DOI:
10.1039/C1MT00144B
(Paper)
Metallomics, 2012,
4, 101-113
DNA, protein binding, cytotoxicity, cellular uptake and antibacterial activities of new palladium(II) complexes of thiosemicarbazone ligands: effects of substitution on biological activity†
Received
31st August 2011
, Accepted 29th September 2011
First published on 3rd November 2011
Abstract
The coordination propensities of 4(N,N′)-diethylaminosalicylaldehyde-4(N)-substituted thiosemicarbazones (H2L1–4) were investigated by reacting with an equimolar amount of [PdCl2(PPh3)2]. The new complexes were characterized by various spectroscopic techniques. The structure determination of the complexes [Pd(DeaSal-tsc)(PPh3)] (1), [Pd(DeaSal-mtsc)(PPh3)] (2) and [Pd(DeaSal-etsc)(PPh3)] (3) by X-ray crystallography showed that ligands are coordinated in a dibasic tridentate ONS donor fashion forming stable five and six membered chelate rings. The binding ability of complexes (1–4) to calf-thymus DNA (CT DNA) has been explored by absorption and emission titration methods. Based on the observations, an electrostatic and an intercalative binding mode have been proposed. The protein binding studies have been monitored by quenching of tryptophan and tyrosine residues in the presence of complexes using lysozyme as a model protein. As determined by MTT assays, complex 3 exhibited a higher cytotoxic effect towards human lung cancer cell line (A549) and liver cancer cells (HepG2). LDH, NO assay and cellular uptake of the complexes have been studied. Further, antibacterial activity studies of the complexes have been screened against the pathogenic bacteria such as Enterococcus faecalis, Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa, MIC50 values of the complexes showed that the complexes exhibited significant activity against the pathogens and among the complexes, 3 exhibited higher activity.
Introduction
Thiosemicarbazones are an important class of mixed hard/soft oxygen/nitrogen–sulfur containing chelates coordinated to the metal ion through the sulfur and one of the hydrazinic nitrogen atoms (N2 or N1).1 Thiosemicarbazones and their metal complexes have aroused considerable interest in coordination chemistry not only because of their versatile ligating properties but also due to their broad profile in biological activities.2–12 These activities are due to their ability to form chelates with metals causing drastic change in the biological properties. The interaction of transition metal complexes with DNA is a vibrant area of research.13–16DNA regulates most aspects of cell life and constitutes an important drug target. The design of compounds able to bind and react with selective nucleotidic sequences is of great importance in probing biological processes and in developing therapeutic drugs. An advantage of using transition metal complexes in such studies is that their ligands and metals can be conveniently varied to suit individual applications. Genomic DNA is most likely the main cellular target for platinum drugs. Especially for cisplatin, it has been demonstrated that the major antitumor activity originates from intrastrand cross links and the formation of DNA kinks.17 Therefore, DNA targeting drugs remain in the limelight and compounds acting towards cancer cells selectively over healthy cells are getting more attention.18–22 The detailed investigation of cisplatin activation, biotransformation, accumulation, coordinative binding, DNA kinking and unwinding explains much of the antitumour activity of this drug, originating from DNA damage.23 In the recent literature, there are some reports on ‘rule-breaker’ platinum compounds, which are highly active against tumor cells.24–26 Nowadays, the approaches are focused on the design of compounds that may form adducts with DNA, differently from the classical cisplatin. The mode of interaction of the newly designed platinum compounds with target biomolecules can be directed either via coordinative (interstrand cross linking), or via non-coordinative (intercalation, electrostatic, groove-binding or hydrogen-bonding) interactions.27,28DNA is the primary intracellular target of an anticancer complex because the interaction between these molecules and DNA can cause DNA damage in cancer cells, blocking the division of cancer cells and resulting in cell death. Recent approaches to anticancer therapy have involved agents that target specific cellular processes that are enhanced or altered in tumors. In particular, the discovery of matrix metalloproteinases (MMPs) as enzymes critically linked to metastasizing tumors has prompted the development of a class of drugs that have the potential to not only be potent, but act selectively on tumors as well.29 MMP inhibitors have reached later phase clinical trials and have much promise not only as anticancer drugs but in treatments for arthritis and other such pathological states.30Nitric oxide (NO) mediates both physiological and pathological processes.31,32 In addition to cardiovascular signaling, NO has been invoked to play a neurochemical role in learning and memory, and it is a powerful necrotic agent wielded by macrophages of the immune system. NO scavenging, where the toxic NO is converted to harmless nitrate, has a wide range of physiological significances.33 Whereas considerable effort has been invested to develop metal-based34–36 and other37,38probes for detecting nitric oxide, there has been significantly less progress in the synthesis of platforms capable of detecting other reactive nitrogen species (RNS).39 With the above objective in mind, we synthesized few platinum mimic palladium complexes containing N-substituted thiosemicarbazones and the new complexes have been characterized by various spectro-analytical and X-ray crystallographic techniques. The DNA, protein binding, cytotoxicity, cellular uptake, LDH, NO scavenging and antibacterial studies have been carried out to test the biological efficacy of the new Pd(II) complexes.
Experimental section
The ligands (H2L1–4) and the palladium precursor [PdCl2(PPh3)2] were prepared according to standard literature procedures.40 All the reagents used were of analar grade, were purified and dried according to the standard procedure.41
Synthesis
Synthesis of 4(N,N′)-diethylaminosalicylaldehydethiosemicarbazone [H2-DeaSal-tsc] (H2L1).
Thiosemicarbazide (0.92 g, 10 mmol) was dissolved in 40 ml of methanol with continuous stirring and it was gently heated for a period of 30 min. To this, a methanolic solution (20 ml) of 4(N,N′)-diethylaminosalicylaldehyde (1.94 g, 10 mmol) was added and the mixture was refluxed with stirring for 2 h. The mixture was then cooled to room temperature whereby a yellow crystalline compound precipitated. This was collected by filtration, washed well with cold methanol and dried under vacuum. The product is soluble in warm acetone, methanol, ethanol, dichloromethane, chloroform, DMF and DMSO.Yield: 45%. Anal. calcd for C12H18N4OS: C, 54.11; H, 6.81; N, 21.03; S, 12.04. Found: C, 54.00; H, 6.90; N, 21.11; S, 11.98%. IR (cm−1) in KBr: 3406 (νOH), 1628 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1286 (νC–O), 792 (νCS); 1H NMR (DMSO-d6, ppm): 11.04 (s, 1H, OH), 9.46 (s, 1H, NHCS), 8.17 (s, 1H, CHN), 7.83 and 7.63 (2br s, 1H each, NH2), 3.28–3.33 (m, 4H, CH2N), 1.08 (t, 6H, CH3), 6.07–7.50 (m, 3H, aromatic).
N), 1286 (νC–O), 792 (νCS); 1H NMR (DMSO-d6, ppm): 11.04 (s, 1H, OH), 9.46 (s, 1H, NHCS), 8.17 (s, 1H, CHN), 7.83 and 7.63 (2br s, 1H each, NH2), 3.28–3.33 (m, 4H, CH2N), 1.08 (t, 6H, CH3), 6.07–7.50 (m, 3H, aromatic).
A similar method as described above was followed to prepare all other thiosemicarbazone ligands.
Synthesis of 4(N,N′)-diethylaminosalicylaldehyde-4(N)-methylthiosemicarbazone [H2-DeaSal-mtsc] (H2L2).
The ligand [H2-DeaSal-mtsc] was prepared from methylthiosemicarbazide (1.05 g, 10 mmol) and 4(N,N′)-diethylaminosalicylaldehyde (1.94 g, 10 mmol). Yield: 55%. Anal. calcd for C13H20N4OS: C, 55.68; H, 7.18; N, 19.98; S, 11.43. Found: C, 55.59; H, 7.07; N, 19.86; S, 11.39%. FT-IR (cm−1) in KBr: 3350 (νOH), 1632 (νCN), 1248 (νC–O), 785(νCS); 1H NMR (DMSO-d6, ppm): 11.05 (s, 1H, OH), 9.46 (s, 1H, NHCS), 8.17 (s, 1H, NHCH3), 8.14 (s, 1H, CHN), 3.31 (q, 4H, CH2N), 6.08–7.53 (m, aromatic), 1.08 (t, 6H, CH3), 2.97 (d, 3H, CH3).
