Sesquiterpene synthases: Passive catalysts or active players?
David J.
Miller
and
Rudolf K.
Allemann
*
School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, United Kingdom. E-mail: allemannrk@cardiff.ac.uk; Fax: (+41) 29-2087-4030
First published on 8th November 2011
Abstract
Covering: up to July 2011
Sesquiterpene synthases catalyse the metal dependent turnover of farnesyl diphosphate to generate a class of natural products characterised by an enormous diversity in structure, stereochemistry, biological function and application. It has been proposed that these enzymes take a passive role in the reactions they catalyse and that they serve mostly as stereochemical templates, within which the reactions take place. Here, recent research into the structure and function of sesquiterpene synthases and the mechanisms of the reactions that they catalyse will be reviewed to suggest that these fascinating enzymes play multifaceted active roles in what are arguably the most complex biosynthetic reactions.
 David J. Miller | David Miller studied chemistry at the University of Oxford (1993). He obtained his PhD (1997) in bioorganic chemistry under the tutelage of Prof. Tim Bugg at the University of Southampton developing inhibitors for enzymes involved in peptidoglycan biosynthesis. Post-doctoral positions followed at the University of St. Andrews and the University of Birmingham under Prof. David Gani and Prof. Rudolf Allemann. He was appointed as a research fellow in the School of Chemistry at Cardiff in 2007. His research interests include terpene biosynthesis, synthetic biology and medicinal chemistry. |
 Rudolf K. Allemann | Rudolf K Allemann studied physics and chemistry at the Eidgenössische Technische Hochschule in Zurich (ETH-Z), where he also received his PhD after studies at Harvard University and ETH-Z. He then moved to the MRC-National Institute for Medical Research as a Postdoctoral Research Fellow of the Royal Society (London) and the Swiss National Science Foundation. After his habilitation at ETH-Z, he took up positions at the University of Birmingham, ETH-Lausanne and Cardiff University, where he holds a Distinguished Research Professorship. The research of the Allemann laboratory focuses on enzyme mechanism, catalysis and design. |
1 Introduction
With tens of thousands of characterised members throughout all forms of life, terpenoids are the largest and most structurally diverse class of natural products.1,2 The chemically most intriguing contribution to the generation of this diversity is made by terpene synthases, enzymes that convert their linear isoprenyl diphosphate substrates into terpenoid hydrocarbons in what are perhaps the most complex biosynthetic reactions. They catalyse the production of a multitude of compounds often containing several stereocentres via carbocationic reaction cascades that involve changes to the connectivity and hybridisation of up to half the carbon atoms of the substrate.Excellent reviews have been published on the biosynthesis of monoterpenes,3–6diterpenes5,7 and triterpenes;8–10 the focus here will be on sesquiterpenes and particularly on sesquiterpene synthases. Sesquiterpene synthases catalyse, within a shared three-dimensional fold, the conversion of farnesyl diphosphate (FDP, 1a) into more than 300 different sesquiterpenes, all with the shared formula C15H24. In the past, monographs have been dedicated to these enzymes from experimental and theoretical perspectives.3,11–13 We will be examining the role played by sesquiterpene synthases in this masterpiece of combinatorial chemistry from a mechanistic physical organic chemistry viewpoint and will aim to describe the general principles shared by all sesquiterpene synthases for catalysis.
The catalytic mechanisms of sesquiterpene synthases can be divided into two phases; the metal ion dependent binding of the substrate and cleavage of the C–O bond to generate an ion pair between diphosphate and a farnesyl cation is followed by a precise carbocationic reaction cascade to the final reaction products. It has previously been suggested that after the initial ionisation, the enzymes act as passive catalysts that simply provide templates to chaperone the intermediates along the reaction pathway. We will suggest a more active role for these enzymes in catalysis, showing that they are intimately involved in the chemistry at all stages of the reaction pathway.
The review will be divided into three sections: (i) investigation of metal ion, substrate binding and generation of the initial carbocation, (ii) analysis of the role of the enzyme in FDP folding, cation stabilisation and coordination of the intermediates and transition states of the cyclisation cascade, and (iii) study of the nature of putative acids and bases in the enzymes' active sites.
2 Metal ion binding and diphosphate cleavage
The formation of sesquiterpenes is initiated by the divalent metal (usually Mg2+) dependent cleavage of diphosphate (PPi) from FDP; three metal ions are bound at the top of the active site and coordinate diphosphate prior to cleavage. Metal coordination is most easily visualised through structural analysis. At the time of writing the protein databank (http://www.pdb.org) contains the crystal structures of seven distinct sesquiterpene synthases that have been solved in forty two forms; both wild type and mutant enzyme structures have been reported in unliganded forms and in complex with various combinations of metal ions, diphosphate, FDP, substrate analogues and/or organic cation mimics. All sesquiterpene synthases contain the class I terpene synthase α-helical fold, within which the reactions takes place; plant sesquiterpene synthases also contain a catalytically silent class II terpene synthase fold (Fig. 1).4 The most conserved motifs in the class I fold are the ‘DDXXD/E’-motif, which is located on helix D and binds Mg2+A and Mg2+C, and the ‘DTE/NSE’ motif on helix H, which binds Mg2+B.4 The recently solved crystal structure of (+)-δ-cadinene (22) synthase from Gossypium arboreum (GA-DCS) (Fig. 1) revealed that the DTE motif in this enzyme is absent and instead takes the form of a second DDXXD motif more reminiscent of the presumed ancestral isoprenyl diphosphate synthases.14(+)-Germacrene D (26) synthase (GDS) from Solidago canadensis also has an altered metal binding motif, although in this case, it is the first DDXXD motif that takes the form of NDTYD.15 Nevertheless, the two metal ion binding sites in all known sesquiterpene synthases are responsible for coordination of three putative metal cations and it is these that coordinate the diphosphate group of the substrate. The binding of the three magnesium ions is highly ordered and triggers the closure of the active site, in which the farnesyl chain adopts a reactive conformation sheltered from water.4,16–26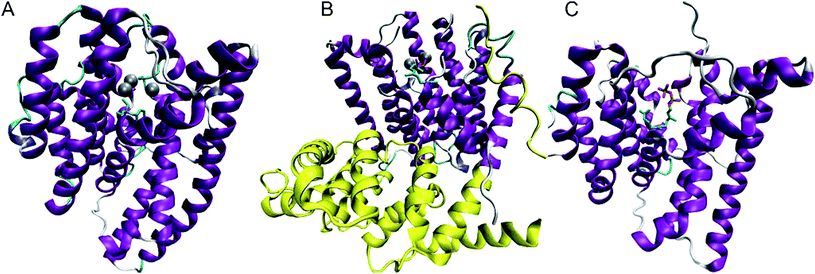 | ||
| Fig. 1 Sketches of the three most recently solved X-ray single crystal structures of sesquiterpene synthases. A: epi-isozizaene (17) synthase from Streptomyces coelicolor A3 (EIZS) in complex with Mg2+3, PPi and benzyltrimethyl ammonium cation (BTAC, 51) (pdb 3KB9);23 B: (+)-δ-cadinene (22) synthase from Gossypium arboreum (GA-DCS) in complex with 3 Mg2+ and 2F-FDP (pdb 3G4F)14 (the catalytically silent class II-like domain is shown in yellow); C: aristolochene (11) synthase from Aspergillus terreus (AT-AS) in complex with FDP (pdb 3BNX).21 | ||
The crystal structures of (+)-aristolochene (11) synthases from Penicillium roquefortii (PR-AS)27 and Aspergillus terreus19 (AT-AS) (Fig. 1) offer an opportunity to compare two separately evolved sesquiterpene synthases that generate the same product. A shared active site contour for the two enzymes has been revealed.19 A series of AT-AS structures were determined in the presence of the substrate and various fluorinated FDP analogues.21 These crystals, which comprised the enzyme in a tetrameric structure bound to Mg2+, PPi and FDP analogues in various combinations provided a series of snapshots of the enzyme in both open and closed form depending upon quaternary structure/crystal packing effects and the coordination state. These structures led to the prediction of a sequence of binding events for catalysis by AT-AS. Binding of the substrate to the open form of the enzyme in the absence of metal ions is followed by the coordination of Mg2+B and Mg2+C and active site closure; the reactive conformation of FDP is formed after Mg2+A binds to initiate the reaction cascade. Aristolochene (11), Mg2+A and Mg2+C release are accompanied by active site opening followed by release of the third magnesium, Mg2+B, and diphosphate to complete the reaction cycle.
