeft and A. F. Kisselev, Chem. Biol., 2009, 16, 1278–1289 CrossRef CAS.
- M. Screen, M. Britton, S. L. Downey, M. Verdoes, M. J. Voges, A. E. M. Blom, P. P. Geurink, M. D. P. Risseeuw, B. I. Florea, W. A. van der Linden, A. A. Pletnev, H. S. Overkleeft and A. F. Kisselev, J. Biol. Chem., 2010, 285, 40125–40134 CrossRef CAS.
- A. C. Mirabella, A. A. Pletnev, S. L. Downey, B. I. Florea, T. B. Shabaneh, M. Britton, M. Verdoes, D. V. Filippov, H. S. Overkleeft and A. F. Kisselev, Chem. Biol., 2011, 18, 608–618 CrossRef CAS.
- M. Verdoes, B. I. Florea, W. A. van der Linden, D. Renou, A. M. C. H. van den Nieuwendijk, G. A. van der Marel and H. S. Overkleeft, Org. Biomol. Chem., 2007, 5, 1416–1426 CAS.
- M. Elofsson, U. Splittgerber, J. Myung, R. Mohan and C. M. Crews, Chem. Biol., 1999, 6, 811–822 CrossRef CAS.
- M. Bogyo, S. Shin, J. S. McMaster and H. L. Ploegh, Chem. Biol., 1998, 5, 307–320 CrossRef CAS.
- M. Bogyo, J. S. McMaster, M. Gaczynska, D. Tortorella, A. L. Goldberg and H. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6629–6634 CrossRef CAS.
- E. Wünsch and F. Drees, Chem. Ber., 1966, 99, 110–120 CrossRef.
- D. E. Nitecki, B. Halpern and J. W. Westley, J. Org. Chem., 1968, 33, 864–866 CrossRef CAS.
- B. I. Florea, M. Verdoes, N. Li, W. A. van der Linden, P. P. Geurink, H. van den Elst, T. Hofmann, A. de Ru, P. A. van Veelen, K. Tanaka, K. Sasaki, S. Murata, H. den Dulk, J. Brouwer, F. A. Ossendorp, A. F. Kisselev and H. S. Overkleeft, Chem. Biol., 2010, 17, 795–801 CrossRef CAS.
- M. Verdoes, L. I. Willems, W. A. van der Linden, B. A. Duivenvoorden, G. A. van der Marel, B. I. Florea, A. F. Kisselev and H. S. Overkleeft, Org. Biomol. Chem., 2010, 8, 2719–2727 CAS.
- G. Eisenbrand, S. Lauck-Birkel and W. C. Tang, Synthesis, 1996, 10, 1246–1258 CrossRef.
- J. E. Sheppeck, H. Kar and H. Hong, Tetrahedron Lett., 2000, 41, 5329–5333 CrossRef CAS.
- J. S. Nowick, D. L. Holmes, G. Noronha, E. M. Smith, T. M. Nguyen and S.-L. Huang, J. Org. Chem., 1996, 61, 3929–3934 CrossRef CAS.
- M. Groll, C. R. Berkers, H. L. Ploegh and H. Ovaa, Structure, 2006, 14, 451–456 CrossRef CAS.
- L. Borissenko and M. Groll, Chem. Rev., 2007, 107, 687–717 CrossRef CAS.
- F. Russo, F. Wångsell, J. Sävmarker, M. Jacobsson and M. Larhed, Tetrahedron, 2009, 65, 10047–10059 CrossRef CAS.
- P. P. Geurink, N. Liu, M. P. Spaans, S. L. Downey, A. M. C. H. van den Nieuwendijk, G. A. van der Marel, A. F. Kisselev, B. I. Florea and H. S. Overkleeft, J. Med. Chem., 2010, 53, 2319–2323 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c1ob06554h |
| This journal is © The Royal Society of Chemistry 2012 |