Synthesis of 4(N,N′)-diethylaminosalicylaldehyde-4(N)-ethylthiosemicarbazone [H2-DeaSal-etsc] (H2L3).
The ligand [H2-DeaSal-etsc] was prepared from ethylthiosemicarbazide (1.19 g, 10 mmol) and 4(N,N′)-diethylaminosalicylaldehyde (1.94 g, 10 mmol). Yield: 53%. Anal. calcd for C14H22N4OS: C, 57.11; H, 7.53; N, 19.02; S, 10.89. Found: C, 57.01; H, 7.48; N, 18.98; S, 10.80%. FT-IR (cm−1) in KBr: 3390 (νOH), 1634 (νCN), 1240 (νC–O), 787 (νCS); 1H NMR (DMSO-d6, ppm): 11.02 (s, 1H, OH), 9.53 (s, 1H, NHCS), 8.22 (s, 1H, NHC2H5), 8.17 (s, 1H, CHN), 6.01–7.66 (m, aromatic), 3.31 (q, 4H, CH2N), 3.51–3.58 (p, CH2), 1.06 (t, 3H, CH3), 1.11 (t, 6H, CH3).
Synthesis of 4(N,N′)-diethylaminosalicylaldehyde-4-phenylthiosemicarbazone [H2-DeaSal-ptsc] (H2L4).
The ligand [H2-DeaSal-ptsc] was prepared from phenylthiosemicarbazide (1.67 g, 10 mmol) and 4(N,N′)-diethylaminosalicylaldehyde (1.94 g, 10 mmol). Yield: 30%. Anal. calcd for C18H22N4OS: C, 63.13; H, 6.47; N, 16.35; S, 9.36. Found: C, 63.09; H, 6.31; N, 16.30; S, 9.34%. IR (cm−1) in KBr: 3380 (νOH), 1633 (νCN), 1239 (νC–O), 780 (νCS); 1H NMR (DMSO-d6, ppm): 11.44 (s, 1H, OH), 9.81 (s, 1H, NHCS), 9.54 (s, 1H, NHPh), 8.29 (s, 1H, CHN), 3.32 (q, 4H, CH2N), 1.09 (t, 6H, CH3), 6.09–7.68 (m, 8H, aromatic).
Preparation of new palladium(II) complexes.
Preparation of [Pd(DeaSal-tsc)(PPh3)] (1).
An ethanolic (25 cm3) solution of [PdCl2(PPh3)2] (0.200 g; 0.285 mmol) was slowly added to 4(N,N′)-diethylaminosalicylaldehydethiosemicarbazone [H2-DeaSal-tsc] (0.076 g; 0.285 mmol) in dichloromethane (25 cm3). The mixture was allowed to stand for 4 days at room temperature. Reddish orange solid formed was filtered, washed with petroleum ether (60–80 °C) and crystallized from dimethylformamide to yield red crystals. Yield: 56%. Mp 120 °C. Anal. calcd for C30H31N4OSPdP: C, 56.91; H, 4.94; N, 8.85; S, 5.06. Found: C, 56.88; H, 4.89; N, 8.79; S, 5.00%. FT-IR (cm−1) in KBr: 1579 (νCN), 1307 (νC–O), 747 (νC–S), 1437, 1096, 696 cm−1 (for PPh3); UV-Vis (CH2Cl2), λmax: 265 (43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 279) nm (dm3 mol−1 cm−1) (intra-ligand transition); 394 (38499), 410 (4971) nm (dm3 mol−1 cm−1) (MLCT); 1H NMR (DMSO-d6, ppm): 8.15 (d, (J = 8.6) 1H, CHN), 7.20–7.68 (m, aromatic), δ 7.75–7.78 and 7.85–7.89 (2br s, –NH2), δ 3.30–3.45 (q, CH2N), δ 1.04 and 1.09 (2t, 6H, –CH3).
279) nm (dm3 mol−1 cm−1) (intra-ligand transition); 394 (38499), 410 (4971) nm (dm3 mol−1 cm−1) (MLCT); 1H NMR (DMSO-d6, ppm): 8.15 (d, (J = 8.6) 1H, CHN), 7.20–7.68 (m, aromatic), δ 7.75–7.78 and 7.85–7.89 (2br s, –NH2), δ 3.30–3.45 (q, CH2N), δ 1.04 and 1.09 (2t, 6H, –CH3).
A similar method was followed to synthesize other complexes.
Preparation of [Pd(DeaSal-mtsc)(PPh3)] (2).
Complex 2 was prepared by the procedure as described for (1) with 4(N,N′)-diethylaminosalicylaldehyde-4(N)-methylthiosemicarbazone [H2-DeaSal-mtsc] (0.080 g; 0.285 mmol). Red solid formed was filtered, washed with petroleum ether (60–80 °C) and crystallized from dichloromethane and acetonitrile to yield red crystals. Yield: 68%. Mp 238 °C. Anal. calcd for C31H33N4OSPdP: C, 57.50; H, 5.14; N, 8.65; S, 4.95. Found: C, 57.41; H, 5.09; N, 8.59; S, 4.91%. FT-IR (cm−1) in KBr: 1626 (νCN), 1272 (νC–O), 748 (νC–S), 1419, 1090, 695 cm−1 (for PPh3); UV-Vis (CH2Cl2), λmax: 263 (25336) nm (dm3 mol−1 cm−1) (intra-ligand transition); 391 (18738) nm (dm3 mol−1 cm−1) (MLCT); 1H NMR (DMSO-d6, ppm): 8.22 (d, (J = 13.6) 1H, CHN), 6.00 (s, 1H, NHCH3), 3.36 (q, CH2N), 6.21–7.66 (m, aromatic), 2.70 (d, (J = 4.8) 3H, –CH3), 1.09 (t, 6H, –CH3).
Preparation of [Pd(DeaSal-etsc)(PPh3)] (3).
Complex 3 was prepared by the procedure as used for (1) with 4(N,N′)-diethylaminosalicylaldehyde-4(N)-ethylthiosemicarbazone [H2-DeaSal-etsc] (0.084 g; 0.285 mmol). Red solid formed was filtered, washed with petroleum ether (60–80 °C) and crystallized from dichloromethane and acetonitrile to yield red crystals. Yield: 65%. Mp 201 °C. Anal. calcd for C32H35N4OSPdP: C, 58.13; H, 5.33; N, 8.47; S, 4.85. Found: C, 58.09; H, 5.28; N, 8.40; S, 4.79%. FT-IR (cm−1) in KBr: 1582 (νCN), 1280 (νC–O), 747 (νC–S), 1435, 1092, 696 cm−1 (for PPh3); UV-vis (CH2Cl2), λmax: 264 (25336) nm (dm3 mol−1 cm−1) (intra-ligand transition); 396 (17538), 414 (14619) nm (dm3 mol−1 cm−1) (MLCT); 1H NMR (DMSO-d6, ppm): 8.22 (d, (J = 13.2) terminal –NH), 8.07 (s, –HCN), 7.15–7.66 (m, aromatic protons), 3.15 (q, CH2N), 3.32 (q, –CH2), 1.04 (t, 3H, –CH3), 1.09 (t, 6H, –CH3).
Preparation of [Pd(DeaSal-ptsc)(PPh3)] (4).
Complex 4 was prepared by the procedure as used for (1) with 4(N,N′)-diethylaminosalicylaldehyde-4(N)-phenylthiosemicarbazone [H2-DeaSal-ptsc] (0.098 g; 0.285 mmol). Yield: 68%. Mp 161 °C. Anal. calcd for C36H35N4OSPdP: C, 60.97; H, 4.97; N, 7.90; S, 4.52. Found: C, 60.91; H, 4.89; N, 7.82; S, 4.49%. FT-IR (cm−1) in KBr: 1604 (νCN), 1248 (νC–O), 749 (νC–S), 1434, 1097, 697 cm−1 (for PPh3); UV-Vis (CH2Cl2), λmax: 267 (33382) nm (dm3 mol−1 cm−1) (intra-ligand transition); 404 (25363), 421 (24377) nm (dm3 mol−1 cm−1) (MLCT); 1H NMR (DMSO-d6, ppm): 9.21 (s, terminal –NH), 8.37(d, J = 13.2 –HCN), 6.85–7.70 (m, aromatic protons), 3.43 (q, CH2N), 1.05 (t, 6H, –CH3).
Measurements
Infrared spectra were measured as KBr pellets on a Nicolet instrument between 400–4000 cm−1. Elemental analyses of carbon, hydrogen, nitrogen, and sulfur were determined using a Vario EL III CHNS at the Department of Chemistry, Bharathiar University, Coimbatore, India. The electronic spectra of the complexes have been recorded in dichloromethane using a Jasco V-630 Spectrophotometer in the 200–800 nm range. 1H NMR spectra were taken in DMSO at room temperature with a Bruker 400 MHz instrument, chemical shift relative to tetramethylsilane. Single crystal data collections and corrections for the new Pd(II) complexes (1–3) were done at 293 K with a CCD kappa Diffractometer using graphite monochromated MoKα (λ = 0.71073 Å) radiation.42 The structural solutions were done by using SHELXS-9743 and refined by full matrix least square on F2 using SHELXL-97.44
DNA binding study
CT DNA solutions of various concentrations (0.05–0.5 μM) dissolved in a phosphate buffer (pH 7) were added to the metal complexes (10 μM dissolved in a DMSO/H2O mixture). Absorption spectra were recorded after equilibrium at 20 °C for 10 min. The intrinsic binding constant Kb was determined by using Stern–Volmer equation (1).45,46| | | ([DNA]/[εa − εf]) = [DNA]/[εb − εf] + 1/Kb[εb − εf] | (1) |
The absorption coefficients εa, εf, and εb correspond to Aobsd/[DNA], the extinction coefficient for the free complex and the extinction coefficient for the complex in the fully bound form, respectively. The slope and the intercept of the linear fit of the plot of [DNA]/[εa − εf] versus [DNA] give 1/[εa − εf] and 1/Kb[εb − εf], respectively. The intrinsic binding constant Kb can be obtained from the ratio of the slope to the intercept (Table 4).45 Emission measurements were carried out by using a JASCO FP-6600 spectrofluorometer. Tris-buffer was used as a blank to make preliminary adjustments. The excitation wavelength was fixed and the emission range was adjusted before measurements. All measurements were made at 20 °C. For emission spectral titrations, complex concentration was maintained constant at 10 μM and the concentration of DNA was varied from 0.05 to 0.5 μM. The emission enhancement factors were measured by comparing the intensities at the emission spectral maxima under similar conditions.
Proteinase binding studies
Lysozyme was purchased from Hi Media, India. Lysozyme solution was prepared in phosphate buffer of pH 7.6 and stored in the dark at 4 °C for use. The concentration of lysozyme was determined spectrophotometrically using ε280 (lysozyme) = 37646 M−1 cm−1.47UV/Vis absorption experiments were performed on a JASCO V-630 spectrophotometer. The excitation wavelength of lysozyme was 280 nm and the emission was monitored at 342 nm. The excitation and emission slit widths (each 5 nm) and the scan rate (500 nm min−1) were maintained constant for all the experiments. A 3 ml solution, containing appropriate concentration of lysozyme (1 × 10−6 M), was titrated with successive additions of the complex. For synchronous fluorescence spectra also the same concentrations of lysozyme and THPP were used and the spectra were measured at two different Δλ (difference between the excitation and emission wavelengths of lysozyme) values such as 15 and 60 nm.
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide) assay
Effects of the complexes (1–4) on the viability of human lung cancer cells (A549) and liver cancer cells (HepG2) were assayed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay.48 The cells were seeded at a density of 10000 cells per well, in 200 μl RPMI 1640 medium, and were allowed to attach overnight in a CO2 incubator and then the complexes (1–4) dissolved in DMSO were added to the cells at a final concentration of 1, 10, 25 and 50 μM in the cell culture media. After 48 hours, the wells were treated with 20 μl MTT (5 mg ml−1PBS) and incubated at 37 °C for 4 hours. The purple formazan crystals formed were dissolved in 200 μl DMSO and read at 570 nm in a micro plate reader.
Release of lactate dehydrogenase (LDH)
LDH activity was determined by the linear region of a pyruvate standard graph using regression analysis and expressed as percentage (%) leakage as described previously.49 Briefly, to a set of tubes, 1 ml of buffered substrate (lithium lactate) and 0.1 ml of the media or cell extract were added and the tubes were incubated at 37 °C for 30 min. After adding 0.2 ml of NAD solution, the incubation was continued for another 30 min. The reaction was then arrested by adding 0.1 ml of DNPH reagent and the tubes were incubated for a further period of 15 min at 37 °C. After this 0.1 ml of media or cell extract was added to blank tubes after arresting the reaction with DNPH. 3.5 ml of 0.4 N sodium hydroxide was added to all the tubes. The intensity of colour developed was measured at 420 nm using a Shimadzu UV/visible spectrophotometer. The amount of LDH released was expressed as percentage.
Nitric oxide (NO) assay
The amount of nitrite was determined by the method of Stueher and Marletta.50Nitrite reacts with Griess Reagent to give a coloured complex measured at 540 nm. To 100 μl of the medium, 50 μl of Griess reagent I was added, mixed and allowed to react for 10 min. This was followed by 50 μl addition of Griess reagent II, the reaction mixture was mixed well and incubated for another 10 min at room temperature. The intensity of pink colour developed was measured at 540 nm using a microquant plate reader (Biotek Instruments).
Cellular uptake study
Cellular uptake of complexes (1–4) was quantified according to the literature method with a slight modification.51 Briefly, the lung cancer cells (A549) and lever cancer cells (HepG2) were treated with the different complexes for 24 hours. The medium was aspirated and cells were washed thrice with ice cold PBS. Then the cells were lysed with PBS containing 1% Triton X-100. The concentration of the complexes in the cell lysates was measured with a fluorescence spectrophotometer (Jasco FP 6600) at their maximum excitation/emission wavelengths of 300/440, 322/428, 302/427 and 350/461 nm, respectively, for complexes 1–4. To offset the background fluorescence from the cellular components, separate standardization curves were prepared using cellular lysates containing a series of known concentrations of different complexes and the intracellular concentrations were found out using the standard curve.
Antibacterial activity studies
MICs (minimum inhibitory concentration) of the compounds against test organisms such as Enterococcus faecalis, Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa were determined by using the broth microdilution method.52 A broth microdilution susceptibility assay was used as recommended by NCCLS for the determination of the MIC. All the tests were performed in Mueller–Hinton Broth (MHB) supplemented with Tween-80 detergent (final concentration of 0.5% (v/v)). Bacterial strains were cultured overnight at 37 °C in MHA. Test strains were suspended in MHB to give a final density of 5 × 105 cfu ml−1 and these were confirmed by viable counts. Geometric dilutions of the compounds were prepared in a 96-well micro titer plate, including one growth control (MHB + Tween 80) and one sterility control (MHB + Tween 80 + Compound). The plates were incubated under normal atmospheric conditions at 37 °C for 24 h.
Results and discussion
The reaction between [PdCl2(PPh3)2] and a series of 4(N)-substituted thiosemicarbazones (H2L)1–4 in 1:1 ethanol/dichloromethane resulted in the formation of new complexes (Scheme 1), where the substituted thiosemicarbazones act as a tridentate ONS ligand. The analytical data of which confirmed the stoichiometry of the complexes (1–4). The structures of the complexes (1–3) were confirmed by the X-ray crystallographic study. Attempts made to grow single crystals for complex 4 in various organic solvents were unsuccessful. The new complexes (1–4) are soluble in common organic solvents such as dichloromethane, chloroform, benzene, acetonitrile, ethanol, methanol, dimethylformamide and dimethylsulfoxide.
 |
| | Scheme 1 Preparation of new palladium(II) complexes. | |
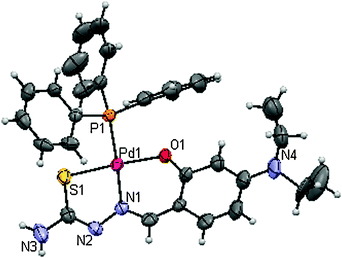 |
| | Fig. 1 ORTEP diagram of 1. | |
 |
| | Fig. 2 ORTEP diagram of 2. | |
 |
| | Fig. 3 ORTEP diagram of 3. | |
| |
[Pd(DeaSal-tsc)(PPh3)] (1) |
[Pd(DeaSal-mtsc)(PPh3)] (2) |
[Pd(DeaSal-etsc)(PPh3)] (3) |
| Empirical formula |
C30H33N4OSPdP
|
C31H33N4OSPdP
|
C32H35N4OSPdP
|
| Formula weight |
635.03 |
647.04 |
661.07 |
| Crystal system |
Orthorhombic |
Orthorhombic |
Orthorhombic |
| Space group |
P2(1)2(1)2(1)
|
P2(1)2(1)2(1)
|
P2(1)2(1)2(1)
|
| Wavelength/Å |
0.71073 |
0.71073 |
0.71073 |
| Temperature/K |
293 |
293 |
293 |
|
a/Å |
9.6836(2) |
9.6590(2) |
9.8285(2) |
|
b/Å |
11.1912(2) |
11.2079(3) |
11.3287(2) |
|
c/Å |
27.4533(5) |
27.4126(7) |
27.6805(7) |
|
α/° |
90 |
90 |
90 |
|
β/° |
90 |
90 |
90 |
|
γ/° |
90 |
90 |
90 |
|
V/Å3 |
2975.14 |
2967.61 |
3082.06(11) |
| Crystal size/mm |
0.10 × 0.20 × 0.20 |
0.10 × 0.14 × 0.18 |
0.10 × 0.10 × 0.20 |
|
Z value |
4 |
4 |
4 |
| Limiting indices |
−12 ≤ h ≤ 13, −15 ≤ k ≤ 10, −35 ≤ l ≤ 28 |
−13 ≤ h ≤ 11, −14 ≤ k ≤ 15, −27 ≤ l ≤ 35 |
−12 ≤ h ≤ 9, −15 ≤ k ≤ 11, −37 ≤ l ≤ 35 |
|
D
calc/Mg m−3 |
1.418 |
1.448 |
1.425 |
| Reflections collected/unique |
22346/5470 [Rint 0.0613] |
18457/3915 [Rint 0.0769] |
27334/4653[Rint 0.0818] |
|
θ range for data collection/° |
2.57 to 28.69 |
2.58 to 28.73 |
2.54to 28.68 |
|
F
000
|
1304 |
1328 |
1360 |
| Goodness-of-fit on F2 |
1.008 |
0.859 |
0.927 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
|
μ(MoKα)/mm−1 |
0.777 |
0.836 |
0.753 |
| Completeness to θ 2θmax/° |
28.69 |
28.73 |
28.68 |
| Data/restraints/parameters |
7447/4/370 |
7345/24/372 |
7503/0/361 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.093, wR2 = 0.0716 |
|
|
| |
R
1 = 0.1336, wR2 = 0.0842 |
R
1 = 0.1061, wR2 = 0.0756 |
|
|
R indices (all data) |
R
1 = 0.0513, wR2 = 0.065 |
R
1 = 0.0484, wR2 = 0.0649 |
R
1 = 0.043, wR2 = 0.0647 |
| Largest diff. peak and hole |
0.807 and −0.791 e A−3 |
0.493 and −0.652 e A−3 |
0.432 and −0.961 e Å−3 |
Table 2 Bond lengths [Å] and angles [°]
| Bond lengths |
[Pd(DeaSal-tsc)(PPh3)]
|
[Pd(DeaSal-mtsc)(PPh3)]
|
[Pd(DeaSal-etsc)(PPh3)]
|
| Pd–S(1) |
2.249(1) |
2.253(1) |
2.245(1) |
| Pd–P(1) |
2.2835(9) |
2.287(1) |
2.278(1) |
| Pd–N(1) |
2.022(2) |
2.015(3) |
2.018(3) |
| Pd–O(1) |
2.013(3) |
2.021(3) |
2.020(2) |
| Bond angles |
| S(1)–Pd(1)–P(1) |
93.40(4) |
93.57(5) |
92.38(4) |
| S(1)–Pd(1)–N(1) |
83.76(8) |
83.9(1) |
84.12(9) |
| P(1)–Pd(1)–N(1) |
177.09(8) |
177.5(1) |
176.39(9) |
| O(1)–Pd(1)–N(1) |
92.7(1) |
92.7(1) |
92.9(1) |
| O(1)–Pd(1)–P(1) |
90.16(8) |
89.85(9) |
90.63(7) |
| O(1)–Pd(1)–S(1) |
176.44(8) |
176.6(1) |
176.98(7) |
Table 3
Hydrogen bonds for 1 [Å and °]
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
<(DHA) |
| Symmetry operation: −1/2 + x, −1/2 − y, 1 − z; −1/2 + x, −1/2 − y, 1 − z; −1 + x, y, z; x, y, z; x, y, z; x, y, z. |
|
N(3A)–H(3A)⋯O(2A) |
0.86(1) |
2.09(8) |
2.87(6) |
149.95 |
|
O(2A)⋯N(2B) |
— |
— |
2.93(6) |
— |
|
N(3B)–H(3B)⋯O(2C) |
0.86(1) |
2.09(8) |
2.87(6) |
149.95 |
|
O(2A)⋯N(2C) |
— |
— |
2.93(6) |
— |
 |
| | Fig. 4
Hydrogen bonding diagram of 1. | |
DNA binding studies
The interaction of transition metal complexes with DNA takes place via both covalent and/or non-covalent interactions.65 In the case of covalent binding, the labile ligand of the complexes is replaced by a nitrogen base of DNA such as guanine N7 while the non-covalent DNA interactions include intercalative, electrostatic and groove binding of metal complexes outside of a DNA helix. A UV absorption titration experiment was carried out to study the DNA binding properties of the new Pd(II) complexes (1–4). The absorption spectra of complexes (1–4) at constant concentration (10 μM) in the presence of different concentrations of CT-DNA (0.05–0.50 μM) are given in Fig. 5. The absorption spectra of complex 1 mainly consist of three resolved bands [intra ligand (IL) and metal-to-ligand (MLCT) transitions] centered at 265 nm (IL), 394 nm and 410 nm (MLCT). As the DNA concentration is increased, the hyperchromism (A = 0.1911 to 0.9081) with a blue shift of 4 nm (up to 261 nm) was observed in the intra ligand band. The MLCT bands at 394 and 410 nm showed modest hypochromism with negligible shifts in the absorption maxima. In addition, the binding of complex 1 to CT DNA led to isosbestic spectral change with the isosbestic point at 302 nm. For complex 2, upon addition of DNA, the intra ligand band at 263 nm exhibited hyperchromism (A = 0.4198 to 1.2219) with a blue shift of 3 nm (up to 260 nm). The MLCT band at 391 nm showed hypochromism without the wavelength shift in the absorption maxima. As shown in Fig. 5, the addition of CT-DNA to complex 2 also led to isosbestic spectral change with the isosbestic point at 303 nm. The binding behaviour of complexes 3 and 4 is also quite similar. The hyperchromism at 264 nm (for 3) and 267 nm (for 4) with a blue shift of 3 nm and 6 nm in the IL bands, respectively, was observed. The CT bands showed hypochromism at 414 nm (for 3) and 404, 421 nm (for 4) without undergoing any shift upon addition of CT-DNA. A distinct isosbestic point appeared at 304 nm for complexes 3 and 4. The observed hyperchromic effect with blue shift suggested that complexes (1–4) bind to CT-DNA by external contact, possibly due to electrostatic binding.66
The intrinsic binding constant Kb is a useful tool to monitor the magnitude of the binding strength of compounds with CT DNA (Table 4). It can be determined by monitoring the changes in the absorbance in the IL band at the corresponding λmax with increasing concentration of DNA and is given by the ratio of the slope to the Y intercept in plots of [DNA]/(εa − εf) versus [DNA] (insets in Fig. 5). From the binding constant values, it is inferred that complex 3 exhibited better binding than other complexes. Based on the Kb value, we can arrange the complexes in the following order with respect to the electron donating ability of the ligand i.e. the substitution on the N-terminal nitrogen atom, 3 (ethyl) > 2 (methyl) > 1 (hydrogen) > 4 (phenyl).
Table 4 Binding constant for interaction of complexes with CT-DNA
| System |
K
b (×106 M−1) |
| CT-DNA + 1 |
0.79272 |
| CT-DNA + 2 |
2.81695 |
| CT-DNA + 3 |
4.1493 |
| CT-DNA + 4 |
0.30303 |
In the emission spectra, complex 1 had fluorescence emission at 351 nm (Fig. 6). Addition of CT DNA to the complex solution resulted in hypochromism by decreasing intensity (I = 31.59 to 14.56) and with bathochromic shift by 2 nm. Similarly, complex 4 showed hypochromism (108.22–58.65) at 352 nm. For complex 2, the enhanced fluorescence was observed at 449 nm (78.87–105.70) without any shift in the absorption maxima. A similar spectral change was observed in complex 3 at 480 nm. The marked decrease in the fluorescence intensity of complexes 1 and 4 indicates the intercalative binding mode of DNA. However, the enhanced fluorescence intensities observed for complexes 2 and 3 symbolize electrostatic binding of DNA with them.
 |
| | Fig. 6 Changes in the emission spectra of 1, 2, 3 and 4 with increasing concentrations (0.05–0.5 μM) of CT-DNA (phosphate buffer, pH 7). | |
Protein binding studies
Fluorescence spectroscopy
.
The binding of the complexes with lysozyme has been estimated from the concentration dependence upon the change in the fluorescence intensity of protein after the addition of complexes. The fluorescence analysis provides information on the binding mechanism, binding mode, binding constant and binding sites of small molecules to protein. The mechanisms of quenching are usually classified by either dynamic quenching or static quenching. Static quenching refers to fluorophore–quencher complex formation and the dynamic quenching refers to a process in which the fluorophore and the quencher come into contact during the transient existence of the excited state. Fig. 7 shows the effect of 3 on the photoluminescence intensity of lysozyme. Upon increasing the concentration of 3 a progressive decrease in the fluorescence intensity accompanied by a blue shift was observed. The observed blue shift may be due to the binding of 3 with the active site in lysozyme.67 This quenching effect indicates the interaction of lysozyme with 3. The fluorescence quenching data have been analysed by the Stern–Volmer equation.where I0 and I are the fluorescence intensities of the fluorophore in the absence and presence of the quencher, KSV is the Stern–Volmer quenching constant and [Q] is the quencher concentration. A plot of I0/I against the concentration of 3 resulted in a linear plot. Similar kinds of linear plots were obtained for all the other three complexes (Fig. 8) and the KSV value is obtained from the slope. Since the lifetime of lysozyme is in the order of 10−9 s, the calculated bimolecular quenching rate constant (kq) using KSV = kqτ0 was found to be higher than the maximum collisional quenching (kq) of various kinds of quenchers to biopolymers (2.0 × 1010 M−1 s−1). Hence, the fluorescence quenching results from the formation of a complex between lysozyme and 3. The other complexes such as 2, 4 and 1 also showed a similar type of fluorescence behavior (S1–S3, ESI†).
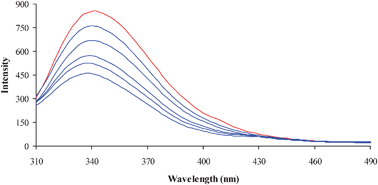 |
| | Fig. 7 Fluorescence quenching of lysozyme (1 × 10−6 M; λexi = 280 nm; λemi = 347 nm) in the absence (red color) and presence (blue color) of various concentrations of 3 (0–5 × 10−5 M). | |
![Plot of I0/I vs.log[Q].](/image/article/2012/MT/c1mt00144b/c1mt00144b-f8.gif) |
| | Fig. 8 Plot of I0/I vs.log[Q]. | |
When small molecules bind to active sites of lysozyme, the equilibrium binding constant and the number of binding sites can be analysed by using the Scatchard equation.
| |  | (3) |
where
K is the binding constant of the quencher with lysozyme,
n is the number of binding sites,
F0 and
F are the fluorescence intensities in the absence and presence of the quencher. The value of
K can be determined from the slope of the plot log[(
F0 −
F)/
F]
versuslog[Q] as shown in
Fig. 9. The calculated value of binding constant (
K) and the number of binding sites (
n) are listed in
Table 5. The binding behaviour decreases in the following order;
3 >
2 >
1 >
4. Complex
3 has high magnitude of binding than complex
2. This indicates that the binding ability to lysozyme increases with the increase in the electron donating ability of the substituent on the terminal
nitrogen of the coordinated thiosemicarbazone. Since, all other groups are the same in both
3 and
2, they differ only in the
alkyl substituent in the
amino group. However, the free amino group containing complex
1 has a slightly higher binding constant than complex
4 which contains a
phenyl substituent.
![Plot of log[(F0 − F)/F] vs.log[Q].](/image/article/2012/MT/c1mt00144b/c1mt00144b-f9.gif) |
| | Fig. 9 Plot of log[(F0 − F)/F] vs.log[Q]. | |
Table 5 Binding constant and number of binding sites for interaction of complexes with lysozyme
| System |
K (×105 M−1) |
n
|
|
Lysozyme + 1 |
0.62 |
1.1357 |
|
Lysozyme + 2 |
0.80 |
1.1103 |
|
Lysozyme + 3 |
1.46 |
1.2134 |
|
Lysozyme + 4 |
0.31 |
1.0236 |
The synchronous fluorescence spectra can give information about the conformational change of the protein molecular environment in the vicinity of the fluorophore functional groups.68 The fluorescence of lysozyme is due to tryptophan and tyrosine residues. Among them tryptophan lies in the active site of protein. In synchronous fluorescence spectroscopy, according to Miller,69 the difference between the excitation wavelength and the emission wavelength (Δλ = λemi − λexc) reflects the spectra of chromophores of a different nature; with large Δλ values such as 60 nm, the synchronous fluorescence of lysozyme is characteristic of tryptophan residue and with small Δλ values such as 15 nm it is characteristic of tyrosine.70 Therefore, to explore the structural change of lysozyme we measured synchronous fluorescence spectra of lysozyme with complexes. The synchronous fluorescence spectra of lysozyme with various concentrations of 3 were recorded at Δλ = 60 nm (Fig. 10) and Δλ = 15 nm. The fluorescence intensity of both tryptophan and tyrosine shows a decrease in intensity but the tryptophan spectrum is accompanied with a blue shift. The other complexes also show the blue shift in the tryptophan region. It reveals that the binding around Trp residues is strengthened. Hence, the results clearly indicate that the complexes bind to active sites in the protein, which makes them potential molecules for biological applications.
 |
| | Fig. 10 Synchronous spectra of lysozyme (1 × 10−6 M) in the absence and presence of 3 (0–5 × 10−5 M) in the wavelength difference of Δλ = 60 nm. | |
MTT(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.
Cancer is a complex disease that is normally associated with a wide range of escalating effects both at the molecular and cellular levels. Synthesized metal complexes have been successfully used as anticancer drugs with high selectivity against malignant cells and with the ability to repress tumor metastasis. As candidates for such drugs, cytotoxic, antitumor or anticancer natural products have been often sought and the synthesized complexes have been used as therapeutics. Effects of the complexes (1–4) on the proliferation of human lung cancer cells (A549) and liver cancer cells (HepG2) were assayed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. All the samples were found to have an antiproliferative effect on A549 and HepG2 cell lines (Fig. 11). Complex 3 showed a higher antiproliferative effect followed by complex 2. Complexes 1 and 4 showed similar IC50 values but the antiproliferative effects of these complexes are less when compared to the other two complexes 3 and 2.
 |
| | Fig. 11 The IC50 values (50% inhibition of cell growth for 48 hours) for complexes 1–4 are depicted. | |
Lactate dehydrogenase release.
Lactate dehydrogenase leakage is an indicator of cell membrane integrity.71 When the cell membrane is disrupted, the membrane bound LDH leaks into the medium and hence LDH leakage is considered as a hallmark of cytotoxicity. Cell death leads to collapse in membrane integrity, thereby releasing LDH into the medium. There was significant increase in the level of LDH released into the medium when the cancer cells were treated with the samples. In the present study, an average LDH leakage was observed in the cell culture medium of A549 and HepG2 cell lines, when they were treated with the respective IC50 concentrations of the complexes for a period of 48 hours (Fig. 12). Our results are comparable to the results shown by Alfa et al., who reported that HepG2 cells treated with Quercetin result in significant release of LDH leakage into the culture medium, which confirms the cytotoxic effect.72 The order of increased induction of lactate dehydrogenase release was 3, 2, 4 and 1.
 |
| | Fig. 12 Percentage of lactate dehydrogenase released by the human cancer cell lines A549 and HepG2 after an incubation period of 48 hours with complexes 1–4. Error bars represent the standard error of mean (n = 6). | |
Nitric oxide assay.
NO is a gaseous signaling molecule, the stable product of which is nitrite. Nitric oxide, a well-known short-lived free radical produced non-enzymatically by NOS, causes damage in most of the biomolecules, including DNA and protein.73Nitric oxide (NO) has been shown to directly inhibit methionine adenosyl transferase, leading to glutathione depletion and its reaction with superoxide generates the strong oxidant peroxynitrite, which can initiate lipid peroxidation or cause a direct inhibition of the mitochondrial respiratory chain.74 Hence, NO is also an important measure of cytotoxicity. Reactions of the free radical nitrogen monoxide with metal–oxygen species of metalloproteins have been recognized as mechanisms relevant to NO metabolism and detoxification in vivo. In the present study NO release by the complexes was evaluated by using A549 and HepG2 cells in which quantification of the nitrite produced in the cell media by the Griess assay is an indirect but cost effective measurement of the amount of NO produced by the cells. It is interesting to note that all the complexes were found to release NO than the control in which 3 is the most effective one (Fig. 13). The NO releasing activity of the complexes can be arranged in the following order 3 > 2 > 4 > 1. All the results of the present study reveal the cytotoxic effect of the complexes (1–4) and their anticancer potency. It is worth to conclude the complexes used in the present investigation as anticancer agents since they show cytotoxicity against both lung and liver cancer cell lines. Further studies are required to reveal their molecular mechanism of cytotoxicity.
 |
| | Fig. 13 nmoles of nitrite released by the human cancer cell lines A549 and HepG2 after an incubation period of 48 hours with complexes 1–4. Error bars represent the standard error of mean (n = 6). | |
Cellular uptake study
The intracellular uptake of a specific drug plays a vital role in ameliorating several diseases. Since the IC50 values are critical when compared to those of the normal cells in the human body, the present study was focused on the concentrations which showed 50% inhibition for A549 and HepG2 cell lines. The intracellular concentrations of complexes 1, 2, 3 and 4 were found out using the method described in the methodology. The intracellular concentration of the complexes after the incubation period of 2 hours is shown in Fig. 14. It is obvious from the results that even low concentrations of the complexes are cytotoxic to the lung cancer cells when they are completely absorbed by the cells. Hence, further studies to improve their cellular uptake may be useful for their clinical relevance.
 |
| | Fig. 14 Percentage of intracellular uptake of complexes 1–4 by human cancer cell lines A549 and HepG2 after an incubation period of 2 hours. Error bars represent the standard error of mean (n = 6). | |
Antibacterial activity studies
Infections due to different pathogenic bacteria are dreadful threat to the human race. Virulent strains of E. coli can cause gastroenteritis, urinary tract infections, and neonatal meningitis. Enterococcus faecalis can cause endocarditis, as well as bladder, prostate, and epididymal infections; nervous system infections are less common. S. aureus infection can cause a severe disease staphylococcal scalded skin syndrome (SSSS) in infants and mastitis in cow. K. pneumoniae leads to the dreadful disease pneumonia and P. aeruginosa is an opportunistic pathogen in immuno-compromised individuals.75–79 Since these bacterial species develop resistance to the existing antibacterial agents, new compounds with more effective cytotoxic/cytostatic effects on the pathogenic bacteria are of urgent need in the medical field. There are hundreds of antibacterial agents but their use is limited due to the low spectrum of activity and good activity only in high concentrations, which may also be toxic to the non-targets. Antibacterial activities for the palladium complexes have not been examined well. Here in our study, we attempted to explore the toxic effects of newly synthesized palladium complexes on five different bacterial species. As described in the materials and methods, different concentrations of the complexes were used to find out their minimum inhibitory concentration on the bacterial species like E. faecalis, S. aureus, E. coli, K. pneumoniae and P. aeruginosa. Complex 3 was found to be more effective on S. aureus, E. coli and K. pneumoniae with lower minimal inhibitory concentrations (Fig. 15). Complex 4 was observed to be more toxic to E. faecalis when compared to the other three complexes. E. faecalis was sensitive to 1, 2 and 3 with almost the same inhibitory concentration. Complex 2 was observed to be more toxic to P. aeruginosa. The activity for the metal chelates can be explained on the basis of chelation theory.80 Chelation considerably reduces the polarity of the metal ion because of the partial sharing of its positive charge with the donor groups and possible π–electron delocalization over the chelate ring. Such chelation could increase the lipophilic character of the central metal atom, which subsequently favors the permeation through the lipid layer of the cell membrane. The mode of action of the complexes may involve the formation of the hydrogen bond through the azomethine group (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CN) with the active centers of the cell constituents resulting in the interference with a normal cell process.81 The variation in the effectiveness of the different complexes against different organisms depends on the impermeability of the cells of microbes or difference in ribosome of the microbial cells.82
CN) with the active centers of the cell constituents resulting in the interference with a normal cell process.81 The variation in the effectiveness of the different complexes against different organisms depends on the impermeability of the cells of microbes or difference in ribosome of the microbial cells.82
 |
| | Fig. 15 Minimal inhibitory concentration of new Pd(II) complexes. The new Pd(II) complexes are exposed at different concentrations for a period of overnight and the minimal inhibitory concentration of each complex is given considering that the control sets gave null values. | |
Conclusions
Reactions of 4-(N,N′)-diethylaminosalicylaldehyde-4(N)-substituted thiosemicarbazones with [PdCl2(PPh3)2] resulted in the formation of four new Pd(II) complexes. The crystal structure determination showed that the anions of thiosemicarbazones coordinate in a planar conformation to a central Pd(II) through the phenolate oxygen, azomethine nitrogen and thiolato sulfur atoms. The fourth coordination site is occupied by phosphorus atoms of the triphenylphosphine unit. The DNA binding through absorption titration studies revealed that the complexes bind to the DNA significantly. From the binding constant values, it is clear that even at very low concentration, the complexes exhibited better activity. A higher value 4.1493 × 106 was found for complex 3 suggesting a very strong affinity of the complex to CT-DNA. From the emission studies, it is observed that the complexes bind to the CT-DNA by either intercalative (1 and 4) or electrostatic (2 and 3) binding modes. From the protein binding studies, it is evident that the complexes underwent considerable binding with the proteins. The potential cytotoxicity of the complexes and their cellular uptake, LDH assay and NO scavenging activity showed that the complexes could be considered as good candidates for their further clinical trials. The antibacterial activity studies of the complexes reveal that the complexes possess a significant degree of activity against various pathogens which were taken. From the results of the biological studies, it is clear that the increase in electron donating ability of the substituent on terminal nitrogen atoms of the thiosemicarbazones in the complexes increases the activity and thus complex 3 was found to be a better candidate in exhibiting better activity than other complexes due to the presence of a more electron rich ethyl group. The activity can be arranged based on the substitution on N-terminal nitrogen atoms as NH-ethyl > NH-methyl > NH2 > NH-phenyl. Further, it will be interesting to go for in vivo and clinical trials of the complexes to explore more on their potential.
Acknowledgements
Authors P.K. and K.N. gratefully acknowledge CSIR India for the financial assistance and author R.P. gratefully acknowledges CNRS, France, for CNRS Fellowship.
References
-
(a) M. J. M. Campbell, Coord. Chem. Rev., 1975, 15, 297–319 CrossRef;
(b) J. S. Casas, M. S. Garcia-Tasenda and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
-
(a) R. Prabhakaran, V. Krishnan, K. Pasumpon, D. Sukanya, E. Wendel, C. Jayabalakrishnan, H. Bertagnolli and K. Natarajan, Appl. Organomet. Chem., 2006, 20, 203–213 CrossRef CAS;
(b) R. Prabhakaran, R. Huang, R. Karvembu, C. Jayabalakrishnan and K. Natarajan, Inorg. Chim. Acta, 2007, 360, 691–694 CrossRef CAS;
(c) R. Prabhakaran, R. Huang, S. V. Renukadevi, R. Karvembu, M. Zeller and K. Natarajan, Inorg. Chim. Acta, 2008, 8, 2547–2552 CrossRef;
(d) M. Muthukumar, S. Sivakumar, P. Viswanathamurthi, R. Karvembu, R. Prabhakaran and K. Natarajan, J. Coord. Chem., 2010, 63, 296–306 CrossRef CAS;
(e) S. Gowri, M. Muthukumar, S. Krishnaraj, P. Viswanathamurthi, R. Prabhakaran and K. Natarajan, J. Coord. Chem., 2010, 63, 524–533 CrossRef CAS;
(f) K. P. Balasubramanian, R. Karvembu, R. Prabhakaran, V. Chinnusamy and K. Natarajan, Spectrochim. Acta, Part A, 2007, 68, 50–54 CrossRef CAS;
(g) V. Chinnusamy and K. Natarajan, Synth. React. Inorg. Met.-Org. Chem., 1993, 23, 889–905 CrossRef CAS;
(h) S. Priyarega, P. Kalaivani, R. Prabhakaran, T. Hashimoto, A. Endo and K. Natarajan, J. Mol. Struct., 2011, 1002, 58–62 CrossRef CAS;
(i) R. Prabhakaran, S. Anantharaman, M. Thilagavathi, M. V. Kaveri, P. Kalaivani, R. Karvembu, N. Dharmaraj, H. Bertagnolli and K. Natarajan, Spectrochim. Acta, Part A, 2011, 78, 844–853 CrossRef CAS;
(j) R. Prabhakaran, P. Kalaivani, R. Jayakumar, M. Zeller, A. D. Hunter, S. V. Renukadevi, E. Ramachandran and K. Natarajan, Metallomics, 2011, 3, 42–48 RSC;
(k) R. Prabhakaran, P. Kalaivani, R. Huang, M. Sieger, W. Kaim, P. Viswanathamurthi, F. Dallemer and K. Natarajan, Inorg. Chim. Acta, 2011, 376, 317–324 CrossRef CAS.
-
(a) C. Q. Debra, A. K. Kathy and R. K. Earl, Antiviral Res., 2006, 71, 24–30 CrossRef;
(b) Y. Teitz, D. Ronen, A. Vansover, T. Stematsky and J. L. Riggs, Antiviral Res., 1994, 24, 305–314 CrossRef CAS.
- M. C. R. Arguelles, E. C. L. Silva, J. Sanmartın, P. Pelagatti and F. Zani, J. Inorg. Biochem., 2005, 99, 2231–2239 CrossRef.
- M. C. R. Arguelles, E. C. L. Silva, J. Sanmartın, A. Bacchi, C. Pelizzi and F. Zani, Inorg. Chim. Acta, 2004, 357, 2543–2552 CrossRef.
- A. Cukurovali, I. Yilmaz, S. Gur and C. Kazaz, Eur. J. Med. Chem., 2006, 41, 201–207 CrossRef CAS.
- A. Murugkar, B. Unnikrishnan, S. Padhye, R. Bhonde, S. Teat, E. Triantafillou and E. Sinn, Met.-Based Drugs, 1999, 6, 177–182 CrossRef CAS.
- Y. Teitz, N. Barko, M. Abramoff and D. Ronen, Chemotherapy (Tokyo), 1994, 40, 195–200 CAS.
- W. X. Hu, W. Zhou, C. Xia and X. Wen, Bioorg. Med. Chem. Lett., 2006, 16, 2213–2218 CrossRef CAS.
- Z. Afrasiabi, E. Sinn, S. Padhye, S. Dutta, S. Padhye, C. Newton, C. E. Anson and A. K. Powell, J. Inorg. Biochem., 2003, 95, 306–314 CrossRef CAS.
- T. Bal, B. Atasever, Z. Solakoglu, S. E. Kuruca and B. U lkuseven, Eur. J. Med. Chem., 2007, 42, 161–167 CrossRef CAS.
- M. Campana, C. Laborie, G. Barbier, R. Assan and R. Milcent, Eur. J. Med. Chem., 1991, 26, 273–278 CrossRef CAS.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS.
- J. Reedijk, Curr. Opin. Chem. Biol., 1999, 3, 236–240 CrossRef CAS.
- Y. W. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS.
- J. Reedijk, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3611–3616 CrossRef CAS.
- J. Reedijk, Platinum Met. Rev., 2008, 52, 2–11 CrossRef CAS.
- M. D. Hall, M. Okabe, D. W. Shen, X. J. Liang and M. M. Gottesman, Annu. Rev. Pharmacol., 2008, 48, 495–535 CrossRef CAS.
- X. Gao, X. Wang, J. Ding, L. Lin, Y. Li and Z. Guo, Inorg. Chem. Commun., 2006, 9, 722–726 CrossRef CAS.
- F. Huq, J. Qing Yu, H. Daghriri and P. Beale, J. Inorg. Biochem., 2004, 98, 1261–1270 CrossRef CAS.
- H. Tayyem, F. Huq, J. Q. Yu, P. Beale and K. Fisher, ChemMedChem, 2008, 3, 145–151 CrossRef CAS.
- D. Richards and A. Rodger, Chem. Soc. Rev., 2007, 36, 471–483 RSC.
- S. Roy, K. D. Hagen, P. U. Maheswari, M. Lutz, A. L. Spek, J. Reedijk and G. P. van Wezel, ChemMedChem, 2008, 3, 1427–1434 CrossRef CAS.
- M. Whittaker, C. D. Floyd, P. Brown and A. J. H. Gearing, Chem. Rev., 1999, 99, 2735–2776 CrossRef CAS.
- M. Hidalgo and S. G. Eckhardt, J. Natl. Cancer Inst., 2001, 93, 178–193 CrossRef CAS.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215–224 RSC.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2796 CrossRef CAS.
- D. S. Sigman, A. Mazumder and D. M. Perrin, Chem. Rev., 1993, 93, 2295–2316 CrossRef CAS.
- F. Murad, Biosci. Rep., 1999, 19, 133–154 CrossRef CAS.
- S. Moncada, R. M. Palmer and E. A. Higgs, Pharmacol. Rev., 1991, 43, 109–142 CAS.
- A. Hausladen, A. Gow and J. S. Stamler, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10108–10112 CrossRef CAS.
- M. H. Lim and S. J. Lippard, Acc. Chem. Res., 2006, 40, 41–51 CrossRef.
- M. H. Lim, B. A. Wong, W. H. Pitcock, D. Mokshagundam, M. H. Baik and S. J. Lippard, J. Am. Chem. Soc., 2006, 128, 14364–14373 CrossRef CAS.
- M. H. Lim, D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375–380 CrossRef CAS.
- H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef CAS.
- J. H. Kim, D. A. Heller, H. Jin, P. W. Barone, C. Song, J. Zhang, L. J. Trudel, G. N. Wogan, S. R. Tannenbaum and M. S. Strano, Nat. Chem., 2009, 1, 473–481 CrossRef CAS.
- M. N. Hughes, Biochim. Biophys. Acta, Bioenerg., 1999, 1411, 263–272 CrossRef CAS.
-
(a) S. Purohit, A. P. Koley, L. S. Prasad, P. T. Manoharan and S. Ghosh, Inorg. Chem., 1989, 28, 3735–3742 CrossRef CAS;
(b) J. L. Burmeister and F. Basolo, Inorg. Chem., 1964, 3, 1587–1593 CrossRef CAS.
-
A. I. Vogel, Text Book of Practical Organic Chemistry, 5th edn, Longman, London, 1989, p. 268 Search PubMed.
-
(a) R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef;
(b) R. H. Blessing, Crystallogr. Rev., 1987, 1, 3–58 CrossRef;
(c) R. H. Blessing, J. Appl. Crystallogr., 1989, 22, 396–397 CrossRef.
-
G. M. Sheldrick, SHELXTL Version 5.1, An Integrated System for Solving, Refining and Displaying Crystal Structures from Diffraction Data, Siemens Analytical X-ray Instruments, Madison, WI, 1990 Search PubMed.
-
G. M. Sheldrick, SHELXL-97, A Program for Crystal Structure Refinement Release 97-2, Institut fur Anorganische Chemie der Universitat Gottingen, Tammanstrasse 4, D-3400, Gottingen, Germany, 1998 Search PubMed.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS.
- G. Cohen and H. Eisenberg, Biopolymers, 1969, 8, 45–55 CrossRef CAS.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS.
- T. Mossman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- W. E. C. Wacker, D. D. Ulmer and B. L. Valee, N. Engl. J. Med., 1956, 255, 449–455 CrossRef CAS.
- D. J. Stueher and M. A. Marletta, J. Immunol., 1987, 139, 518–525 Search PubMed.
- X. B. Xiong, Z. Ma, R. Lai and A. Lavasanifar, Biomaterials, 2010, 31, 757–768 CrossRef CAS.
- NCCLS, National Committee for Clinical Laboratory Standards, Performance Standards for Antimicrobial Disk Susceptibility Test, 6th ed., Approved Standard, Wayne, PA, M2-A6, 1997.
- L. M. Fostiak, I. Gracia, J. K. Swearinger, E. Bermejo, A. Castineivas and D. X. West, Polyhedron, 2003, 2, 83–92 CrossRef.
- D. X. West, I. S. Billeh, J. P. Jasinski, J. M. Jasinski and R. J. Butcher, Transition Met. Chem., 1998, 23, 209–214 CrossRef CAS.
- R. Prabhakaran, S. V. Renukadevi, R. Karvembu, R. Huang, J. Mautz, G. Huttner, R. Subashkumar and K. Natarajan, Eur. J. Med. Chem., 2008, 43, 268–273 CrossRef CAS.
- R. Karvembu, S. Hemalatha, R. Prabhakaran and K. Natarajan, Inorg. Chem. Commun., 2003, 6, 486–490 CrossRef CAS.
- Y. P. Tiam, C. Y. Duan, Z. L. Lu and X. Z. You, Polyhedron, 1996, 15, 2263–2271 CrossRef.
- S. Dey, V. K. Jain, A. Kwoedler and W. Kaim, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2003, 42, 2339–2343 Search PubMed.
- D. M. Boghaei and S. Mohebi, J. Chem. Res., 2001, 6, 224–226 CrossRef.
- F. Basuli, M. Ruf, C. G. Pierpont and S. Bhattacharya, Inorg. Chem., 1998, 37, 6113–6116 CrossRef CAS.
- L. M. Fostiak, I. Gracia, J. K. Swearinger, E. Bermejo, A. Castineivas and D. X. West, Polyhedron, 2003, 22, 83–92 CrossRef CAS.
- F. Basuli, M. Ruf, C. G. Pierpont and S. Bhattacharya, Inorg. Chem., 1998, 37, 6113–6116 CrossRef CAS.
- R. Prabhakaran, R. Sivasamy, J. Angayarkanni, R. Huang, P. Kalaivani, R. Karvembu, F. Dallemer and K. Natarajan, Inorg. Chim. Acta, 2011, 374, 647–653 CrossRef CAS.
- D. L. Klayman, J. P. Scovill, J. F. Brtosevich and J. Bruce, J. Med. Chem., 1983, 26, 35–39 CrossRef CAS.
- Q. L. Zhang, J. G. Liu, H. Chao, G. Q. Xue and L. N. Ji, J. Inorg. Biochem., 2001, 83, 49–55 CrossRef CAS.
-
(a) E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271–273 CrossRef CAS;
(b) R. F. Pasternack, E. J. Gibbs and J. J. Villafranca, Biochemistry, 1983, 22, 251–255 CrossRef.
- Y. J. Hu, Y. O. Yang, C. M. Dai, Y. Liu and X. H. Xiao, Biomacromolecules, 2010, 11, 106–112 CrossRef CAS.
-
G. Z. Chen, X. Z. Huang, J. G. Xu, Z. B. Wang and Z. Z. Zhang, Method of Fluorescent Analysis, Science Press, Beijing, 2nd edn, 1990, ch. 4, pp. 123–126 Search PubMed.
- J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 203–208 CAS.
- E. a. Brustein, N. S. Vedenkina and M. N. Irkova, Photochem. Photobiol., 1973, 18, 263–279 CrossRef.
- G. Belmadani, A. M. Tramu, P. S. Betbeder and E. E. Steyn, Arch. Toxicol., 1998, 72, 656–662 CrossRef.
- E. Bonfoco, D. Krainc, M. Ankarcrona, P. Nicotera and S. A. Lipton, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7162–7166 CrossRef CAS.
- C. Legrand, J. M. Bour, C. Jacob, J. Capiaumont, A. Martial, A. Marc, M. Wudtke, G. Kretzmer, C. Demangel, D. Duval and J. Hache, J. Biotechnol., 1992, 25, 231–243 CrossRef CAS.
- L. Yu, P. E. Gengaro, M. Niederberger, T. J. Burke and R. W. Schriert, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1691–1695 CrossRef CAS.
-
K. Todar, Pathogenic E. coli. Online Textbook of Bacteriology, University of Wisconsin–Madison Department of Bacteriology Search PubMed.
-
L. L. Pelletier, Microbiology of the Circulatory System, in Baron's Medical Microbiology, ed. S. Baronet al., Univ. of Texas Medical Branch, 4th edn, 1996, ISBN 0-9631172-1-1 Search PubMed.
-
K. J. Ryan and C. G. Ray, Sherris Medical Microbiology, 4th edn, McGraw Hill, 2004, p. 294, ISBN 0-8385-8529-9 Search PubMed.
- J. P. Curran and F. L. Al-Salihi, Pediatrics, 1980, 66, 285 CAS . PMID 6447271.
- B. T. Cenci-Goga, M. Karama, P. V. Rossitto, R. A. Morgante and J. S. Cullor, J. Food Prot., 2003, 66, 1693 CAS . PMID 14503727.
- B. G. Tweedy, Phytopathology, 1964, 55, 910–914 Search PubMed.
- R. Prabhakaran, A. Geetha, M. Thilagavathi, R. Karvembu, V. Krishnan, H. Bertagnolli and K. Natarajan, J. Inorg. Biochem., 2004, 98, 2131–2140 CrossRef CAS.
- P. G. Lawrence, P. L. Harold and O. G. Francis, Antibiot. Chemother., 1957, 4, 1980 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 