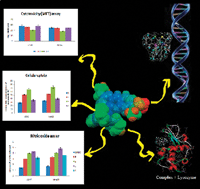
The coordination propensities of 4(N,N′)-diethylaminosalicylaldehyde-4(N)-substituted thiosemicarbazones (H2L1–4) were investigated by reacting with an equimolar amount of [PdCl2(PPh3)2]. The new complexes were characterized by various spectroscopic techniques. The structure determination of the complexes [Pd(DeaSal-tsc)(PPh3)] (1), [Pd(DeaSal-mtsc)(PPh3)] (2) and [Pd(DeaSal-etsc)(PPh3)] (3) by X-ray crystallography showed that ligands are coordinated in a dibasic tridentate ONS donor fashion forming stable five and six membered chelate rings. The binding ability of complexes (1–4) to calf-thymus DNA (CT DNA) has been explored by absorption and emission titration methods. Based on the observations, an electrostatic and an intercalative binding mode have been proposed. The protein binding studies have been monitored by quenching of tryptophan and tyrosine residues in the presence of complexes using lysozyme as a model protein. As determined by MTT assays, complex 3 exhibited a higher cytotoxic effect towards human lung cancer cell line (A549) and liver cancer cells (HepG2). LDH, NO assay and cellular uptake of the complexes have been studied. Further, antibacterial activity studies of the complexes have been screened against the pathogenic bacteria such as Enterococcus faecalis, Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae and Pseudomonas aeruginosa, MIC50 values of the complexes showed that the complexes exhibited significant activity against the pathogens and among the complexes, 3 exhibited higher activity.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
