Large scale synthesis of carbon nanospheres and their application as electrode materials for heavy metal ions detection†
Keming
Pan
,
Hai
Ming
,
Yang
Liu
* and
Zhenhui
Kang
*
Institute of Functional Nano & Soft Materials (FUNSOM) and Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices, Soochow University, Suzhou 215123, China. E-mail: zhkang@suda.edu.cn; yangl@suda.edu.cn; Fax: +86-512-65882846; Tel: +86-512-65880957
First published on 31st October 2011
Abstract
Large-scale and monodisperse colloidal carbon nanospheres (CNSs) were synthesized with the assistance of polyoxometalates (POMs) under hydrothermal conditions. The POMs could act not only as a catalyst to promote the glucose dehydration process, but also as a stabilizer to prevent the aggregation of CNSs. The products were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and Fourier-transform infrared spectroscopy (FT-IR). The diameters of CNSs from 100 nm to 900 nm can be controlled by different acidic and oxidizing properties of POMs. This method was demonstrated to be simple, reliable and reproducible. The conversion yield of CNSs prepared with the addition of phosphomolybdic acid was up to 37%. Based on the experiment, the possible mechanism for CNSs formation was proposed. After modification of CNSs with cysteine, the samples were utilized as electrode materials for sensitive heavy metal ions detection. The detection limits for lead(II) and cadmium(II) were 1 × 10−8 M and 2 × 10−7 M, respectively. The results highlight the potential application of CNSs as electrode materials for biosensors and catalysis.
Introduction
Over the past few decades, carbon nanomaterials with tunable size and shape have been paid tremendous attention owing to their unique properties and potential applications in physics, chemistry and electronic fields.1–5 Among them, carbon nanospheres (CNSs) have attracted considerable attention due to their excellent properties and potential applications in the fields of gas storage,6–8 sensing,9,10catalyst supports,11,12lithium-ion batteries13–15 and so on. Numerous methods have been employed for the synthesis of CNSs, such as chemical vapor deposition (CVD),16–19pyrolysis of carbohydrate and polymers,20–23 hydrothermal and solvothermal treatment,24–26 ultrasonic treatment27,28 and arc-discharge process.29 However, most of these methods require complicated and expensive equipments. Some carbon precursors are toxic, expensive and non-renewable. And it is difficult to obtain the CNSs with the desired sizes. Therefore, the synthesis of CNSs with desired diameters and shapes seems to be very important and urgent for the development of micro-/nano-physics and chemistry.In concern of the environment, economy and energy, biomass resources are now becoming important as alternative resources.30–32Glucose is an important source of physiological energy. It can be obtained from decomposition of various renewable biomass resources, such as polysaccharose, starch and cellulose. It is also cheap, easily available experimental material. Considering all of the above advantages, the preparation of CNSs from glucose is very promising.33 However, the CNSs formed by glucose were prone to merge together under hydrothermal conditions, and the productivity was low. These problems make it difficult to meet the demand of large scale CNSs with high quality in potential applications.
Polyoxometalates (POMs) are a valuable class of inorganic compounds,34,35 and have been extensively investigated due to their tunability of size, charge of a polyoxometallic cage, acidic and oxidizing properties.36,37 In the area of catalysis, they were used as both Brønsted acid catalysts and oxidation catalysts since the late 1970s. For example, Keggin-type POMs have been widely used as homogeneous and heterogeneous catalysts for the oxidation of alcohols, aldehydes, isobutyric acid, or alkanes.38,39
In light of the merit of previous works, we believe POMs may exhibit excellent catalytic abilities for synthesis of high quality CNSs. In this work, we demonstrate the large scale synthesis (the yield of CNSs was up to 37%) of well-defined CNSs with the assistance of POMs. Three kinds of Keggin-type POMs were employed to catalyze the synthetic process, to improve the productivity of CNSs and control the diameter of CNSs from 100 to 900 nm. This method was proved to be simple, reliable and reproducible. The CNSs (100 nm in diameter) were modified with cysteine (Cys-CNSs), and utilized as electrode materials for detection of heavy metal ions (Pb2+, Cd2+), which were widely distributed in water, plants, air, and have been the fatal threat to the environment and human health due to their accumulative characters. The cysteine can especially bind heavy metals and further enhance the detection sensitivity. The result indicated that the Cys-CNSs were sensitive electrode materials.