Recently, a crystallographic analysis of epi-isozizaene (17) synthase (EIZS) from Streptomyces coelicolorA3(2) (Fig. 1) showed similar conformational changes upon binding of ligands.23 The structure was solved in complex with three Mg2+ ions, PPi and benzyltrimethyl ammonium cation (BTAC, 51, Fig. 2) as well as in complex with Hg2+; in addition, a structure of the ligand-free EIZS with a mutation to the DDXXD motif was also obtained. This work revealed a significant conformational change of helix G to cap the active site upon metal binding. The structure of trichodiene (20) synthase from Fusarium sporotrichioides (FS-TS) in complex with Mg2+, PPi and BTAC showed similar conformational changes upon binding to metal ions.22 In all these cases, it is the combination of binding to three Mg2+ and PPi (FDP in the actual reaction) that triggers closure of the active site, followed by the cleavage of the C–O bond in FDP. Clearly, such a coordinated sequence of molecular events to achieve the reactive conformation of the substrate must be considered active enzyme participation.
Cation formation and carbocation cyclisation are of course not separate but intimately linked events. For example, mutation of the D100DSKD motif of FS-TS to E100DSKD resulted in an increase in the active site volume and hence a more promiscuous product spectrum indicating that metal ion binding and overall active site contours are intimately linked.17 A site directed mutagenesis (SDM) study aimed at investigating the role of the NSE motif (typical of bacterial/fungal derived enzymes) in FS-TS, where it was sequentially converted to a plant-like DTE motif, showed a gradual reduction of product fidelity and catalytic activity of the enzyme.20 Finally, Arg 304 of FS-TS forms a hydrogen bond to PPi to stabilise the anion in the active site and to ensure formation of the correct active site contour.18 These observations underline the notion that metal binding is central to the fidelity of FS-TS catalysis.25,26,28
Recent work has shown that cation generation by sesquiterpene synthases need not necessarily precede the cyclisation reactions, but the two processes often occur in a concerted fashion. Interrogation of the mechanism of action of PR-AS using 12,13-difluoro-farnesyl diphosphate (12,13-FF-FDP, 1n, Fig. 4) showed that this compound was a competitive inhibitor of PR-AS (vide infra for an explanation of the effects of fluoro substituents).29 The fluorine atoms of 1n are distant from C1 and should not inhibit formation of the farnesyl cation. Linear difluoro-farnesene analogues were formed upon incubation of 1n with the mutant enzyme F112A-PR-AS suggesting that in the absence of the phenyl ring of Phe 112 a farnesyl cation was formed followed by proton loss to generate the farnesene products. This indicated that the role of Phe 112 in PR-AS is (together with other bulky residues in the active site) to direct the distal double bond of FDP into a reactive conformation to promote the formation of germacrenyl cation (4) directly from FDP through an SN2 like reaction and not via a farnesyl cation.29 This is consistent with the observed inversion of configuration at C1 when (R)- and (S)-1-[2H]-FDP (1b and 1c, Fig. 3) were tested as substrates of this enzyme.30 In further corroboration of the direct displacement of PPi, 2-fluoro-farnesyl diphosphate (2F-FDP, 1k) was turned over efficiently by PR-AS to a fluoro-germacrene A analogue.31 Interestingly, 2F-FDP has been reported not to be a substrate for tobacco 5-epi-aristolochene (12) synthase (TEAS), an enzyme that also proceeds through the germacrenyl cation (4), suggesting that a farnesyl cation is an intermediate in the production of 12 and that the presence of fluorine prevents its formation in this case. PR-AS plays a crucial active role in determining the nature of the cation that is formed upon cleavage of PPi – the catalysed reaction does not always proceed through farnesyl cation and beyond for all sesquiterpene synthases.
Isomerisation of FDP to nerolidyl diphosphate (NDP 3) to facilitate the production of products arising from α-bisabolyl (6), cycloheptenyl (7), helminthogermacrenyl (8) or cis-humulenyl (9) cations (Scheme 1) has been a difficult process to observe experimentally. A study of the pre-steady state kinetics of FS-TS revealed that formation of NDP is slow but once formed it reacts onwards too quickly for NDP to be observed experimentally.32 A recent study has revealed a single residue switch in the maize terpene synthase 10 (TPS10) for NDP formation. TPS10 produces (mostly) a mixture of E-β-farnesene (30) and E-α-bergamotene (21).33 The farnesene product arises from elimination of H+ and PPi from FDP but the second product arises from a downstream reaction after isomerisation to NDP. SDM of TPS10 showed that Leu 356 controls the rate of isomerisation from trans to cisfarnesyl cations.33
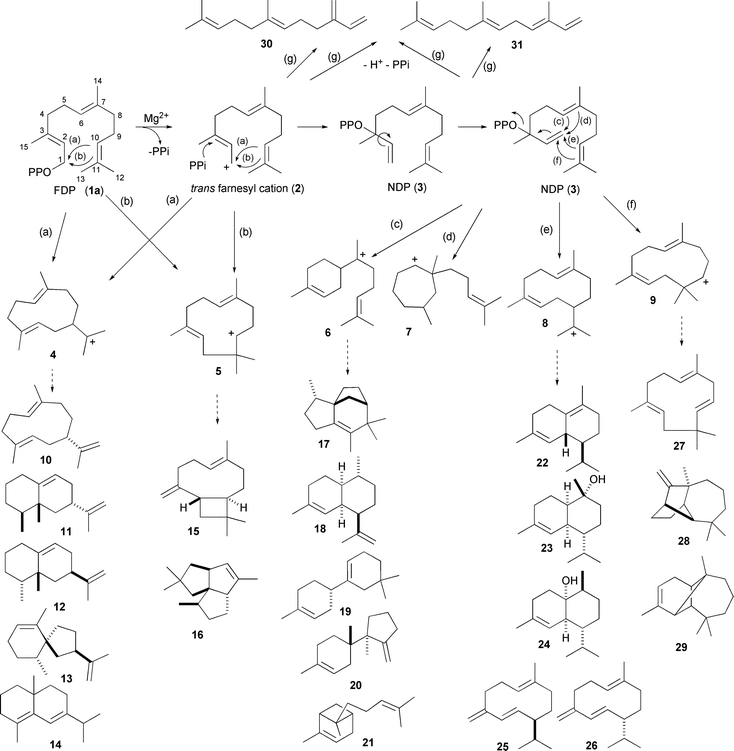 | ||
| Scheme 1 Early steps in the formation of cyclic sesquiterpenoids from FDP (1a) catalysed by sesquiterpene synthases and some of the resulting products discussed in the text. Loss of H+ and PPi from FDP (1a) or NDP (3) can also lead to the linear farnesenes. | ||
3 Carbocationic reaction cascades
Following cleavage of PPi and the generation of the initial carbocation, sesquiterpene synthases steer the reaction through often complex cyclisation cascades that contain additional carbocationic as well as neutral intermediates. In contrast to promiscuous sesquiterpene synthases, such as γ-humulene (27) (GHS) and δ-selinene (14) synthase (DSS) from Abies grandis34 or the recently discovered cadalane producing enzyme MtTPS5 from Medicago truncatula,35 high fidelity enzymes like AT- and PR-AS or GA-DCS differentiate rigorously against alternative reaction paths that will normally be of similar energies.Directing the reaction along a specific reaction coordinate is achieved through the provision of an active site template largely made up of aromatic and aliphatic residues, where the enzyme's active site surface contour aligns orbitals in productive spatial orientations, and through the stabilisation of the mostly carbocationic intermediates and transition states. The latter effect has been studied using a variety of experimental approaches, including SDM of aromatic amino acid residues, the use of aza-analogues, ammonium containing species, which mimic carbocationic intermediates and substrate analogues designed to intercept the reaction at specific points or to examine the regio- and/or stereochemistry of specific steps in the catalytic cycle.
3.1 Site directed mutagenesis studies
PR-AS contains four aromatic residues in its active site (Tyr 92, Phe 112, Phe 178 and Trp 334) and the influence of these have been analysed in a series of SDM experiments.36–40 Mutants of Trp 334 produce varying amounts of (−)-germacrene A (10), the putative intermediate of PR-AS catalytic activity.41 This result was interpreted as evidence that Trp 334 stabilises the positive charge on the eudesmane cation, the species that is formed upon protonation of germacrene A (10).38 Replacement of Trp 334 with para-substituted phenylalanines of increasing electron-withdrawing properties led to a progressive accumulation of 10 with a good correlation with the interaction energies of simple cations, such as Na+, with substituted benzenes.42 These results provide compelling evidence for the importance of Trp 334 in cation stabilization during the energetically demanding transformation of the neutral germacrene A to eudesmane cation in PR-AS catalysis. Tyr 92 may, in part, play a similar role to that played by Trp 334 since incubation of FDP with PR-AS-Y92V gave a similar product profile to that observed for PR-AS-W334V.39 In addition, the bulkiness of Tyr 92 clearly contributes to the accurate folding of the substrate in the active site; a series of replacements of Tyr 92 revealed that the amounts of aristolochene (11) increased with the van der Waals volume of the side chain. When the bulkiness of residue 92 was reduced, linear farnesenes were observed as the main products.43 While these results suggest that cyclisation of FDP might proceed through the farnesyl cation, experiments with a fluorinated substrate analogue indicated that the formation of the germacryl cation was indeed a concerted process due to optimal overlap of the p-orbital of the C2,C3 double bond with the C1–O bond facilitated by Tyr 92 (together with i.a. Phe 112 and Phe 178) (vide infra).29In studies on the bacterial enzyme pentalenene (16) synthase (PS), the roles of the active site residues Phe 76 and Phe 77 were examined by separate replacement with alanine, resulting in massive reductions of the catalytic activity in both cases. Anomalous germacrene A production by these mutants might have been considered to be due to alternate cation stabilisation but instead was proposed, on energetic grounds, to be a derailment of the cyclisation rather than perturbation of the stabilities of carbocations by altered amino acid residues.16 Alteration of Mg2+ binding motifs in FS-TS has also been observed to lead to increased promiscuity in the product spectrum.20,28
EIZS has been the subject of SDM experiments, both of the Mg2+ binding motifs44 and the aromatic portions of the active site.23 In the former case, as with other sesquiterpene synthases, altered products were formed, presumably due to altered coordination geometries of the metal ions at the active site cleft.44 Alteration of the active site aromatic residues Phe 95, Phe 96 and Phe 198 resulted in alternate products but the formation of these could not be assigned purely to either perturbation of cation–π stabilisation or a change of the active site contour and the role of these residues remains ambiguous.23
TEAS and Hysocyamus premnaspirodiene (13) synthase (HPS) are two evolutionarily related enzymes that share a common carbocationic intermediate but catalyse different rearrangements of this cation (Scheme 2). In an impressive demonstration of artificial control of sesquiterpene synthase activity, various amino acid residues in spheres of increasing radii around the active site contours of these two enzymes were identified and changed reciprocally to those of the other enzyme. Thereby each enzyme was converted to the activity of the other, indicating that close control over substrate folding and structural dynamics are crucial to product specificity and demonstrating the potential for modification of these enzyme activities for bespoke synthetic biology purposes. The enzyme is clearly not an uninvolved spectator as amino acid residues in and around the active sites of TEAS and HPS determine which of two alkyl groups migrates from a common carbcationic intermediate.45 It should, however, be noted here that for several sesquiterpene synthases from plants like amorpha-4,11-diene (18)46 or δ-cadinene (22) synthases (V. Gonzalez and R. K. Allemann, unpublished results), such an exchange of activities through the replacement of residues in or around the active sites were unsuccessful, in that only a reduction of the activities were observed. Saturation mutagenesis of GA-DCS using error-prone PCR was only able to produce a small number of clones that produced germacrene D-4-ol instead of δ-cadinene (22).47
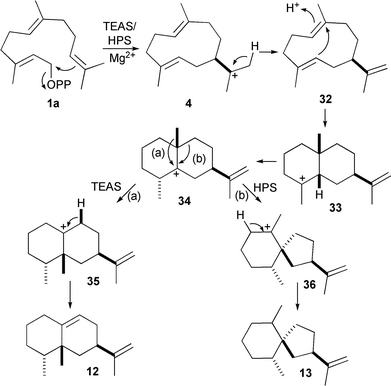 | ||
| Scheme 2 Bifurcation in sesquiterpenoid cyclisation pathways. Through the alterations of active site residues, tobacco 5-epi-aristolochene synthase (TEAS) and Hysocyamus premnaspirodiene synthase (HPS) can be tuned to transform the shared carbocationic intermediate 34 into either 5-epi-aristolochene (12) or premnaspirodiene (13). | ||
Another successful example of man-made control of sesquiterpene cyclisation is the manipulation of the highly promiscuous GHS from Abies grandis (52 known products34) towards much higher fidelity production of some of its minor products; after determination of residues in the two spheres around the active site of GHS a computer algorithm was used to predict the mutations required to alter the activity of GHS in the desired directions. With only five mutation GHS was converted from a promiscuous sesquiterpene synthase into a variety of highly active enzymes with reduced product spectra (Scheme 3).48
 | ||
| Scheme 3 Site directed mutagenesis of γ-humulene synthase from G. aroreum (GHS) has been used to alter the enzyme to a specific synthase for each of the above sesquiterpene products. | ||
Recently a group of sesquiterpene synthases from the fungus Coprinus cinereus have been examined in detail.49,50 Cop6 produces predominantly (−)-α-cuprenene (42). Cop4 is much more promiscuous, with (+)-δ-cadinene (22), β-copaene (43) and β-cubebene (44) as major products, while Cop3 makes predominantly α-muurolene (45) and (+)-germacrene A (32) (Scheme 4). The binding pockets of Cop3 and Cop4 (as judged by homology models based on the crystal structure of AT-AS) seem to be much larger than that of Cop6, which might explain the greater promiscuity. A SDM study looked at the exchange of the H1-α loop between these sesquiterpene synthases, mirroring a previous study with an α-farnesene synthase from an apple species.51 The H1-α loop closes over the active site upon binding, moving the NSE motif into the correct position for catalysis. Various mutants altered the ability of the loop to close over the active site leading to premature quenching of the cations and hence production of germacrenes.
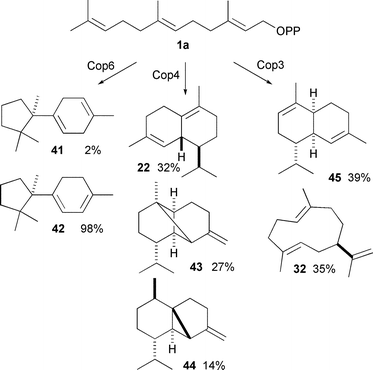 | ||
| Scheme 4 The major products generated by sesquiterpene synthases Cop3 and Cop6 from Coprinus cinereus. | ||
An interesting case of the flexibility of sesquiterpene synthase catalysed chemistry has been provided by studies of the biosynthesis of the trisnor-sesquiterpene geosmin (50). Although not strictly a sesquiterpene, this compound is formed by a sesquiterpene synthase that eliminates acetone from an intermediate sesquiterpenoid alcohol to generate its product (Scheme 5). Geosmin synthase is a bifunctional enzyme from Streptomyces coelicolor52 that possesses functionally different C-and N-terminal catalytic domains with significant sequence similarity to pentalenene synthase. Cleavage of the protein into the two separate domains gave two functional enzymes; the N-terminal half was found to convert FDP to germacradienol (48) and (−)-germacrene D (25),52 while the C-terminal half is responsible for the conversion of germacradienol to geosmin (50).53 A series of mutant geosmin synthases with changed DDXXD and NSE Mg2+ binding motifs provided insight into the mechanism; most notably the S233A mutant produced wild-type products together with 11% of isolepidozene (47), a neutral putative cyclopropyl intermediate that is attacked by water under acid catalysis to give germacradienol prior to conversion to geosmin.53
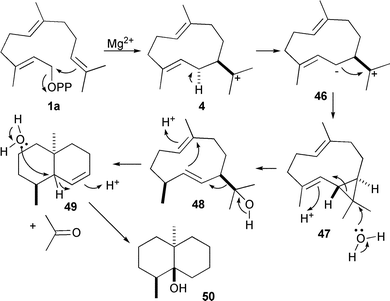 | ||
| Scheme 5 The mechanism for the biosynthesis of geosmin (50). | ||
3.2 Aza-analogues
An alternative approach to SDM for the study of cation stabilisation and conformational control in sesquiterpene synthase catalysis is the use of synthetic analogues of the proposed cationic intermediates. Nitrogen containing compounds that are either quaternised or simply protonated in aqueous solution to generate a cation, have been used in kinetic studies and as structural analogues for single crystal X-ray analyses (for examples of aza analogues see Fig. 2).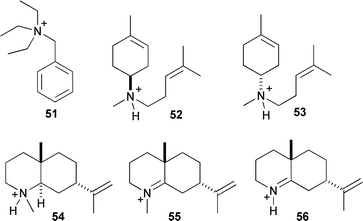 | ||
| Fig. 2 Aza-analogues of sesquiterpenoid cationic intermediates. | ||
In the crystal structure of wild-type EIZS, BTAC (51) was observed to orient itself with aromatic residues.23 The crystal structure of EIZS-F198A coordinated to three Mg2+ ions, PPi and BTAC revealed a different orientation of the cation relative to the wild type structure, implying that the resulting expansion of the active site volume allowed other orientations of cation binding and hence altered product specificity.23
A recent crystal structure of FS-TS-Y305F in complex with the potent inhibitor 53 indicated that this aza-analogue bound to the enzyme in a conformation that did not mimic the proposed orientation of α-bisabolyl cation (6) during the catalytic cycle.17,54 Favourable interactions with PPi seemed to cause the observed binding orientation rather than cation–π interactions with aromatic residues, suggesting that sesquiterpene synthases enforce cation location and formation through kinetic rather than thermodynamic control.17
Use of BTAC in crystal structure analysis of mutant FS-TS provided the first definitive proof that a cation can bind in the active site of a sesquiterpene synthase since aza-bisabolyl cations were of unknown protonation state in other known structures.22 An apparent ‘loosening’ of the coordination of PPi and Mg2+ in FS-TS structures led to the interpretation that this leads to greater versatility of PPi to act as a general base (vide infra) and for the observed diversity of products for this enzyme.
Recently, aza-analogues designed to resemble eudesmane cation have been prepared and tested as inhibitors of PR-AS. In contrast to the observations discussed above for FS-TS, one of these compounds (54, Fig. 2) proved to be a potent inhibitor of PR-AS in both the presence and absence of PPi. This suggested that this compound may be a genuine mimic of eudesmane cation since no obvious favourable interactions with PPi were detected.55 The trigonal aza-analogues 55 and 56, designed to mimic the likely transition state between (−)-germacrene A (10) and eudesmane cation (33), inhibit PR-AS synergistically with PPi, implying a role for PPi in carbocation formation, perhaps as the active site acid.56
3.3 Substrate analogues
Many of the finer points in the mechanism of formation of inter alia(+)-aristolochene (11),30,57trichodiene (20),32,58δ-cadinene (22),59,60epi-cubenol (24),61–64pentalenene (16),65–67 and (−) and (+)-germacrene D (25 and 26)68 were uncovered using deuterated, tritiated and carbon-14 labelled analogues of FDP and NDP. Much of this work has been reviewed previously.3,4,11More recently, the catalytic mechanism of amorpha-4,11-diene synthase from Artemisia annua (ADS) has been studied using labelled FDP analogues. In contrast to the antimalarial compound artemisinin, which is synthesised from amorpha-4,11-diene, ADS has not been studied extensively from a mechanistic point of view. Two parallel studies with (R)- and (S)-[1-2H]-FDP (1b and 1c) established a 1,3-shift of HSi on C1 of FDP to C7 implying a mechanism that proceeds viaα-bisabolyl cation (6), which is formed by 1,6 ring closure of NDP (Scheme 6).69,70 Interestingly, deuterium incorporation was observed upon incubation in >75% 2H2O. Although the position of incorporation was not determined, the authors speculated that an intermediate cation may be quenched and reprotonated as a side reaction from the main catalytic pathway.70 This may hint at the alternate reaction pathways to some of the minor products that have been observed for this enzyme and present opportunities for engineering altered reactivities into this enzyme.71 It will be interesting to see how the enzyme promotes 1,6 ring closure of cis farnesyl cation rather than the 1,10 ring closure catalysed by cadalane producing sesquiterpene synthases.
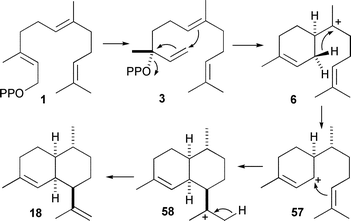 | ||
| Scheme 6 Mechanisms for the biosynthesis of amorpha-4,11-diene (18). | ||
Further to the SDM studies on the related enzymes TEAS and HPS, deuterium labelling experiments were used to analyse the stereochemistry and timing of protonation, rearrangement and elimination steps for these two enzymes.72 Incubation with both enantiomers of [8-2H]-FDP (1d and 1e), tris- and hexa-deuterated FDP analogues at C12 and C13 (1f, 1g and 1h) and incubation of unlabelled FDP in 2H2O gave results that were broadly in line with mechanistic observations for the fungal aristolochene synthases,57,72,73 namely that the proton lost upon formation of the intermediate germacrene A is from C12; the intermediate is then reprotonated from the Re face and HSi is lost from C8 to form the final product.
A new trend in sesquiterpene synthase research is the hunt for new activities using genome mining. Two presumptive sesquiterpene synthase genes from Streptomyces clavuligerus were recently identified and codon optimised synthetic genes constructed and overexpressed in E. coli. The gene products were identified as related sesquiterpene synthases producing the cadalane family sesquiterpenoids (-)-δ-cadinene (22) and (+)-T-muurolol (23).74 Incubation of the enzymes with 1b and 1c and analysis of the products by 1H and 2H NMR spectroscopy revealed a [1,3]-hydride shift of HSi on C1 of FDP to C11 in the reactions catalysed by both enzymes. This is in line with a similar stereochemical prediction for the hydride shift observed for GA-DCS.59 Further to this, a promiscuous cadalane producing sesquiterpene synthase MtTPS5 from Medicago truncatula has recently been identified.35 Incubations of unlabelled FDP in 2H2O showed C15 proton/deuteron exchange, which was predicted through an alternative mechanism for cadalane production that was first postulated many years ago but until this report had no experimental support.75 Some of the products appeared to be generated viagermacrene D, which after protonation at C15 reacts to a variety of bicyclic products. Interestingly, the alteration of GA-DCS by saturation mutagenesis led to an enzyme that produced a close analogue of germacrene D,47 implying that only subtle differences in enzyme structure and dynamics differentiate between production of germacrene D or germacrene D-4-ol and δ-cadinene from a common intermediate.
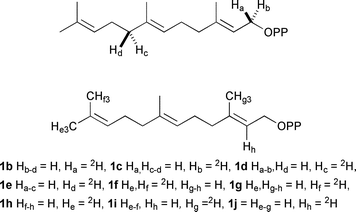 | ||
| Fig. 3 Deuterated substrate analogues discussed in the text. | ||
Another enzyme that produces germacrene D as a by-product is geosmin synthase. Interrogation of the mechanism of this enzyme using 1b and 1c and analysis of the products by GC-MS established that HSi on C1 of FDP is the species that undergoes [1,3]-hydride transfer in this case.76 Despite producing the same enantiomer of germacrene D, (-)-GDS from Solidago canadensis proceeds via [1,3]-migration of HRe on C1 of FDP indicating that the two enzyme must convert FDP to (-)-germacrene D (25) through different catalytic mechanisms.68,76 The mechanism of the dominant geosmin production was established to result from retro-Prins fragmentation of germacradienol by labelling studies. Incubation of unlabelled FDP in 2H2O resulted in specific incorporation of several deuterium atoms in geosmin and the by-products although not in the (-)-germacrene D.77 The use of tris-deuterated FDP at C13 (1h) resulted in trapped acetone containing the label. This confirmed the retro-Prins fragmentation of 47 and the use of 1j as a substrate and analysis of the resulting product demonstrated a predicted [1,2]-hydride shift from C2 of FDP.78
Genome mining studies first identified EIZS from Streptomyces coelicolor. Incubation with 1b and 1c, [1-2H2]-FDP (1j) and the trisdeutero-FDP analogues 1g and 1h established the current understanding of the mechanism of this reaction including the stereochemistry of the methyl migration in the penultimate step (Scheme 7).44,79
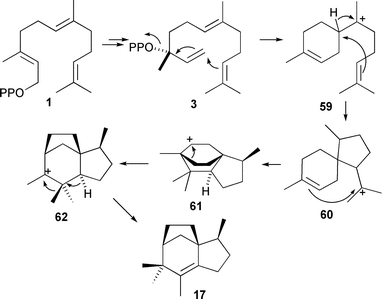 | ||
| Scheme 7 Biosynthesis of epi-isozizaene (17). | ||
Another genome mining study, this time of Streptomyces avermitilis, identified a gene encoding a previously undiscovered sesquiterpene synthase that produces the eponymous avermitilol (66).80 The stereochemistry of the ring closures was elucidated using 1b and 1c. The enzyme also produces small amounts of (+)-germacrene A (32), germacrene B (63) and viridiflorol (67) (Scheme 8).80
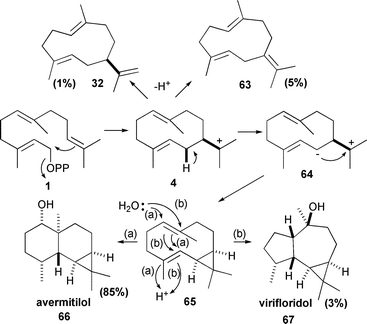 | ||
| Scheme 8 The biosynthesis of avermitilol (66). Virifloridol (67), germacrene A (32) and germacrene B (63) are minor byproducts. | ||
The product of presilphiperfolan-8β-ol (72) synthase from Botrytis cinerea is a common precursor in the biosynthesis of a series of toxins produced by this necrotic plant pathogen.81 With the use of deutero-FDP analogues it was established that the reaction sequence proceeds viahumulyl cation (5) and does not require the intermediacy of NDP (3) (Scheme 9), although this compound is an alternate substrate of this enzyme.
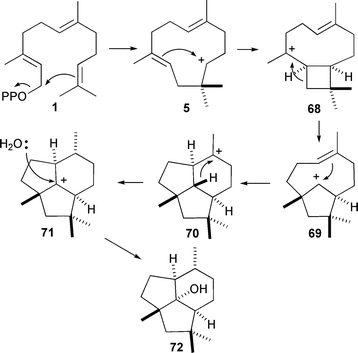 | ||
| Scheme 9 Mechanism for the biosynthesis of presilphiperfolan-8β-ol (72). | ||
Fluorinated isoprenyl diphosphates have been used in the past to study the mechanism of enzymes involved in terpene biosynthesis,82–84 but only recently have they been extensively employed for the analysis of sesquiterpene synthases. Their behaviour as either inhibitors or substrates has revealed several subtle mechanistic details that may not otherwise have come to light. The crystal structures of AT-AS and GA-DCS have now been solved in the presence of several fluorinated sesquiterpene analogues (vide supra).14,21Fig. 4 illustrates the fluorinated FDP analogues that have been used in this work and also shows several of the fluorinated sesquiterpenoids produced with various sesquiterpene synthases.
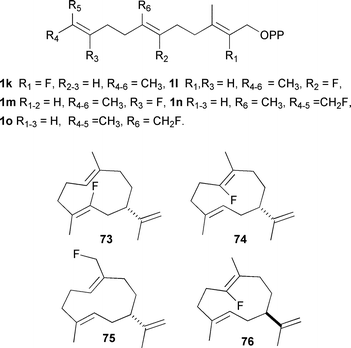 | ||
| Fig. 4 Fluorinated FDP analogues referred to in the text and some of the germacrene A analogues resulting from turnover of these analogues by sesquiterpene synthases. | ||
Fluorinated FDP analogues have been used to interrogate the mechanism of the PR-AS; 12,13-FF-FDP (1n) was instrumental in showing the role of Phe 112 (and other bulky active site residues) in aligning the distal double bond for attack at C1 during PRAS catalysis (vide supra).29 In addition, 2F-FDP (1k),31 6F-FDP(1l)85 and 14F-FDP (1o)85 have proved to be efficient substrates of PR-AS, which converts these fluorinated FDPs to analogues of (−)-germacrene A (73–75, Fig. 4). This provides further evidence of the intermediacy of germacrene A in the production of eremophilenyl sesquiterpenes such as (+)-aristolochene (11) since the presence of fluorine on C2 or C6 prevents the cyclisation of (−)-germacrene A by reduction of electron density on the C2,C3 or C6,C7 double bonds, respectively, and also by preventing the build up of positive charge on C3 in the eudesmane cation (34). In the case of fluorine on C14, there is again a depletion of electron density in the C6,C7-double bond, which reduces the π-basicity and so prevents protonation of 10. It also shows that these enzymes can be utilised to produce analogues of natural products that may be exploitable in the future for medicinal or agrochemical uses. 6F-FDP (1l) is also an efficient substrate of TEAS, giving the corresponding fluorinated (+)-germacrene A analogue (76) as the sole product.86
The inclusion of TEAS amongst the so-called high-fidelity sesquiterpene synthases has recently been challenged by the observation of several minor products in its product pool originating from both transoid and cisoid ring closures of FDP.87 The amount of cisoid products increased dramatically when the enzyme was incubated with the 2Z isomer of FDP.88 In addition, crystals of TEAS and a promiscuous mutant (M4 TEAS) were soaked with 2F-FDP and 2-fluoro-2E-FDP to provide snapshots of the binding modes of substrates in conformations leading to both of these cyclisation pathways.89 Comparison of the X-ray structures showed that both substrates were bound in the active site with the farnesyl chain in a U-shaped conformation but with differing orientations (inversions) of the first two isoprene units – hence showing complete occupancy of the active site and also displaying conformations consistent with the expected orientation that would lead to their observed product pools. The cisoid substrate analogue, however, was not bound in an active conformation since the C–O bond was coplanar with the C2,C3 bond, in contrast to the transoid substrate where the C–O bond was perpendicular to this and hence oriented optimally for the reaction. These results show that while the enzyme active site surface contour is important for the control of product outcome, it appears that an interplay exists between enzymatic enforcement of substrate conformation and the intrinsic conformational possibilities of the substrate; the use of a cisoid C2,C3 double bond can produce dramatically altered products. Interestingly, it was also observed that several of the mutations in the M4 mutant TEAS cause little change to the contour of the active site. Since this mutant is much more promiscuous than the wild-type enzyme, this hints strongly at dynamic effects playing an important part in the product outcome for this enzyme. The electron density of the farnesyl chain in the crystal structures of this mutant soaked with the two substrate analogues showed discontinuities, perhaps indicating increased motional flexibility of the substrate in the active site relative to the wild type enzyme; this is fully consistent with the promiscuity observed for this mutant when incubated with FDP.
In agreement with results revealing active site plasticity in GHS,48 the active site contour of PR-AS has been shown to be highly plastic for binding FDP analogues. Three phenyl-FDP analogues are potent competitive inhibitors of this enzyme.90 This reveals potential for synthetic biology applications, since it may be possible to engineer a larger active site cavity to allow productive binding of such bulky substrate analogues. It also reveals that the active sites of these enzymes are not static and rigid cavities but are able to adopt different shapes to accommodate bulkier ligands hinting at the dynamic role played by these proteins during catalysis.
4 Acid/base catalysis
Many sesquiterpene synthases require participation of acids and bases during their catalytic cycle. For example, the two enantiomers of germacrene A are thought to be neutral intermediates in the pathways to (+)-aristolochene (11) (via10) and 5-epi-aristolochene (via32) requiring an acid to regenerate a cation; a base is then required in the final step of the reaction.11 Various candidates have been proposed to act as active site acids/bases including amino acid side chains, active site water molecules as well as the diphosphate generated from the substrate itself.Based on the proposal that Tyr 520 may be the active site acid responsible for protonation of germacrene A in catalysis by TEAS,91 it was suggested that the analogous function is fulfilled by Tyr 92 in the active site of PR-AS.27 However, since replacements of Tyr 92 in PR-AS lead to enzymes that still produce significant quantities of (+)-aristolochene (11), Tyr 92 cannot be the active site acid.40PPi has since been suggested to act as the acid/base for AT-AS on the basis of crystallographic analysis but there remains little other than structural evidence for this.19 Meanwhile, a theoretical study proposed that instead of being a reaction intermediate, (−)-germacrene A (10) could be a by-product of PR-AS produced via a different catalytic mechanism to the major product.92 Instead, it was proposed based on gas-phase calculations that there may be a direct hydride transfer between C12 and C6, or a proton transfervia an intermediary water molecule. Such a mechanism would bypass the neutral germacrene A and avoid an energetically expensive sequence of cation quenching followed by cation regeneration.92 This attractive mechanistic proposal had to be rejected, however, since in deuterium labelling experiments the stereochemistry of protonation at C6 was inconsistent with either direct hydride transfer or transfer by way of a water molecule.73 This labelling study established that the protonation of 10 occurs on the same face of the molecule as the later deprotonation event that produces the final product, implying that the same species is involved in both reaction steps.73 A similar study on TEAS showed that protonation occurs on the same face of (+)-germacrene A in TEAS catalysis.72
The idea of tyrosine as an active-site acid in TEAS has proved enticing for other sesquiterpene synthases also. Y295 has been proposed as the active site base responsible for final deprotonation in FS-TS catalysis – however, SDM of this residue ruled out this possibility, since mutants had similar activity to the wild type enzyme. Once more the PPi group most likely acts as the proton donor/acceptor.20
Further enzymes where tyrosine has been investigated as an active site acid are the (S)-β-macrocarpene (19) synthases TPS6 and TPS11 from maize, which possess a tyrosine residue (Tyr 522 in both) that appears to be an evolutionarily conserved residue (cf. Tyr 520 in TEAS).33(S)-β-Macrocarpene was shown by deuterium labelling studies to arise from protonation of a neutral intermediate ((S)-β-bisabolene, 77) and SDM of Y522 suppressed this protonation activity yielding β-bisabolene as product (Scheme 10). Additionally, protonation of (S)-β-bisabolene was found to be both Mg2+ and pH dependent, as has also been reported for TEAS protonation of (+)-germacrene A (32).93 A similar pH sensitivity has been observed to affect the product profile of Cop4 from Coprinus cinereus. This, however, appears to be a protein folding effect since SDM experiments where H235 and N239 were altered to proline and leucine, respectively, resulted in abolition of the pH dependence of product formation - probably due to hydrogen bonding changes within the enzyme.49 Whatever the specific identity of active site acids/bases in various sesquiterpene synthases, it is clear that amino acid residues are at least in part responsible for such processes in some cases – notably TEAS and in TPS6 and TPS 11 from maize and that the enzyme thereby plays a crucial active chemical role in the catalysis.
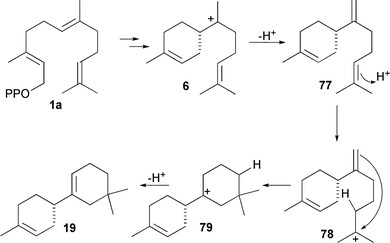 | ||
| Scheme 10 The mechanism of formation of (S)-β-bisabolene (77) and (S)-β-macrocarpene (19) catalysed by TPS6 and TPS11 from maize. | ||
Finally, a recent interrogation of patchouli alcohol (80, Scheme 11) synthase from Pogostemon cablin using the singly labelled [2-2H]-FDP 1j showed a surprising result. In addition to the expected singly labelled patchouli alcohol product (∼65% of isolated alcoholic material), a doubly deuterated product was also observed (∼35% of isolated alcohols).94 From the positions of deuteration it was concluded that an intermediate alkene structure becomes protonated (or deuterated when 1j is used as substrate) to give the final product. Minor hydrocarbon by-products β-patchoulene (81) and α-guaiene (82) were depleted of deuterium, indicating the source of the second deuteron. Comparison of the amino acid sequence with other sesquiterpene synthases and use of an active site model identified Leu 410 as an active site residue that may undergo reorientation during the catalytic cycle, effectively creating a second pocket in the active site were the observed deuteration events occur.94 This may be another example of active enzyme participation in catalysis.
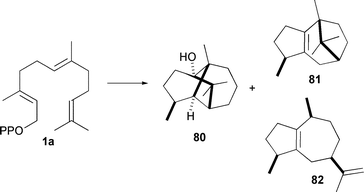 | ||
| Scheme 11 Patchouli alcohol (80) and two of the minor hydrocarbon products, β-patchoulene (81) and α-guaiane (82) produced by patchouli alcohol synthase from Pogostemon cablin. | ||
5 Conclusions
All of the work described here has helped unravel the general principles that control the mode of action of sesquiterpene synthases. Even though not the subject of this review, it is worth noting that the same principles will also operate during catalysis by mono- and diterpene synthases. Despite a lack of significant sequence homology between sesquiterpene synthases in plants, insects, bacteria and fungi, the chemistry that they facilitate within the shared class I terpene synthase fold is the basis for the generation of the enormous diversity in structure and stereochemistry found in natural sesquiterpenes. The plasticity of the sesquiterpene synthases appears to provide a framework for the combinatorial production of many natural terpenoids though subtle alterations in the composition of the active site during evolution. The enzymes bind their substrates in reactive conformations that take advantage of the chemical reactivity inherent in FDP. This first binding step is central for the initiation of the Mg2+-dependent expulsion of diphosphate and the product profile. Perhaps not surprisingly, there is complexity in the formation of the Michaelis complex of PR-AS21 and perhaps all sesquiterpene synthases, in that after the initial molecular recognition of FDP, coordination to Mg2+B reorients the substrate diphosphate group. Subsequently, binding of Mg2+C triggers a more substantive conformational change leading to active site closure. Finally, binding of Mg2+A completes the formation of the trinuclear metal ion cluster and triggers further structural changes to complete active site closure and to generate the cyclisation-competent conformation of FDP. In the Michaelis complex the three negative charges of the PPi leaving group are fully neutralized to facilitate ionization and initiation of the cyclisation cascade. It is likely that diphosphate stays bound throughout the reaction thereby providing stability to the carbocationic transition states and intermediates. The parts of the active site that bind the isoprenoid tail of the substrate are largely made up of aliphatic and aromatic amino acids to allow additional stabilisation of the carbocations of the reaction cascades through cation–π or charge–quadrupole interactions. The specific orientation of the active site residues allows the enzymes to channel the reaction along a specific pathway to generate the desired outcome and at the same time strictly controls water access.Finally, general acid and base catalysis is involved in terpene synthase reactions. There has so far been little evidence that the enzymes provide amino acids to act as acids and bases and, by exclusion, it is now assumed that in many cases the bound diphosphate ion helps move protons. The selectivities of the protonation and deprotonation reactions depend on the correct orientation of acid and bases with in the active sites.
Sesquiterpene synthases play an active controlling role in catalysis; they provide a fascinating example of how enzymes have evolved to take advantage of the chemical properties inherent in their substrates to generate a biologically desirable outcome with remarkable efficiency and specificity. (Sesqui)-terpene synthases, the chemistry they facilitate, their biology and their applications will no doubt continue to astound us through a combination of structural complexity and chemical simplicity.
6 Acknowledgments
We are grateful for the financial support of the Biotechnology and Biological Sciences Research Council (grants 6/B17177; BB/G003572/1 and BB/H01683X/1), the Engineering and Physical Sciences Research Council (grant EP/D06958/1) and Cardiff University. We thank Dr Juan A. Faraldos, Dr Sabrina Touchet, Dr Mahmoud Akhtar, Dr Robert Mart and Dr E. Joel Loveridge for critical comments on the manuscript and past and present members of the terpene cyclase group for stimulating discussions.7 References
- J. D. Connolly and R. A. Hill, Dictionary of Terpenoids, Chapman and Hall, London, 1991 Search PubMed.
- J. S. Glasby, Encyclopedia of Terpenoids, Wiley, Chichester, 1982 Search PubMed.
- J. Degenhardt, T. G. Kollner and J. Gershenzon, Phytochemistry, 2009, 70, 1621 CrossRef CAS.
- D. W. Christianson, Chem. Rev., 2006, 106, 3412 CrossRef CAS.
- E. M. Davis and R. Croteau, Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Springer, Berlin Heidelberg, 2000, vol. 209, pp. 53–95 Search PubMed.
- E. M. Davis, in Comprehensive Natural Products II, ed. L. Mander and H.-W. Liu, Elsevier, 2010, vol. 1, pp. 585–608 Search PubMed.
- T. Toyomasu and T. Sassa, in Comprehensive Natural Products II, ed. L. Mander and H.-W. Liu, Elsevier, 2010, vol. 1, pp. 643–672 Search PubMed.
- T. Kushiro and Y. Ebizuka, in Comprehensive Natural Products II, ed. L. Mander and H.-W. Liu, Elsevier, 2010, vol. 1, pp. 673–708 Search PubMed.
- D. R. Phillips, J. M. Rasbery, B. Bartel and S. P. T. Matsuda, Curr. Opin. Plant Biol., 2006, 9, 305 CrossRef CAS.
- R. Xu, G. C. Fazio and S. P. T. Matsuda, Phytochemistry, 2004, 65, 261 CrossRef CAS.
- D. Cane, Chem. Rev., 1990, 90, 1089 CrossRef CAS.
- D. J. Tantillo, Nat. Prod. Rep., 2011, 28, 1035 RSC.
- J. Chappell and R. M. Coates, in Comprehensive Natural Products II, ed. M. L. and H.-W. Liu, Elsevier, 2010, vol. 1, pp. 609–641 Search PubMed.
- H. A. Gennadios, V. Gonzalez, L. Di Costanzo, A. A. Li, F. L. Yu, D. J. Miller, R. K. Allemann and D. W. Christianson, Biochemistry, 2009, 48, 6175 CrossRef CAS.
- I. Prosser, I. G. Altug, A. L. Phillips, W. A. Konig, H. J. Bouwmeester and M. H. Beale, Arch. Biochem. Biophys., 2004, 432, 136 CrossRef CAS.
- M. Seemann, G. Z. Zhai, J. W. de Kraker, C. M. Paschall, D. W. Christianson and D. E. Cane, J. Am. Chem. Soc., 2002, 124, 7681 CrossRef CAS.
- L. S. Vedula, M. J. Rynkiewicz, H. J. Pyun, R. M. Coates, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 6153 CrossRef CAS.
- L. S. Vedula, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 12719 CrossRef CAS.
- E. Y. Shishova, L. Di Costanzo, D. E. Cane and D. W. Christianson, Biochemistry, 2007, 46, 1941 CrossRef CAS.
- L. S. Vedula, J. Y. Jiang, T. Zakharian, D. E. Cane and D. W. Christianson, Arch. Biochem. Biophys., 2008, 469, 184 CrossRef CAS.
- E. Y. Shishova, F. L. Yu, D. J. Miller, J. A. Faraldos, Y. X. Zhao, R. M. Coates, R. K. Allemann, D. E. Cane and D. W. Christianson, J. Biol. Chem., 2008, 283, 15431 CrossRef CAS.
- L. S. Vedula, Y. X. Zhao, R. M. Coates, T. Koyama, D. E. Cane and D. W. Christianson, Arch. Biochem. Biophys., 2007, 466, 260 CrossRef CAS.
- J. A. Aaron, X. Lin, D. E. Cane and D. W. Christianson, Biochemistry, 2010, 49, 1787 CrossRef CAS.
- C. A. Lesburg, G. Z. Zhai, D. E. Cane and D. W. Christianson, Science, 1997, 277, 1820 CrossRef CAS.
- M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Biochemistry, 2002, 41, 1732 CrossRef CAS.
- M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13543 CrossRef CAS.
- J. M. Caruthers, I. Kang, M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, J. Biol. Chem., 2000, 275, 25533 CrossRef CAS.
- D. E. Cane, Q. Xue and J. E. VanEpp, J. Am. Chem. Soc., 1996, 118, 8499 CrossRef CAS.
- F. L. Yu, D. J. Miller and R. K. Allemann, Chem. Commun., 2007, 4155 RSC.
- D. E. Cane, P. C. Prabhakaran, E. J. Salaski, P. H. M. Harrison, H. Noguchi and B. J. Rawlings, J. Am. Chem. Soc., 1989, 111, 8914 CrossRef CAS.
- D. J. Miller, F. L. Yu and R. K. Allemann, ChemBioChem, 2007, 8, 1819 CrossRef CAS.
- D. E. Cane, H. T. Chiu, P. H. Liang and K. S. Anderson, Biochemistry, 1997, 36, 8332 CrossRef CAS.
- T. G. Kollner, J. Gershenzon and J. Degenhardt, Phytochemistry, 2009, 70, 1139 CrossRef.
- C. L. Steele, J. Crock, J. Bohlmann and R. Croteau, J. Biol. Chem., 1998, 273, 2078 CrossRef CAS.
- S. Garms, T. G. Kollner and W. Boland, J. Org. Chem.75, p. 5590 Search PubMed.
- S. Forcat and R. K. Allemann, Org. Biomol. Chem., 2006, 4, 2563 CAS.
- S. Forcat and R. K. Allemann, Chem. Commun., 2004, 2094 RSC.
- A. Deligeorgopoulou, S. E. Taylor, S. Forcat and R. K. Allemann, Chem. Commun., 2003, 2162 RSC.
- M. J. Calvert, S. E. Taylor and R. K. Allemann, Chem. Commun., 2002, 2384 RSC.
- B. Felicetti and D. E. Cane, J. Am. Chem. Soc., 2004, 126, 7212 CrossRef CAS.
- M. J. Calvert, P. R. Ashton and R. K. Allemann, J. Am. Chem. Soc., 2002, 124, 11636 CrossRef CAS.
- J. A. Faraldos, A. K. Antonczak, V. G. González, R. Fullerton, E. M. Tippmann and R. K. Allemann, J. Am. Chem. Soc., 2011, 133, 13906 CrossRef CAS.
- A. Deligeorgopoulou and R. K. Allemann, Biochemistry, 2003, 42, 7741 CrossRef CAS.
- X. Lin and D. E. Cane, J. Am. Chem. Soc., 2009, 131, 6332 CrossRef CAS.
- B. T. Greenhagen, P. E. O'Maille, J. P. Noel and J. Chappell, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9826 CrossRef CAS.
- M. K. Julsing, PhD thesis, University of Groningen, 1975.
- Y. Yoshikuni, V. J. J. Martin, T. E. Ferrin and J. D. Keasling, Chem. Biol., 2006, 13, 91 CrossRef CAS.
- Y. Yoshikuni, T. E. Ferrin and J. D. Keasling, Nature, 2006, 440, 1078 CrossRef CAS.
- F. Lopez-Gallego, G. T. Wawrzyn and C. Schmidt-Dannert, Appl. Environ. Microbiol., 2010, 76, 7723 CrossRef CAS.
- F. Lopez-Gallego, S. A. Agger, D. Abate-Pella, M. D. Distefano and C. Schmidt-Dannert, ChemBioChem, 2010, 11, 1093 CrossRef CAS.
- S. Green, C. J. Squire, N. J. Nieuwenhuizen, E. N. Baker and W. Laing, J. Biol. Chem., 2009, 284, 8652 Search PubMed.
- D. E. Cane and R. M. Watt, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1547 CrossRef CAS.
- J. Y. Jiang, X. F. He and D. E. Cane, Nat. Chem. Biol., 2007, 3, 711 CrossRef CAS.
- D. E. Cane, G. H. Yang, R. M. Coates, H. J. Pyun and T. M. Hohn, J. Org. Chem., 1992, 57, 3454 CrossRef CAS.
- J. A. Faraldos, B. Kariuki and R. K. Allemann, J. Org. Chem., 2010, 75, 1119 CrossRef CAS.
- J. A. Faraldos and R. K. Allemann, Org. Lett., 2011, 13, 1202 CrossRef CAS.
- D. E. Cane, P. C. Prabhakaran, J. S. Oliver and D. B. McIlwaine, J. Am. Chem. Soc., 1990, 112, 3209 CrossRef CAS.
- D. E. Cane and Q. Xue, J. Am. Chem. Soc., 1996, 118, 1563 CrossRef CAS.
- C. R. Benedict, J. L. Lu, D. W. Pettigrew, J. G. Liu, R. D. Stipanovic and H. J. Williams, Plant Physiol., 2001, 125, 1754 CrossRef CAS.
- I. Alchanati, J. A. A. Patel, J. G. Liu, C. R. Benedict, R. D. Stipanovic, A. A. Bell, Y. X. Cui and C. W. Magill, Phytochemistry, 1998, 47, 961 CAS.
- K. Nabeta, M. Fujita, K. Komuro, K. Katayama and T. Takasawa, J. Chem. Soc., Perkin Trans. 1, 1997, 2065 RSC.
- K. Nabeta, K. Kigure, M. Fujita, T. Nagoya, T. Ishikawa, H. Okuyama and T. Takasawa, J. Chem. Soc., Perkin Trans. 1, 1995, 1935 RSC.
- D. E. Cane, M. Tandon and P. C. Prabhakaran, J. Am. Chem. Soc., 1993, 115, 8103 CrossRef CAS.
- D. E. Cane and M. Tandon, Tetrahedron Lett., 1994, 35, 5355 CrossRef CAS.
- D. E. Cane and S. W. Weiner, Can. J. Chem., 1994, 72, 118 CrossRef CAS.
- D. E. Cane, J. S. Oliver, P. H. M. Harrison, C. Abell, B. R. Hubbard, C. T. Kane and R. Lattman, J. Am. Chem. Soc., 1990, 112, 4513 CrossRef CAS.
- P. H. M. Harrison, J. S. Oliver and D. E. Cane, J. Am. Chem. Soc., 1988, 110, 5922 CrossRef CAS.
- C. O. Schmidt, H. J. Bouwmeester, S. Franke and W. A. Konig, Chirality, 1999, 11, 353 CrossRef CAS.
- S. Picaud, P. Mercke, X. F. He, O. Sterner, M. Brodelius, D. E. Cane and P. E. Brodelius, Arch. Biochem. Biophys., 2006, 448, 150 CrossRef CAS.
- S. H. Kim, K. Heo, Y. J. Chang, S. H. Park, S. K. Rhee and S. U. Kim, J. Nat. Prod., 2006, 69, 758 CrossRef CAS.
- P. E. O'Maille, J. Chappell and J. P. Noel, Anal. Biochem., 2004, 335, 210 CrossRef CAS.
- D. J. Schenk, C. M. Starks, K. R. Manna, J. Chappell, J. P. Noel and R. M. Coates, Arch. Biochem. Biophys., 2006, 448, 31 CrossRef CAS.
- D. J. Miller, J. L. Gao, D. G. Truhlar, N. J. Young, V. Gonzalez and R. K. Allemann, Org. Biomol. Chem., 2008, 6, 2346 CAS.
- Y. Hu, W. K. W. Chou, R. Hopson and D. E. Cane, Chem. Biol., 2011, 18, 32 CrossRef CAS.
- D. Arigoni, Pure Appl. Chem., 1975, 41, 219 CrossRef CAS.
- X. F. He and D. E. Cane, J. Am. Chem. Soc., 2004, 126, 2678 CrossRef CAS.
- J. Y. Jiang, X. F. He and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 8128 CrossRef CAS.
- J. Y. Jiang and D. E. Cane, J. Am. Chem. Soc., 2008, 130, 428 CrossRef CAS.
- X. Lin, R. Hopson and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 6022 CrossRef CAS.
- W. K. W. Chou, I. Fanizza, T. Uchiyama, M. Komatsu, H. Ikeda and D. E. Cane, J. Am. Chem. Soc., 2010, 132, 8850 CrossRef CAS.
- C. M. Wang, R. Hopson, X. Lin and D. E. Cane, J. Am. Chem. Soc., 2009, 131, 8360 CrossRef CAS.
- C. D. Poulter, J. C. Argyle and E. A. Mash, J. Biol. Chem., 1978, 253, 7227 CAS.
- R. Croteau, Arch. Biochem. Biophys., 1986, 251, 777 CrossRef CAS.
- D. E. Cane, G. H. Yang, Q. Xue and J. H. Shim, Biochemistry, 1995, 34, 2471 CrossRef CAS.
- D. J. Miller, F. L. Yu, D. W. Knight and R. K. Allemann, Org. Biomol. Chem., 2009, 7, 962 CAS.
- J. A. Faraldos, Y. X. Zhao, P. E. O'Maille, J. P. Noel and R. M. Coates, ChemBioChem, 2007, 8, 1826 CrossRef CAS.
- P. E. O'Maille, J. Chappell and J. P. Noel, Arch. Biochem. Biophys., 2006, 448, 73 CrossRef CAS.
- J. A. Faraldos, P. E. O'Maille, N. Dellas, J. P. Noel and R. M. Coates, J. Am. Chem. Soc., 2010, 132, 4281 CrossRef CAS.
- J. P. Noel, N. Dellas, J. A. Faraldos, M. Zhao, B. A. Hess, L. Smentek, R. M. Coates and P. E. O'Maille, ACS Chem. Biol., 2010, 5, 377 CrossRef CAS.
- D. J. Miller, F. L. Yu, N. J. Young and R. K. Allemann, Org. Biomol. Chem., 2007, 5, 3287 CAS.
- K. A. Rising, C. M. Starks, J. P. Noel and J. Chappell, J. Am. Chem. Soc., 2000, 122, 1861 CrossRef CAS.
- R. K. Allemann, N. J. Young, S. H. Ma, D. G. Truhlar and J. L. Gao, J. Am. Chem. Soc., 2007, 129, 13008 CrossRef CAS.
- P. E. O'Maille, B. Greenhagen, J. Chappell, Y. Zhao, R. M. Coates and J. P. Noel, in Terpnet 2003, Lexington, Kentucky USA, 2003 Search PubMed.
- J. A. Faraldos, S. Q. Wu, J. Chappell and R. M. Coates, J. Am. Chem. Soc., 2010, 132, 2998 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |