Multifunctional ferritin cage nanostructures for fluorescence and MR imaging of tumor cells†
Ke
Li‡
ab,
Zhi-Ping
Zhang
a,
Ming
Luo
ab,
Xiang
Yu
ab,
Yu
Han
ab,
Hong-Ping
Wei
a,
Zong-Qiang
Cui
*a and
Xian-En
Zhang
*a
aState Key Laboratory of Virology, Wuhan Institute of Virology, Chinese Academy of Sciences, No.44, Xiaohongshan, Wuhan, 430071, P. R. China. E-mail: x.zhang@wh.iov.cn; Fax: +86 27 87199492; Tel: +86 10 58881508; czq@wh.iov.cn; Fax: +86 27 87199492; Tel: +86 27 87199115
bGraduate School, Chinese Academy of Sciences, No.19, Yuquan Road, Shijingshan, Beijing, 100049, P. R. China
First published on 14th November 2011
Abstract
Bionanoparticles and nanostructures have attracted increasing interest as versatile and promising tools in many applications including biosensing and bioimaging. In this study, to image and detect tumor cells, ferritin cage-based multifunctional hybrid nanostructures were constructed that: (i) displayed both the green fluorescent protein and an Arg–Gly–Asp peptide on the exterior surface of the ferritin cages; and (ii) incorporated ferrimagnetic iron oxide nanoparticles into the ferritin interior cavity. The overall architecture of ferritin cages did not change after being integrated with fusion proteins and ferrimagnetic iron oxide nanoparticles. These multifunctional nanostructures were successfully used as a fluorescent imaging probe and an MRI contrast agent for specifically probing and imaging αvβ3 integrin upregulated tumor cells. The work provides a promising strategy for tumor cell detection by simultaneous fluorescence and MR imaging.
1. Introduction
In recent years, nanoparticles (NPs) have attracted increasing interest as versatile and promising tools in many applications including biosensing and bioimaging.1–3 Bionanoparticles and nanostructures are naturally produced with nanometre dimensions and can be easily modified to acquire a multifunctional capacity by fusion with various peptides, proteins, and nucleic acids.4–8 The protein cage architecture, as a type of fascinating bionanostructure and nanoreactor, is a good platform to develop new devices for biosensing, bioimaging and cargo delivery.9–13Protein cage architectures, such as virus capsids and ferritins, are assembled from a limited number of protein subunits and present three distinct interfaces (the interior, exterior and the interface between the subunits) that can be exploited to impart functionality by design.14 The subunits are ideal as building blocks to construct new devices because they can be easily modified using genetic and chemical approaches and their assemblies are often controllable. It is therefore possible to simultaneously impart multiple functionalities (such as targeting and mineralization) into a single protein cage.15,16
Ferritins are a family of proteins found in all domains of life, and are composed of 24 subunits that assemble to form a cage with a 12 nm diameter and an interior cavity of 8 nm in diameter.17 The interior cavity of the ferritin (especially ferritin consisting of only the heavy chain) can act as a superior size-constrained reaction vessel for the synthesis of Fe3O4 nanoparticles with a narrow size distribution.18,19 Biological molecules with specific functions can be easily fused to the N-terminal or C-terminal of human ferritin by fusion of the genes encoding human ferritin and the functional molecule. For example, GFP can be fused to the ferritin by genetic engineering and provide the ferritin nanoparticle with stable fluorescence and good biocompatibility.20–22
So far, the early detection of tumors is still a big challenge. The combination of fluorescence and MR imaging may provide new methodology to deal with the problem.23 In the present work, based on the human ferritin cage, we created a kind of new multifunctional organic/inorganic hybrid nanosystem to target and image tumor cells. Fig. 1 shows the scheme. Human H-chain ferritin was exploited as a template for superparamagnetic iron oxide nanoparticle synthesis and was engineered with GFP and a tumor cell-specific targeting moiety (RGD) on its exterior.24–26 These multifunctional nanostructures not only target tumor cells specifically, but also serve as MRI contrast and fluorescent imaging agents. The constructed nanostructures will be a good tool to be use for tumor detection.
 | ||
| Fig. 1 Schematic illustration of the construction process of the multifunctional nanostructure. | ||
2. Materials and methods
2.1. Plasmid construction
The pET-GFP-rHF plasmid was constructed by inserting the green fluorescent protein gene (GFP) and the human H chain ferritin (HFn) gene using the NcoI and XhoI restriction sites of the pET-28a(+) plasmid (Novagen, Madison, WI). The gene encoding the green fluorescent protein (GFP) was amplified by PCR from pEGFP-C1 (Clontech, Palo Alto, CA) using the forward (5′ CTA TCC ATG GTG AGC AAG GGC GAG GAG 3; bold-type defines the NcoI site) and reverse (5′TCAGGA TCC CTT GTA CAG CTC GTC CAT G 3; bold-type defines the BamHI site) primers. The plasmid pET-rHF, coding for the human H chain ferritin gene, was kindly provided by Dr Paolo Santambrogio (Milan, Italy). The gene encoding the human H chain ferritin was amplified by PCR using the forward (5′ CTTGGA TCC GGT AGC GGT AGC ACG ACC GCG TCC ACC TCG 3; bold-type delineates a BamHI site) and reverse (5′ C GAT CTC GAG TTA GCT TTC ATT ATC ACT GTC T 3; bold-type delineates an XhoI site) primers. This plasmid was used to produce the GFP-human H ferritin cage.The plasmid pET-RGD-GFP-rHF was constructed to produce the GFP-human H ferritin cage with an additional targeting peptide at the N-terminus. Here, a RGD peptide and a linker sequence (MGRGDSPSSSGGSGSGS) were incorporated at the N-terminus of the GFP-human H ferritin fusion protein. The sequences encoding the RGD peptide and the linker, and the DNA sequences encoding GFP and human H chain ferritin were subsequently inserted into the NdeI and XhoI restriction sites of the pET-20b(+) plasmid (Novagen, Madison, WI). The sequences of the RGD peptide and the linker were annealed by the oligonucleotides (5′T ATG GGT CGT GGT GAT AGC CCG TCG AGC TCA GGT GGT AGC GGC TCA GGT TC 3′) and (5′ CAT GGA ACC TGA GCC GCT ACC ACC TGA GCT CGA CGG GCT ATC ACC ACG ACC CA 3). The DNA sequences of GFP and human H chain ferritin were the product of the digest of the pET-GFP-ferritin plasmid with NcoI and XhoI restriction enzymes. All the inserted DNA sequences were confirmed by sequencing.
2.2. Purification of ferritin
The ferritin (rHF), GFP-ferritin (GFP-rHF) and RGD-GFP-ferritin (RGF) constructs were expressed in E. coli. Cultures of 1 L each of E. coli (BL21 (DE3), Novagen, Madison, WI) containing either the pET-rHF or pET-GFP-rHF or pET- RGD-GFP-rHF plasmids were grown overnight at 37 °C in LB medium with required antibiotics acting as selection markers. The rHF, GFP-rHF or RGF protein production was induced by the addition of IPTG (1 mM), and cells were incubated for an additional 10 h at 25 °C. After the incubation, cells were collected by centrifugation and the pellets were resuspended in 50 mL of buffer A (20 mM Tris-HCl, 50 mM NaCl, pH 8.0). The solution was sonicated on ice and centrifuged to remove E. coli debris. The supernatant was heated at 60 °C for 10 min. This heating step precipitated many of the E. coliproteins, which were removed by centrifugation. The supernatant was subjected to size exclusion chromatography (SEC, GE Healthcare, Uppsala, Sweden) using a Superose 6 column to purify rHF, GFP-rHF or RGF protein.2.3. Iron oxide mineralization of ferritin
Thirty millilitres of 0.4 μM rHF protein (or GFP-rHF and RGF) in a degassed solution (100 mM NaCl) was added to a jacketed reaction vessel under an N2 atmosphere. The temperature of the vessel was maintained at 65 °C using a water bath. The pH was titrated to 8.5 using 50 mM NaOH (T50 Auto Titrator, Mettler Toledo, Greifensee, Switzerland). Fe(II) was added (12.5 mM (NH4)2Fe(SO4)·6H2O) to attain a theoretical loading factor of 5000 Fe per protein cage. Stoichiometric equivalents (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 H2O2:Fe(II)) of freshly prepared degassed H2O2 (4.17 mM) was also added as an oxidant. The Fe(II) and H2O2 solutions were added simultaneously for 30 min at a constant rate of 160 μL min−1 using a pump. The reaction was considered complete 5 min after the addition of all the iron and oxidant solutions. After the completion of the reaction, 600 μL of a 300 mM sodium citrate solution was added to chelate any free iron. The mineralized sample (rHF/Fe3O4, GFP-rHF/Fe3O4 or RGF/Fe3O4) was purified by size exclusion chromatography (SEC; GE Healthcare, Uppsala, Sweden) with a Superdex 200 column. Absorbance values at 280 and 410 nm were simultaneously monitored for protein and mineral content, respectively.
:3 H2O2:Fe(II)) of freshly prepared degassed H2O2 (4.17 mM) was also added as an oxidant. The Fe(II) and H2O2 solutions were added simultaneously for 30 min at a constant rate of 160 μL min−1 using a pump. The reaction was considered complete 5 min after the addition of all the iron and oxidant solutions. After the completion of the reaction, 600 μL of a 300 mM sodium citrate solution was added to chelate any free iron. The mineralized sample (rHF/Fe3O4, GFP-rHF/Fe3O4 or RGF/Fe3O4) was purified by size exclusion chromatography (SEC; GE Healthcare, Uppsala, Sweden) with a Superdex 200 column. Absorbance values at 280 and 410 nm were simultaneously monitored for protein and mineral content, respectively.
2.4. Characterization of ferritin and mineralized ferritin
Gel electrophoresis: SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was performed to verify the presence of the ferritin cage monomer. Native agarose gel electrophoresis was also performed to characterize the ferritin cage protein and mineralized ferritin cage. The ferritin cage protein and mineralized ferritin cage were loaded onto the 1% agarose gel in 1 × TAE running buffer (40 mM Tris-acetate, pH 8.0). Gels were stained for protein using Coomassie blue and stained for iron using Prussian blue.Transmission electron microscopy (TEM): The purified ferritin and mineralized ferritin samples were imaged by TEM. A sample (15 μL) was dropped onto a carbon-coated copper grid, blotted after 5 min and then unstained or negatively stained. For negative staining, samples were stained with 0.5% phosphotungstate for 30 s. All samples were imaged by an FEI Tecnai G2 20 TWIN electron microscope operating at 200 kV and equipped with an Olympus Cantega G2 bottom-mounted CCD TEM camera. TEM images were processed and analyzed using iTEM (Olympus, Tokyo, Japan).
Spectrum analysis: Fluorescence spectra of all samples containing GFP were performed on a fluorescence spectrophotometer (LS55, PerkinElmer, Waltham, MA) using the excitation wavelength of 480 nm.
2.5. Tumor cell targeting and fluorescence imaging of multifunctional protein nanoparticles
Human glioblastoma U87MG cells and human lung adenocarcinoma A549 cells maintained in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) at 37 °C in 5% CO2 were plated onto 24-well cell culture plates to about 50% confluence, 24–36 h before the experiments. Mineralized GFP-rHF and RGF were diluted in binding buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM Ca2+, 1 mM Mg2+, 1% bovine serum albumin, pH 7.4) to a final concentration of 50 nM and added to the cells after three washes with PBS. After incubation for 1 h at 37 °C, the cells were washed three times with PBS and imaged with an epifluorescence microscope system (Carl Zeiss, Oberkochen, Germany) using a cooled CCD camera (Photometrics, Tucson, AZ). Images were acquired and manipulated using the MetaMorph software (version 6.0; Molecular Devices, Sunnyvale, CA).2.6. MR imaging of tumor cells by ferritin hybrid nanoparticles
Human glioblastoma U87MG cells and human lung adenocarcinoma A549 cells were incubated with mineralized GFP-rHF and RGF for 1 h at 37 °C, and control cells were incubated in the absence of nanoparticles. The cells were then washed with PBS, collected after digestion by a trypsin solution, and washed once more by centrifugation and resuspension in 0.5 mL of PBS. The cells were transferred to NMR tubes. MR studies were performed at 4.7 T on a Bruker Biospec imager. T2 MR relaxometry of these cells and nanoparticles were performed using a multiecho spin-echo pulse sequence (TR, 5000 ms; TE, 80–960 ms, 12 echoes; FOV, 40 mm; matrix, 128 × 128; slice thickness, 1 mm; voxel size, 0.3125 × 0.3125 × 1 mm3).3. Results and discussion
To construct the multifunctional nanoparticle, fusion proteins ferritin (rHF), GFP-ferritin (GFP-rHF) and RGD-GFP-ferritin (RGF) were firstly heterologously expressed in E. coli. The yields of rHF, GFP-rHF and RGF fusion protein are ∼100, ∼60 and ∼50 mg L−1 cultures, respectively. The proteins were purified by size exclusion chromatography and then analyzed by SDS-PAGE (Fig. 2A). The estimated molecular weights of the three purified proteins were almost identical to the theoretical molecular weights, which are 21.2, 48.6 and 49.9 kDa, respectively. These results indicate that GFP and RGD-GFP were successfully fused to the N-terminus of the ferritin subunit. | ||
| Fig. 2 Construction and characterization of ferritin cages displaying GFP and RGD peptide. (A) SDS-PAGE analysis of the proteins rHF, GFP-rHF and RGF. (B) TEM images of protein cages of rHF, GFP-rHF and RGF, upper insets show protein cages with a zoomed version. (C) Size distribution histograms as measured by TEM of rHF, GFP-rHF and RGF. | ||
Transmission electron microscopy of the purified proteins revealed that rHF, GFP-rHF and RGF adopt the expected spherical cage-like structures (Fig. 2B). These results indicate that fusion of the GFP or RGD-GFP to the N-terminus of the ferritin subunit does not interfere with the self-assembly of the subunits to form the characteristic 24 subunit protein cage architecture of ferritin. Fig. 2C shows the diameter distributions of the rHF, GFP-rHF and RGF proteins. The diameter of the three protein cages exhibited narrow distributions, which were 12.0 ± 0.76 nm, 12.9 ± 0.74 nm and 13.0 ± 0.74 nm for rHF, GFP-rHF and RGF, respectively (statistical analysis from 200–300 particles). Both GFP-rHF and RGF have diameters about 1 nm larger than ferritin alone due to the fusion of GFP and RGD-GFP to the N-terminus of the ferritin subunits.
Ferritin is a superior platform for the homogeneous nucleation of iron oxide inside of the protein cage. The purified protein cages were subjected to synthetic iron oxide mineralization under conditions of elevated pH and temperature to direct the formation of the iron oxide nanoparticles. A homogeneous brown colored solution was obtained following the reaction in the presence of the rHF protein, whereas a brown–green coloration was observed in the presence of the GFP-rHF and RGF proteins. About 70% of fusion proteins were retained after the mineralization. The TEM images of the negatively-stained samples of protein-mediated mineralization (Fig. 3A) showed that most particles appeared as protein shells with a dark black dot. Only electron-dense nanoparticles (iron oxide) could be seen in all the unstained samples of protein-mediated mineralization (Fig. 3B). Note that in un-mineralized protein cages (Fig. 2B) there were no electron-dense nanoparticles inside the protein shells (data not shown). Fig. 3C shows the diameter distributions of the particles mineralized in the presence of rHF, GFP-rHF and RGF protein cages. The diameter of the resulting particles in cages were 6.0 ± 0.80 nm, 6.1 ± 0.93 nm and 6.0 ± 0.84 nm, respectively (200–300 particles). There was no significant difference in particle size between the iron oxide products mediated by the rHF, GFP-rHF and RGF protein cages. This indicates that GFP and RGD-GFP, on the exterior surface of the cage, had little effect on the mineralization process.
 | ||
| Fig. 3 Iron oxide mineralization of ferritin cages. (A) TEM images of stained rHF/Fe3O4, GFP-rHF/Fe3O4, and RGF/Fe3O4 nanoparticles, upper insets show a zoomed-in image of a single nanoparticle. (B) TEM images of unstained ferritin cage hybrid nanoparticles. (C) Size distribution histograms of Fe3O4 in rHF, GFP-rHF and RGF cage as measured by TEM. (D) Native agarose gel electrophoresis of rHF, GFP-rHF and RGF cage before and after Fe3O4 mineralized. (a) (c) (e) were Coomassie blue stained; (b) (d) (f) were Prussian blue stained. Lane 1 was rHF cage, lane 2 was Fe3O4 mineralized rHF cage, lane 3 was GFP-rHF cage, lane 4 was Fe3O4 mineralized GFP-rHF cage, lane 5 was RGF cage, lane 6 was Fe3O4 mineralized RGF cage. | ||
Both the empty protein cage and the iron oxide mineralized protein cages were electrophoresed on 1% agarose gels under native (non-denaturing) conditions (Fig. 3D). Gels were selectively stained for Fe using Prussian blue and for protein using Coomassie blue. As expected, the mineralized protein cages stained with both the Coomassie and Prussian blues, while the empty protein cage stained only with the Coomassie blue. The co-migration of the assembled protein cage in both mineralized samples and in the empty protein cage samples indicated that the mineralized cage remained intact and essentially unchanged by the synthesis. In addition, these data suggest that the overall charge of the protein and the size of mineralized protein samples has not been measurably altered during the synthesis (also see analysis of size exclusion chromatograms in Fig. S1†).
The fluorescence spectral properties of the empty and the iron oxide mineralized GFP-rHF and RGF were examined with GFP as a control (Fig. 4A). It was found that the GFP on the exterior of the ferritin cages preserved their fluorescence spectral properties. Neither the ferritin cage nor the iron oxide inside the ferritin cage affected the GFP fluorescence spectral properties.
 | ||
| Fig. 4 Fluorescence spectrum of the hybrid nanostructures and fluorescence imaging of tumor cells. (A) Emission spectra of GFP-rHF and RGF cage before and after Fe3O4 mineralized, with GFP as a control. (B) In vitro staining of human epithelial lung A549 and human glioblastoma U87MG cell using 40 nM RGF, 40 nM GFP-rHF as a control. | ||
To test the tumor cell fluorescence imaging efficacy of the nanoparticles, binding of the nanoparticles to the αvβ3 integrin-positive U87MG cells and A549 cells were compared. Both U87MG cells and A549 cells exhibited high intensity fluorescence after binding with RGF/Fe3O4, but showed undetectable fluorescence when incubated with GFP-rHF/Fe3O4 (Fig. 4B). This indicates that RGF/Fe3O4 can be used for fluorescence imaging of the αvβ3 integrin-positive tumor cells. These results provide a new protocol for the genetic incorporation of cell targeting peptides onto the exterior surface of protein cages for developing different kinds of cell specific targeting and imaging delivery systems. Furthermore, multi-fluorescent imaging systems could be developed by incorporating different fluorescent proteins (e.g., CFP, YFP, RFP) into the protein cage.
To investigate the MR signal enhancement effects of the nanoparticles, the aqueous as-prepared nanoparticles at different Fe concentrations were measured on a 4.7 T MRI scanner. As shown in Fig. 5A, T2 weighted images changed drastically in signal intensity as the amount of nanoparticles present increased. Fig. 5B shows the T2 relaxation times as a function of the iron concentration. These results indicate that the synthesized nanoparticles can affect the MR signal enhancement. For MRI, 106 U87MG and A549 cells were treated with RGF/Fe3O4, GFP-rHF/Fe3O4 or no contrast agent. T2 maps of these treated cells were generated with MRI (Fig. 5C). The U87MG and A549 cells incubated with RGF/Fe3O4 had T2 values equal to 283.8 and 314.0 ms, respectively, while the cells incubated with GFP-rHF/Fe3O4 without the RGD peptide had T2 values equal to 457.3 and 451.3 ms, respectively. Cells that were not incubated with nanoparticles had T2 values equal to 454.4 and 447.6 ms. These data showed that the RGF/Fe3O4 binding to U87MG and A549 cells resulted in a significant decrease in the T2 relaxation times. These data also demonstrated that RGF/Fe3O4 can be used in MR imaging of αvβ3 integrin-positive tumor cells. A ferritin protein cage–iron oxide nano-composite material had previously been used as an ultra-small superparamagnetic iron oxide (USPIO) MRI contrast agent to label macrophages.27 In this work, we have shown that multifunctional nanostructures based on ferritin can be used as an MRI contrast agent to target and image tumor cells.
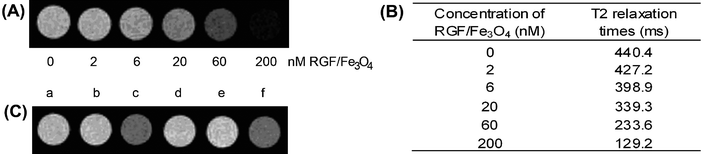 | ||
| Fig. 5 MR imaging of tumor cells by ferritin hybrid nanostructures. (A) T2-weighted images of RGF/Fe3O4 in PBS at Fe concentrations of 0, 2, 6, 20, 60 and 200 nM. (B) The T2 relaxation times as a function of the iron concentration. (C) T2-weighted MR image of A549 cell in PBS (a), and after 30-min incubation with GFP-rHF/Fe3O4 (b), RGF/Fe3O4 (c). T2-weighted MR image of U87-MG cell in PBS (d), and after 30-min incubation with GFP-rHF/Fe3O4 (e), RGF/Fe3O4 (f). | ||
4. Conclusions
A multifunctional protein nanostructure was constructed by incorporating the GFP and RGD peptide to a ferritin cage and synthesizing iron oxide nanoparticles in the ferritin interior cavity. These multifunctional nanostructures were successfully used as a fluorescent imaging probe and an MRI contrast agent for specifically targeting and imaging αvβ3 integrin upregulated tumor cells. The work provides a new nanoprobe and a promising strategy for simultaneous fluorescence and MR imaging of tumor cells. The proposed protocol can also be extended to construct other multifunctional organic/inorganic hybrid nanosystems.Acknowledgements
We thank Dr Paolo Santambrogio (Milan, Italy) for providing the pET-rHF plasmid. We also thank Dr Hao Lei, and Dr Xu-Xia Wang for their help on the MR analysis. Z.-Q. Cui is supported by the National Nano Project (no. 2011CB933600). The authors also thank the support from the Chinese Academy of Sciences (no. KJCX2-YW-M15 and XDA01020403).Notes and references
- W. Cai and X. Chen, Small, 2007, 3, 1840–1854 CrossRef CAS.
- K. Douma, L. Prinzen, D. W. Slaaf, C. P. Reutelingsperger, E. A. Biessen, T. M. Hackeng, M. J. Post and M. A. van Zandvoort, Small, 2009, 5, 544–557 Search PubMed.
- Y. W. Jun, J. H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122–5135 CrossRef CAS.
- M. N. Rhyner, A. M. Smith, X. Gao, H. Mao, L. Yang and S. Nie, Nanomedicine, 2006, 1, 209–217 Search PubMed.
- A. Chatterji, L. L. Burns, S. S. Taylor, G. P. Lomonossoff, J. E. Johnson, T. Lin and C. Porta, Intervirology, 2002, 45, 362–370 CrossRef CAS.
- E. Strable, J. E. Johnson and M. G. Finn, Nano Lett., 2004, 4, 1385–1389 CrossRef CAS.
- K. M. Taylor, T. Lin, C. Porta, A. G. Mosser, H. A. Giesing, G. P. Lomonossoff and J. E. Johnson, J. Mol. Recognit., 2000, 13, 71–82 CrossRef CAS.
- Q. Wang, K. S. Raja, K. D. Janda, T. Lin and M. G. Finn, Bioconjugate Chem., 2003, 14, 38–43 CrossRef CAS.
- A. Jaaskelainen, R. R. Harinen, T. Soukka, U. Lamminmaki, T. Korpimaki and M. Virta, Anal. Chem., 2008, 80, 583–587 Search PubMed.
- F. Li, K. Li, Z. Q. Cui, Z. P. Zhang, H. P. Wei, D. Gao, J. Y. Deng and X. E. Zhang, Small, 2010, 6, 2301–2308 CrossRef CAS.
- F. Li, Z. P. Zhang, J. Peng, Z. Q. Cui, D. W. Pang, K. Li, H. P. Wei, Y. F. Zhou, J. K. Wen and X. E. Zhang, Small, 2009, 5, 718–726 CrossRef CAS.
- T. Wang, Z. Zhang, D. Gao, F. Li, H. Wei, X. Liang, Z. Cui and X.-E. Zhang, Nanoscale, 2011, 3, 4275–4282 RSC.
- X. Lin, J. Xie, G. Niu, F. Zhang, H. Gao, M. Yang, Q. Quan, M. A. Aronova, G. Zhang, S. Lee, R. Leapman and X. Chen, Nano Lett., 2011, 11, 814–819 CrossRef CAS.
- T. Douglas and M. Young, Science, 2006, 312, 873–875 CrossRef CAS.
- M. L. Flenniken, D. A. Willits, S. Brumfield, M. J. Young and T. Douglas, Nano Lett., 2003, 3, 1573–1576 CrossRef CAS.
- M. L. Flenniken, D. A. Willits, A. L. Harmsen, L. O. Liepold, A. G. Harmsen, M. J. Young and T. Douglas, Chem. Biol., 2006, 13, 161–170 CrossRef CAS.
- M. L. Flenniken, L. O. Liepold, B. E. Crowley, D. A. Willits, M. J. Young and T. Douglas, Chem. Commun., 2005, 447–449 RSC.
- M. Uchida, M. L. Flenniken, M. Allen, D. A. Willits, B. E. Crowley, S. Brumfield, A. F. Willis, L. Jackiw, M. Jutila, M. J. Young and T. Douglas, J. Am. Chem. Soc., 2006, 128, 16626–16633 CrossRef CAS.
- K. K. W. Wong, T. Douglas, S. Gider, D. D. Awschalom and S. Mann, Chem. Mater., 1998, 10, 279–285 CrossRef CAS.
- S.-E. Kim, K.-Y. Ahn, J.-S. Park, K. R. Kim, K. E. Lee, S.-S. Han and J. Lee, Anal. Chem., 2011, 83, 5834–5843 Search PubMed.
- F. Missirlis, S. Holmberg, T. Georgieva, B. C. Dunkov, T. A. Rouault and J. H. Law, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5893–5898 Search PubMed.
- G. S. Waldo, B. M. Standish, J. Berendzen and T. C. Terwilliger, Nat. Biotechnol., 1999, 17, 691–695 CrossRef CAS.
- Q. T. Nguyen, E. S. Olson, T. A. Aguilera, T. Jiang, M. Scadeng, L. G. Ellies and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4317–4322 CrossRef CAS.
- W. Arap, R. Pasqualini and E. Ruoslahti, Science, 1998, 279, 377–380 CrossRef CAS.
- J. D. Hood and D. A. Cheresh, Nat. Rev. Cancer, 2002, 2, 91–100 CrossRef.
- H. Jin and J. Varner, Br. J. Cancer, 2004, 90, 561–565 CrossRef CAS.
- M. Uchida, M. Terashima, C. H. Cunningham, Y. Suzuki, D. A. Willits, A. F. Willis, P. C. Yang, P. S. Tsao, M. V. McConnell, M. J. Young and T. Douglas, Magn. Reson. Med., 2008, 60, 1073–1081 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1nr11132a |
| ‡ Present address: Shenzhen Mindray Bio-Medical Electronics Co., Ltd. Mindray Building, Keji 12th Road South, High-tech Industrial Park, Nanshan, Shenzhen 518057, P. R. China |
| This journal is © The Royal Society of Chemistry 2012 |