Experimental section
Synthesis of CNSs
All reagents were of analytical grade. The preparation of CNSs was quite straightforward. In a typical procedure, 2.252 g of C6H12O6 was dissolved in 25 mL distilled water to form 0.5 M glucose solution. Then 0.2 g of phosphotungstic acid (H3PW12O40·xH2O) was added into the glucose solution, and stirred with a magnetic blender for 5 min. The mixture was transferred to a 30 mL Teflon-sealed autoclave, and maintained at 160 °C for 8 h. After reaction, the products were collected from the autoclave, washed with absolute ethanol and distilled water consecutively, and finally dried under vacuum at 60 °C for 5 h. The conversion yields of CNSs were calculated, the equation is shown as follows: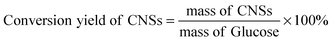 | (1) |
Modification of CNSs with cysteine
The modification of CNSs with cysteine can be found elsewhere.40,41 In a typical procedure, 5 mg of CNSs was suspended in a solution containing 0.1 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and 0.1 M N-hydroxysuccinimide (NHS) in 0.1 M phosphate buffer (pH 5.5)42 with constant agitation for 1 h. Then the products were centrifuged, washed with deionized water. The samples were resuspended into a solution containing 0.1 M cysteine in phosphate buffer overnight with constant stirring. Finally, the CNSs were collected by centrifugation and washed with deionized water. The cysteine-modified CNSs are referred to as “Cys-CNSs” in this document.Electrode preparation
A glassy carbon electrode (GCE) was polished carefully with smaller sizes of alumina slurries (1.0 and 0.05 μm), and sonicated in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 nitric acid, acetone, and pure water after each stage of polishing, successively. A 0.5 mg mL−1 suspension of Cys-CNSs in water was sonicated for 15 minutes. 5 μL of Cys-CNSs suspension was cast on the GCE surface and left to dry under ambient conditions. And then a drop of the Nafion solution (5 μL, 0.5 wt%) was placed on the electrode surface and the solvents were left to evaporate at room temperature.
:1 nitric acid, acetone, and pure water after each stage of polishing, successively. A 0.5 mg mL−1 suspension of Cys-CNSs in water was sonicated for 15 minutes. 5 μL of Cys-CNSs suspension was cast on the GCE surface and left to dry under ambient conditions. And then a drop of the Nafion solution (5 μL, 0.5 wt%) was placed on the electrode surface and the solvents were left to evaporate at room temperature.
Characterization
Scanning electron microscopy (SEM) images were taken on a FEI-quanta 200F scanning electron microscope with acceleration voltage of 20 kV. Transmission electron micrographs (TEM) were taken on a FEI-Tecnai F20 (200 kV) transmission electron microscope (FEI). Fourier transform infrared spectroscopy (FT-IR) was investigated by Hyperion 3000 (Bruker). The particle size distribution of as-synthesized CNSs was performed on Malvern Zen 3690. All electrochemical measurements were performed using a CHI 750B (Shanghai CH Instruments, China) Electrochemical Workstation.Results and discussion
Characterization of CNSs
In this paper, the CNSs were prepared from glucose with the assistance of POMs at 160 °C. The temperature is higher than the normal glycosidation temperature and would lead to polymerization and carbonization.43,44 Typical SEM and TEM images of CNSs prepared with the addition of different POMs are shown in Fig. 1, exhibiting the perfectly spherical morphology and monodisperse diameter. Detailed TEM images, insets in Fig. 1a and b, clearly show that the CNSs prepared with the addition of phosphomolybdic acid (H3PMo12O40·xH2O) and phosphotungstic acid (H3PW12O40·xH2O) have an average diameter of 100 and 230 nm, respectively. The particle size distribution of these samples is shown in Fig. S1 (ESI†), and the Brunauer–Emmett–Teller adsorption isotherms of these samples are shown in Fig. S2 (ESI†). Interestingly, we found that the diameter of CNSs increased to 800–900 nm when using silicotungstic acid (H4SiW12O40·xH2O) as the additive, shown in Fig. 1c. That is to say, the diameter of CNSs could be controlled by different kinds of POMs. This phenomenon may be attributed to different acidic and oxidizing properties of POMs. | ||
| Fig. 1 SEM images of the obtained CNSs, 0.5 M glucose solution with the addition of (a) H3PMo12O40·xH2O, (b) H3PW12O40·xH2O acid, (c) H4SiW12O40·xH2O. The scale bars of TEM images inset in (a) and (b) are 100 nm and 200 nm, respectively. | ||
A series of experiments were carried out to investigate the influence of POMs, concentration of glucose and reaction time. The SEM images of CNSs obtained in the absence of POMs while keeping other conditions unchanged are shown in Fig. 2a and b. It could be observed that these as-prepared CNSs were small in size, but they were prone to adhere together, forming chain structures, and at the same time, the CNSs transformed into irregular vesicles in some cases (Fig. 2b). This phenomenon could be ascribed to the formation mechanism of CNSs, which was previously reported.33,44 As glucose aqueous solution (without additive POMs) under hydrothermal conditions, they were dehydrated and polymerized together, forming a kind of amphiphilic compounds. With the polymerization of glucose molecules, these amphiphilic compounds would form micelles, with the hydrophobic groups making up the core of the micelles and the hydrophilic hydroxyls occupying the surface. This is followed by the growth of the micelle particles through the combination of the surface hydroxyls with the nearest free molecules until the glucose molecules are exhausted. As the shape of micelles may vary under different reaction conditions, the CNSs obtained in this way would exhibit different shapes. The result indicated that the usage of POMs is critical to obtain CNSs with perfect spherical morphology and monodisperse diameter.
 | ||
| Fig. 2 SEM images of CNSs prepared at 160 °C, (a,b) with 0.5 M glucose but without POMs, (c) with 1 M glucose, 0.2 g H3PW12O40·xH2O, and (d) with 0.5 M glucose, 0.3 g H3PW12O40·xH2O. | ||
Upon increasing the concentration of glucose from 0.5 M to 1 M while keeping other conditions unchanged (e.g., 160 °C, 0.2 g H3PW12O40·xH2O), the size distribution of CNSs was getting worse, shown in Fig. 2c. It could be observed that although the CNSs still maintained the spherical morphology, the diameters of CNSs ranged from 400 nm to 4 μm. In comparison, as the amount of H3PW12O40·xH2O increased from 0.2 to 0.3 g, the similar result was observed, as shown in Fig. 2d. The unexpected phenomenon may be ascribed to the change of pH value and high concentration of POM anions in the mixed solution, which accelerated the CNSs formation process. From the above experimental result, it is obvious that the proper control on the concentration of glucose and POMs (e.g., 0.5 M, 0.2 g) is very essential for the preparation of monodisperse CNSs.
The XRD pattern of the as-prepared CNSs is shown in Fig. 3. In the XRD pattern, only a wide peak at 2θ ≈ 20° was observed, indicating the highly amorphous nature of the CNSs. No diffraction peaks belonging to impurity were observed, indicating the high purity of the CNSs.
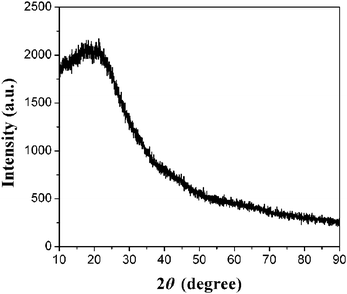 | ||
| Fig. 3 XRD pattern of CNSs. | ||
The FT-IR spectrum of CNSs is shown in Fig. 4. The peaks at 3423 and 2972 cm−1 are characteristic peaks of O–H and C–H bonds, and the bands at 1703 and 1625 cm−1 are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC vibrations, respectively. The bands at 1000–1300 cm−1 are due to the existence of the C–C stretching, C–OH stretching and −OH bending vibration. These residual hydroxyl groups (−OH), aldehyde groups (−CHO) and carboxyl groups (−COOH) were covalently bonded to the carbon frameworks, improving the hydrophilicity and stability of CNSs in aqueous systems, and greatly widen their potential applications in diagnostics, drug delivery and as templates for hybrid core/shell structures.45–48
O and CC vibrations, respectively. The bands at 1000–1300 cm−1 are due to the existence of the C–C stretching, C–OH stretching and −OH bending vibration. These residual hydroxyl groups (−OH), aldehyde groups (−CHO) and carboxyl groups (−COOH) were covalently bonded to the carbon frameworks, improving the hydrophilicity and stability of CNSs in aqueous systems, and greatly widen their potential applications in diagnostics, drug delivery and as templates for hybrid core/shell structures.45–48
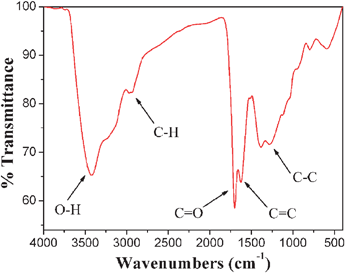 | ||
| Fig. 4 FT-IR spectrum of CNSs. | ||
Formation mechanism
According to the literature,33,44,49,50 the transformation of glucose molecules into CNSs would involve several processes, such as dehydration, polymerization, and carbonization. First of all, the intramolecular dehydration would occur easily under hydrothermal conditions, since glucose is a polyhydroxy compound. The major product of glucose dehydration was hydroxymethylfurfural (HMF), which was composed of hydroxyl, aldehyde group and furan ring.51–53 There were other byproducts such as levulinic acid, dihydroxyacetone, and formic acid,54,55 which were formed by decomposition of glucose and HMF. Also, there were a few amphiphilic compounds formed by intermolecular dehydration of glucose.Then, the hydration of the aldehyde group of HMF may result in the further elimination of formic acid,54 and the consequent protonation of the α-position C atom with another furan ring may occur. The elimination of the hydroxyl group on HMF and followed by nucleophilic addition would generate CH2 groups, which act as linkers between furan rings.56 The formation of specific CH3 and COOH end groups was previously reported.56 These reactions of HMF that occurred at high temperature would form carbonaceous scaffolds,49 which were composed of the mutually connected polyfuran network and cross-linked by aliphatic carbons. It is possible that the initial stages of nucleation occurred as the concentration of carbonaceous scaffolds reached a critical micelle concentration.44,57 The “core” is decorated with aliphatic chains and carbonyl groups on the surface.
At last, the growth of the “core” is through the combination of the surface hydroxyls, aldehydes and carboxyl groups with the nearest free molecules until the molecules are exhausted.33,44 Finally, the monodisperse CNSs were obtained.
Based on the previous reports,36–38 the effect of POMs during the CNSs formation could be attributed to the following: (1) they could catalyze/promote the reaction process; (2) they could effectively control the diameters of CNSs, the possible mechanism is shown in Scheme 1.
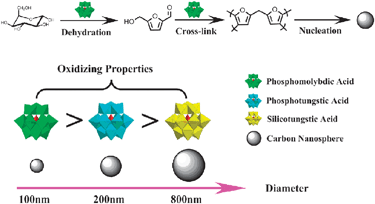 | ||
| Scheme 1 The possible mechanism for the formation of CNSs prepared with the addition of POMs. | ||
In order to understand the process vividly, the appearance of resulting solutions obtained at different reaction times is shown in Fig. 5. It is obvious that colors of solution turned from colorless, orange, red to brown and reddish-black gradually. The solutions obtained at 1 h, 2 h were colorless and orange, and no precipitates were observed after centrifugation, shown in Fig. 5. The result was in accordance with our proposition: the additive POMs could accelerate the dehydration of glucose and catalyze the polymerization of HMF.58 The orange solution obtained at 2 h indicated the initial stage of the cross-link process of HMF, since glucose, HMF levulinic acid, and formic acid were colorless in solution. The dehydration and polymerization processes occurred continuously and upon reaching a critical point, a short single burst of nucleation resulted; the nucleation occurred between 2 h and 4 h. As a result, some precipitates were observed after centrifugation, the conversion yield was calculated and shown in Fig. 5. As there was an abundance of hydroxyl, aldehyde and carboxyl groups on the surface of particles, the cross-link process between particle to particle would occur easily, which leads to the aggregation of CNSs, as shown in Fig. 2a. This situation will be changed with the addition of POMs, which could oxidize the hydroxyl and aldehyde groups to carboxyl groups. The high ratio of carboxyl groups on the surface of particles results in separation of each particle, owing to the negative charge of carboxyl groups. And this result was in accordance with the diameters of CNSs that could be tuned by POMs with different oxidizing properties. The particles accumulated molecules from solution and grew up to CNSs eventually. It is worth noting that the conversion yield of CNSs prepared with the addition of phosphomolybdic acid was up to 37% (shown in Fig. 5, curve a), significantly higher than that of H3PW12O40·xH2O (curve b) and silicotungstic acid (curve c). It means that the CNSs with a diameter of 100 nm could be obtained in a large scale, and have potential for further industrial applications.
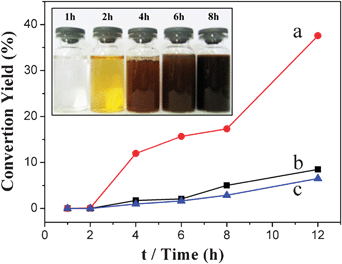 | ||
| Fig. 5 The conversion yields of CNSs with (a) H3PMo12O40·xH2O, (b) H3PW12O40·xH2O, (c) H4SiW12O40·xH2O, and appearance of resulting solution with time (inset). | ||
Electrochemical characteristics of the electrode surface
As cysteine, a nonessential amino acid, has a very high binding constant for some toxic heavy metal ions, the modification of CNSs with cysteine could enhance the detection of heavy metal ions.40,41 The FT-IR spectrum of Cys-CNSs is shown in Fig. 6. The characteristic C–N stretching of secondary aromatic amine appears at 1384 cm−1, which is attributed to the unique amidation reaction. The FT-IR spectrum provided evidence that cysteine was covalently bonded to the CNSs. With the help of modified cysteine, the Cys-CNSs may be especially modified with heavy metals which could further enhance the detection sensitivity.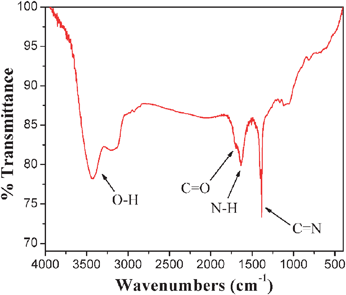 | ||
| Fig. 6 FT-IR spectrum of Cys-CNSs. | ||
A Cys-CNSs modified glassy carbon electrode (GCE) was investigated viaSWASV methods. Calibration plots for determination of Pb2+ and Cd2+ on the Cys–CNSs were achieved in 0.1 M acetate buffer (pH 4.5) under the same conditions. SWASV for different concentrations of Pb2+ and Cd2+ are shown in Fig. 7a and c, respectively. The calibration curves were linear over the range 1 × 10−8 to 1 × 10−6 M for Pb2+ and 2 × 10−7 M to 2 × 10−6 M for Cd2+ (Fig. 7b and d).
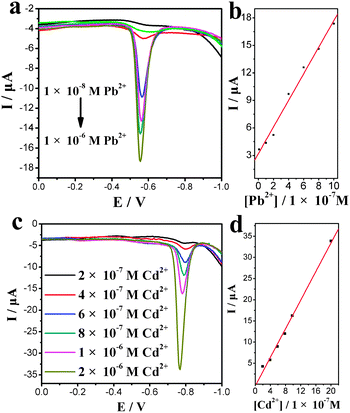 | ||
| Fig. 7 SWASV analysis of Pb2+ and Cd2+ with various concentrations. (a) SWASV of Pb2+ accumulated on the Cys–CNSs electrode, (b) calibration plot of anodic current versus different concentrations, 1 × 10−8–1 × 10−6 M Pb2+, (c) SWASV of Cd2+ accumulated on the Cys-CNSs electrode, (d) calibration plot of anodic current versus different concentrations, 2 × 10−7–2 × 10−6 M Cd2+. | ||
The calibration curves were Pb2+ with y = 3.202 + 1.45x (R = 0.9858, N = 7) and Cd2+ with y = −0.34347 + 1.68672x (R = 0.99164, N = 5). The detection limits were 1 × 10−8 M and 2 × 10−7 M for Pb2+ and Cd2+, respectively. Compared with Cd2+, Pb2+ possessed a wider linear range and lower detection limits, while the linear correlation coefficient was poorer. These results revealed that Cys-CNSs could be used to effectively detect Pb2+ and Cd2+, especially Pb2+.
Conclusions
In conclusion, we demonstrate that well-defined CNSs could be obtained on a large-scale with the assistance of POMs. The POMs could catalyze the CNS formation process and control the diameters. This method is proved to be reproducible, of low cost and environmentally friendly. The conversion yield of CNSs prepared with the addition of H3PW12O40·xH2O was up to 37%. The electrochemical studies show that the Cys-CNSs were sensitive electrode materials. The detection limits for Pb2+ and Cd2+ were 1 × 10−8 M and 2 × 10−7 M, respectively. The result indicated that the CNSs may hold wide potential applications for development of new biosensors and catalysis.Acknowledgements
This work is supported by the National Basic Research Program of China (973 Program) (No. 2010CB934500), National Natural Science Foundation of China (NSFC) (No. 51132006, 91027041, 21073127, 21071104, 20801010, 20803008), A Foundation for the Author of National Excellent Doctoral Dissertation of P R China (FANEDD)(No. 200929) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- G. L. Che, B. B. Lakshmi, E. R. Fisher and C. R. Martin, Nature, 1998, 393, 346 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. X. Cui, I. Khayrullin, S. O. Dantas, I. Marti and V. G. Ralchenko, Science, 1998, 282, 897 CrossRef CAS.
- M. Kocirik, J. Brych and J. Hradil, Carbon, 2001, 39, 1919 CrossRef CAS.
- L. Tosheva, J. Parmentier, V. Valtchev, C. Vix-Guterl and J. Patarin, Carbon, 2005, 43, 2474 CrossRef CAS.
- Q. Wang, F. Y. Cao, Q. W. Chen and C. L. Chen, Mater. Lett., 2005, 59, 3738 CrossRef CAS.
- Y. D. Xia and R. Mokaya, Adv. Mater., 2004, 16, 886 CrossRef CAS.
- X. J. Chen, K. Zhang, J. J. Zhou, J. Xuan, W. Yan, L. P. Jiang and J. J. Zhu, Biosens. Bioelectron., 2010, 25, 1130 CrossRef CAS.
- L. Q. Cheng, Y. L. Liu, J. X. Zhang, D. S. Yuan, C. W. Xu and G. H. Sun, Prog. Chem., 2006, 18, 1298–1304 CAS.
- Z. C. Zhou, Q. F. Yan, F. B. Su and X. S. Zhao, J. Mater. Chem., 2005, 15, 2569 RSC.
- R. Alcantara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222 CrossRef CAS.
- K. T. Lee, J. C. Lytle, N. S. Ergang, S. M. Oh and A. Stein, Adv. Funct. Mater., 2005, 15, 547–556 CrossRef CAS.
- W. Qian, L. Z. Wei, F. Y. Cao and Q. W. Chen, Carbon, 2006, 44, 1303–1307 CrossRef CAS.
- H. S. Qian, F. M. Han, B. Zhang, Y. C. Guo, J. Yue and B. X. Peng, Carbon, 2004, 42, 761 CrossRef CAS.
- J. Y. Miao, D. W. Hwang, C. C. Chang, S. H. Lin, K. V. Narasimhulu and L. P. Hwang, Diamond Relat. Mater., 2003, 12, 1368 CrossRef CAS.
- J. Y. Miao, D. W. Hwang, K. V. Narasimhulu, P. I. Lin, Y. T. Chen, S. H. Lin and L. P. Hwang, Carbon, 2004, 42, 813 CrossRef CAS.
- Y. D. Xia and R. Mokaya, Adv. Mater., 2004, 16, 1553 CrossRef CAS.
- Y. Z. Jin, C. Gao, W. K. Hsu, Y. Q. Zhu, A. Huczko, M. Bystrzejewski, M. Roe, C. Y. Lee, S. Acquah, H. Kroto and D. R. M. Walton, Carbon, 2005, 43, 1944 CrossRef CAS.
- B. Friedel and S. Greulich-Weber, Small, 2006, 2, 859 CrossRef CAS.
- S. O. Zhao, C. Y. Wang, M. M. Chen, Z. Q. Shi and N. Liu, New Carbon Mater., 2010, 25, 438 CrossRef CAS.
- N. Koprinarov and M. Konstantinova, J. Mater. Sci., 2011, 46, 1494 CrossRef CAS.
- Y. Yan, H. F. Yang, F. Q. Zhang, B. Tu and D. Y. Zhao, Carbon, 2007, 45, 2209 CrossRef CAS.
- S. X. Liu and J. Sun, J. Inorg. Mater., 2009, 24, 1132 CrossRef CAS.
- C. Y. Chen, X. D. Sun, X. C. Jiang, D. Niu, A. B. Yu, Z. G. Liu and J. G. Li, Nanoscale Res. Lett., 2009, 4, 971 CrossRef CAS.
- Z. X. Wang, L. P. Yu, W. Zhang, Z. Y. Zhu, G. W. He, Y. Chen and G. Hu, Phys. Lett. A, 2003, 307, 249 CrossRef CAS.
- B. Jokic, S. Drmanic, T. Radetic, J. Krstic, R. Petrovic, A. Orlovic and D. Janackovic, Mater. Lett., 2010, 64, 2173 CrossRef CAS.
- W. M. Qiao, Y. Song, S. Y. Lim, S. H. Hong, S. H. Yoon, I. Mochida and T. Imaoka, Carbon, 2006, 44, 187 CrossRef CAS.
- S. Zhao, C. Y. Wang, M. M. Chen and Z. Q. Shi, Mater. Lett., 2008, 62, 3322 CrossRef CAS.
- H. Q. Wang, Q. F. Dai, Q. Y. Li, J. H. Yang, X. X. Zhong, Y. G. Huang, A. N. Zhang and Z. X. Yan, Solid State Ionics, 2009, 180, 1429 CrossRef CAS.
- A. M. Herring, J. T. McKinnon, B. D. McCloskey, J. Filley, K. W. Gneshin, R. A. Pavelka, H. J. Kleebe and D. J. Aldrich, J. Am. Chem. Soc., 2003, 125, 9916 CrossRef CAS.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 597 CrossRef.
- D. E. Katsoulis, Chem. Rev., 1998, 98, 359 CrossRef CAS.
- T. Yamase, Chem. Rev., 1998, 98, 307 CrossRef CAS.
- E. Coronado and C. J. Gomez-Garcia, Chem. Rev., 1998, 98, 273 CrossRef CAS.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS.
- M. Misono, Chem. Commun., 2001, 1141 RSC.
- L. Plault, A. Hauseler, S. Nlate, D. Astruc, J. Ruiz, S. Gatard and R. Neumann, Angew. Chem., Int. Ed., 2004, 43, 2924 CrossRef CAS.
- B. Sljukic, G. G. Wildgoose, A. Crossley, J. H. Jones, L. Jiang, T. G. J. Jones and R. G. Compton, J. Mater. Chem., 2006, 16, 970 RSC.
- G. G. Wildgoose, H. C. Leventis, A. O. Simm, J. H. Jones and R. G. Compton, Chem. Commun., 2005, 3694 RSC.
- D. H. Chen, B. Hu, M. He and C. Z. Huang, Microchem. J., 2010, 95, 90 CrossRef CAS.
- G. C. A. Luijkx, F. Vanrantwijk, H. Vanbekkum and M. J. Antal, Carbohydr. Res., 1995, 272, 191 CrossRef CAS.
- Q. Wang, H. Li, L. Q. Chen and X. J. Huang, Solid State Ionics, 2002, 152, 43 CrossRef.
- M. L. Hans and A. M. Lowman, Curr. Opin. Solid State Mater. Sci., 2002, 6, 319 CrossRef CAS.
- S. C. Tang, S. Vongehr and X. K. Meng, J. Phys. Chem. C, 2010, 114, 977 CAS.
- F. B. Su, Z. Q. Tian, C. K. Poh, Z. Wang, S. H. Lim, Z. L. Liu and J. Y. Lin, Chem. Mater., 2010, 22, 832 CrossRef CAS.
- S. H. Sun, F. Jaouen and J. P. Dodelet, Adv. Mater., 2008, 20, 3900 CrossRef CAS.
- N. Baccile, G. Laurent, F. Babonneau, F. Fayon, M. M. Titirici and M. Antonietti, J. Phys. Chem. C, 2009, 113, 9644 CAS.
- Y. Shin, L. Q. Wang, I. T. Bae, B. W. Arey and G. J. Exarhos, J. Phys. Chem. C, 2008, 112, 14236 CAS.
- M. J. Antal, W. S. L. Mok and G. N. Richards, Carbohydr. Res., 1990, 199, 91–109 CrossRef CAS.
- K. Lourvanij and G. Rorrer, Ind. Eng. Chem. Res., 1993, 32, 11 CrossRef CAS.
- K. Lourvanij and G. L. Rorrer, Appl. Catal., A, 1994, 109, 147 CrossRef CAS.
- B. Girisuta, L. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339 CrossRef CAS.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 2163 CrossRef CAS.
- A. Gandini and M. Belgacem, Prog. Polym. Sci., 1997, 22, 1203 CrossRef CAS.
- M.-M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204 RSC.
- W. B. Kim, T. Voitl, G. J. Rodriguez-Rivera and J. A. Dumesic, Science, 2004, 305, 1280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1nj20756c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |