Iron-based redox centres of reductase and oxygenase components of phenol hydroxylase from A. radioresistens: a redox chain working at highly positive redox potentials
Francesca
Valetti
a,
Andrea
Fantuzzi
b,
Sheila J.
Sadeghi
a and
Gianfranco
Gilardi
*a
aDepartment of Human and Animal Biology, University of Torino, via Accademia Albertina 13, 10123, Torino, Italy. E-mail: gianfranco.gilardi@unito.it; Fax: +39 0116704643; Tel: +39 0116704593
bDivision of Molecular Biosciences, Imperial College, London, UK
First published on 10th October 2011
Abstract
This is the first report of the direct electrochemistry of the reductase (PHR) and oxygenase (PHO) components of phenol hydroxylase from Acinetobacter radioresistensS13 studied by cyclic and differential pulse voltammetry. The PHR contains one 2Fe2S cluster and one FAD that mediate the transfer of electrons from NAD(P)H to the non-heme diiron cluster of PHO. Cyclic and differential pulse voltammetry (CV and DPV) on glassy carbon showed two redox pairs with midpoint potentials at +131.5 ± 13 mV and −234 ± 3 mV versusnormal hydrogen electrode (NHE). The first redox couple is attributed to the FeS centre, while the second one corresponds to free FAD released by the protein. DPV scans on native and guanidinium chloride treated PHR highlighted the presence of a split signal (ΔE ≈ 100 mV) attributed to heterogeneous properties of the 2Fe2S cluster interacting with the electrode, possibly due to the presence of two protein conformers and consistently with the large peak-to-peak separation and the peak broadening observed in CV. DPV experiments on gold electrodes performed on PHO confirm a consistently higher reduction potential at +396 mV vs. NHE. The positive redox potentials measured by direct electrochemistry for the FeS cluster in PHR and for the non-heme diiron cluster of PHO show that the entire phenol hydroxylase system works at higher potentials than those reported for structurally similar enzymes, for example methane monooxygenases.
Introduction
Phenol hydroxylase (PH) is the first enzyme of the catabolic pathway that confers to Acinetobacter radioresistens S13 the ability to efficiently degrade phenol1,2 at a rate of 100 mg L−1 h−1. The hydroxylation of phenol to catechol leads to intradiolic cleavage, producing cis,cis-muconic acid which is finally converted to obtain succinyl- and acetyl-CoA.3 The enzyme comprises a short redox chain, from NAD(P)H to a reductase (PHR) and then to an oxygenase component (PHO) that is able to bind phenol and activate molecular oxygen.4–6PHR exhibits a MW of 38.8 kDa and it contains one 2Fe2S cluster and one FAD. It recognises both NADH (KM = 48.3 μM, kcat = 95.1 s−1) and NADPH (KM = 489.8 μM, kcat = 44.9 s−1) as electron donors.4 The redox centres identified in PHR suggest that the electron transfer occurs from the NAD(P)H to the FAD and then to the 2Fe2S cluster. PHO is a (αβγ)2 multimeric protein, harboring a non-heme diiron cluster Fe–O–Fe on the α subunit. A third intermediate component, PHI, is also necessary for catalysis, it does not contain redox centres and its role is still unclear.6The arrangement of the PH multicomponent system is very similar to that found in PseudomonasCF600,7–10Acinetobacter calcoaceticus,11Methylococcus capsulatus and Methylosinus trichosporium methane monooxygenase,12–21 where reductases similar to PHR have been found.13,22,23 3D structures are available for the methane monooxygenase reductase from Methylococccus capsulatus24,25 and for the benzoate dioxygenase reductase from Acinetobacter sp. strain ADP1.26 N-terminal and internal amino acid sequencing of PHR4 shows a high percentage of identity with analogous reductases from Acinetobacter calcoaceticus11 and Pseudomonas CF6007 and with the available sequences of putative reductases from Acinetobacter radioresistensSK82 (ZP_05359728.1) and Acinetobacter sp.G16 (ACS74440.1). Multiple-sequence alignment suggests a putative arrangement of PHR with an N-terminal thioredoxin-like [2Fe2S] binding domain and a FAD/NAD-binding domain spanning the C-terminal third of the protein. The consensus motif is C(X)4C(X)2C(X)31C and the consensus fold also correlates with the structure of terpredoxin.27–30
The interest of investigating structural–functional properties of 2Fe2S as well as other iron–sulfur proteins in connection with analysis of their redox potentials is reflected in the wide literature published and reviewed on the subject.31–35 Influence of solvent exposure,32hydrogen bonding,36 electrostatic effects (individual charged residues, dipole, amide stabilisation of cluster charge),37 pH38,39 and coordination sphere32,40 have been addressed to account for the observed redox properties of iron–sulfur proteins. While the results appear clearer for certain classes such as Rieske type 2Fe2S clusters and HiPiP, 3Fe4S or 4Fe4S clusters,32 for 2Fe2S ferredoxins and ferredoxin-type domains in multicentre redox proteins the correlation is less clear, though a trend of dependence upon charges or solvent exposure has been suggested.41 In 2Fe2S clusters of the ferredoxin type the redox potential varies from very negative values (around −450 mV vs.NHE) of plant-type ferredoxins to −230 mV of putidaredoxin.42 Moreover, in the case of the latter, the interaction with redox partners seems to modulate the potential to even higher values.43 The modulation of a protein embedded redox-centre potential upon interaction with protein or electrode surfaces is of speculative and practical interest.44–48
Here we report the first direct electrochemistry of PHR and PHO studied by cyclic and differential pulse voltammetry.
Materials and methods
PHR and PHO sample preparation
The A. radioresistensLMG S-13648 strain1,2 was grown in Sokol and Howell minimal medium49 with phenol as the sole carbon source. A fed-batch fermentation procedure was employed in a 10 litre vessel containing 7 litres of medium and with a supply of phenol of 100 ppm h−1 over 16 hours after acclimation of the cells.3Cells were harvested and the biomass was lysed by sonication (Microsonix Ultrasonic Liquid Processor XL2020) and the clarified crude extract purified by FPLC as described.4 The purity of the protein was assessed by SDS-PAGE and confirmed by calculating the ratios of PHR characteristic absorbances at 345, 450 and 470 nm against the protein absorbance contribution at 271 (ε271 = 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 cm−1, ε345 = 15500 M−1 cm−1, ε450 = 18000 M−1 cm−1, ε470 = 17000 M−1 cm−1); the values found were all equal or slightly above the values obtained from the calculated ε ratios. PHO was purified as previously described,5 the protein purity was assessed by SDS-PAGE and concentrations were determined by the absorbance at 280 nm (ε280 = 643800 M−1 cm−1) and 350 nm (typical shoulder due to the presence of an oxo-bridged di-iron centre, ε350 = 6000 M−1 cm−1) on an Agilent 8453 UV/Visible spectrophotometer. Both PHR and PHO activity in the reconstituted PH complex was assessed polarographically using a Clark-type electrode.5 The activity of the isolated PHR subunit was measured by following at the spectrophotometer (Agilent 8453 UV/Visible) the increase in absorbance at 550 nm of horse heart cyctochrome c (Sigma) used as an artificial electron acceptor. Initial reaction rates were calculated using an absorption coefficient at 550 nm of 19.5 mM−1 cm−1. All experiments were performed in potassium phosphate buffer 5 mM, pH 7.05. PHR concentrations ranged from 2 to 20 nM with NADH 0.5 mM.
500 M−1 cm−1, ε345 = 15500 M−1 cm−1, ε450 = 18000 M−1 cm−1, ε470 = 17000 M−1 cm−1); the values found were all equal or slightly above the values obtained from the calculated ε ratios. PHO was purified as previously described,5 the protein purity was assessed by SDS-PAGE and concentrations were determined by the absorbance at 280 nm (ε280 = 643800 M−1 cm−1) and 350 nm (typical shoulder due to the presence of an oxo-bridged di-iron centre, ε350 = 6000 M−1 cm−1) on an Agilent 8453 UV/Visible spectrophotometer. Both PHR and PHO activity in the reconstituted PH complex was assessed polarographically using a Clark-type electrode.5 The activity of the isolated PHR subunit was measured by following at the spectrophotometer (Agilent 8453 UV/Visible) the increase in absorbance at 550 nm of horse heart cyctochrome c (Sigma) used as an artificial electron acceptor. Initial reaction rates were calculated using an absorption coefficient at 550 nm of 19.5 mM−1 cm−1. All experiments were performed in potassium phosphate buffer 5 mM, pH 7.05. PHR concentrations ranged from 2 to 20 nM with NADH 0.5 mM.
Mediated cyclic voltammetry
Mediated electrochemistry in the presence of horse heart cytochrome c (hhc, Sigma) was performed in the same microcell under the following conditions: hhc 75 μM, NADH 5 mM, PHR from 0.515 to 4.12 μM at 25 °C, using a nitric acid activated glassy carbon electrode polished with aluminium oxide 0.3 and 0.015 μm. The buffer used was 5 mM potassium phosphate (pH 7.05) at ionic strength (I) of 0.00963 M. The ionic strength was increased by addition of NaCl to reach I = 0.084 M. The voltammograms were analysed after correction for buffer background (5 mM KPi pH 7.05 with NADH 5 mM). The current was evaluated by automated peak search with a linear front baseline. The ratio ik/id was related to the parameter kf/a considering the working curve published by Nicholson and Shain.50The slope of the linear fitting of kf/a versus 1/scan rate at different concentrations of PHR was used to calculate the kf considering that a = nFV/RT with n = 2, R = 8.31 J K−1 mol−1, F = 96485 C mol−1 and T = 298 K). The r2 of the fittings were all above 0.9. The second order rate constant was calculated from the slope of the linear fitting of all calculated kfversus [PHR], the value obtained was divided by 2 according to the following: if kf = k2 [PHR–hhc complex] then to relate kf and [PHR] a correction is needed to account for the stoichiometry of eqn (1).
| (2hhcox + PHRred ↔ 2hhcred + PHRox) | (1) |
Electrochemical measurements
The electrochemical measurements were carried out using an Autolab 10 electrochemical analyser controlled by GPES software (Eco Chemie Utrecht).Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were performed at 20 °C under nitrogen-saturated atmosphere in a three electrode Hagen cell52 on a sample droplet volume of 40 μl. The working electrode was a 15 mm diameter per 2 mm height glassy carbon disc (Le Carbon Lorraine, NL). A platinum wire was used as a counter electrode and a saturated calomel electrode (SCE) with a potential of 0.246 V (according to the SCE electrode manufacturer) versus the normal hydrogen electrode (NHE) was taken as the reference in all measurements.
CV and DPV experiments of PHR were carried out with working electrodes polished in sequence with diamond paste and aqueous slurry of aluminium oxide of decreasing granularity, namely 0.3 and 0.015 μm; after polishing an extensive rinse was performed with redistilled water, followed by 5 minutes of mild sonication in redistilled water. The electrode was activated by exposure to a methane flame53,54 until red hot, and the face more closely exposed to the flame was always the opposite to the polished one chosen to be in contact with the protein droplet.
The set-up was extensively flushed with nitrogen and the protein droplet (40 μl) applied to the electrode after lowering the nitrogen flow to avoid excessive drying. The PHR concentration was 195 μM, as calculated from the absorbance at 271 nm using an ε271 of 69500 M−1 cm−1 in HEPES 50 mM pH 7.0.
CV scans were recorded from 0.25 V to −1 or −0.8 V (vs.SCE) at a step potential of 0.00195 V and at the scan rates indicated in the text and figures. DPV scans were obtained at step potential and modulation amplitude of 4.05 mV (modulation time 0.05 s, interval time 0.25 s). Consistent results were obtained at 2 to 10 mV step potential. Denaturation experiments were carried out under the same conditions in the presence of 2M guanidinium chloride in the protein droplet. Promoted electrochemistry was carried out by addition of a drop of 110 mM neomycin sulfate (Sigma) in water applied to the electrode surface, left in contact for 30 seconds and dried with a tissue prior to the protein deposition.54
Differential pulse voltammetry experiments on PHO were performed in the same cell set-up described above at a polycrystalline gold electrode (15 mm diameter, 0.5 mm thickness) supported by a brass disc (15 mm diameter, 2 mm thickness). The gold electrode was polished with alumina and with diamond cloth and thoroughly washed by sonication. A further cleaning was performed by cycling from −0.3 to 1.4 V vs.SCE at a scan rate of 50 mV s−1 in 0.1 M H2SO4. The peptide Lys–Cys–Thr–Cys–Cys–Ala (purchased from Sigma) was then applied to the gold surface at a concentration of 5 mM in water and immobilised by reductive cycling under nitrogen atmosphere (at least 10 scans from 0 to −0.6 V vs.SCE at 50 mV s−1, step 2 mV).55 The electrode was then rinsed with nitrogen saturated buffer (MOPS 100 mM pH 7.0) within the anaerobic atmosphere of the cell set-up and the scans performed on buffer and on PHO (protein concentration: 13.35 μM) at a step potential and modulation amplitude of 4.05 mV (modulation time 0.05 s, interval time 0.25 s). All values of potential reported in the text are versusNHE.
Results and discussion
PHR displays a well defined cyclic voltammetry (CV) at a glassy carbon electrode (GC, Fig. 1A) and the signal is stable up to a scan rate of 400 mV s−1. The CVs show two redox pairs, the first with oxidation and reduction peaks at −2 and +265 mV (peaks 1 and 2 of Fig. 1A), the second at −246 and −204 mV (peaks 3 and 4). The first redox pair displays a linear dependence of the peak current with the square root of the scan rate (Fig. 1B), with a large peak-to-peak separation of 267 mV and a midpoint potential of 131.5 ± 13 mV. The quasi-reversible diffusion controlled process of this redox couple is consistent with the behaviour of a protein-embedded redox centre and it confirms that the protein diffuses in solution to/from the electrode, rather than being adsorbed onto the electrode in a denatured form. The redox couple with Em of −234 mV is attributed to free FAD that is partially released from PHR and it is adsorbed onto the electrode surface, showing a linear dependence of the peak current with the scan rate (Fig. 1C). This effect has been previously observed for the direct electrochemistry of other FAD-containing reductases56 and for flavodoxins,53 and the value of its Em is in good agreement with that of −219 mV reported for free flavins. CV scans recorded with up to 10 mM of the promoter neomycin did not show improvement of the signal intensity.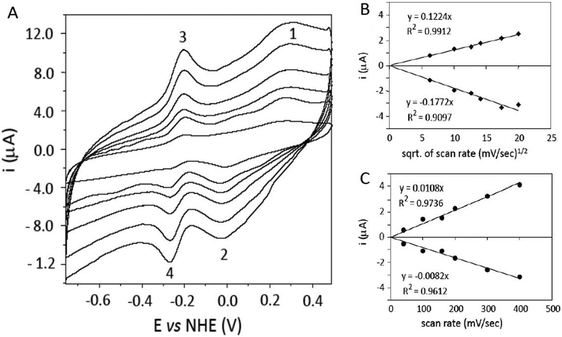 | ||
| Fig. 1 Direct CV of 195 μM PHR in 50 mM HEPES pH 7.0 on a glassy carbon electrode activated by methane flame. (A) Voltammograms obtained at scan rates of 40, 100, 160, 200, 300 and 400 mV s−1. (B) Plot of peak current versus the square root (sqrt) of the scan rate and relative linear fitting for peaks 1 and 2. (C) Plot of peak current versus the scan rate and linear fitting for peaks 3 and 4. | ||
Differential pulse voltammetry (DPV) was used to resolve the overlapping signals at Em 131.5 mV (Fig. 2A). The results show the presence of two signals at +69 and +201 mV. Upon addition of 2M guanidinium chloride, the free FAD signal at −0.250 mV sharply increases, as expected for the release of FAD by the unfolded PHR (Fig. 2B, dotted line). On the other hand, the signals at +69 and +201 mV are replaced by a single peak at +119 mV. This effect is attributed to the influence of the protein matrix on the two Fe atoms of the 2Fe2S cluster: NMR experiments on ferredoxin-type clusters have shown that a difference of about 100 mV can be expected for the potential of the iron cluster according to different exposures of either iron atom within proteins.32 The observed splitting can be attributed to the presence of two protein conformers differing to some extent in the cluster environment and solvent exposure, which then merge to a single form upon denaturation.
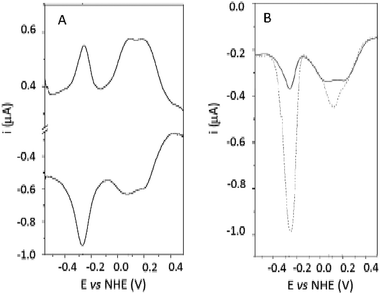 | ||
| Fig. 2 (A) DPV of 195 μM PHR in 50 mM HEPES pH 7.0 on a glassy carbon electrode. Step potential was 4.05 mV, modulation amplitude 4.05 mV, modulation time 0.05 s, interval time 0.25 s. (B) DPV (reducing scan only) of 195 μM M PHR before (continuous line) and after (dotted line) treatment with 2M guanidinium chloride. | ||
The relatively high midpoint potential observed for the PHR iron–sulfur cluster can be ascribed to the effect of the protein matrix, through amide interaction stabilisation and hydrogen bonding.34 The expected potentials for the Fe–S clusters of the ferredoxin type (4Cys, 2Fe 2S) are in the range −250 to −450 mV, much lower than the observed peak. The Em in the reductase of the methane monooxygenase determined by spectrophotometric/EPR titration with NADH are −150 mV for the FAD/FAD˙ couple, −220 mV for the Fe2S2/ Fe2˙S2 couple and −260 mV for the FAD˙/FAD˙˙ couple.57 These potentials have been determined by potentiometric titration and therefore the range of values obtained might not be the best reference for direct cyclic voltammetry.
High potentials in iron–sulfur centres have been previously observed in cyclic voltammetry, and the shift to positive potentials has been attributed to the formation of a complex at the electrode surface, which would lower the dielectric constant around the iron–sulfur cluster microenvironment. This would mimic the effect of surface interaction on protein complexes, as suggested for example by putidaredoxin anodic potential shift from −240 to −196 mV upon interaction with P450cam58 and by the analogous effect observed upon interaction of the same protein with polylysine coated electrodes (+64 mV anodic shift).43 A high potential for ferredoxin type redox centres (−7 mV) has also been reported for the mitochondrial succinate dehydrogenase.59 The remarkably high E° value of PHR could be the result of an unusually large exposure of the dinuclear Fe–S center to solvent which would stabilize the more negatively charged reduced ‘valence-trapped’ Fe2+/Fe3+ state over the oxidized Fe3+Fe3+ state, namely [Fe2S2(Cys)4]−3vs. [Fe2S2(Cys)4]−2.60
The spectral properties of PHR were tested on the sample recovered from the electrochemical cell after the CV and DPV measurements in order to ensure that no denaturation processes affected the protein following the interaction with the electrode. The spectra are reported in Fig. 3A: no differences were observed between the original PHR sample and the one used for the electrochemical experiment. The same does not hold for the guanidinium chloride treated sample used in DPV (Fig. 3B). The activity of the folded PHR was also assessed by spectrophotometrical measurements of the reduction of horse heart cytochrome c (hhc) by PHR in solution in the presence of NADH. Rates of hhc reduction by PHR before and after the electrochemical measurements corresponding to the same specific activity were experimentally recorded (60 ± 8 s−1, in line with ref. 4).
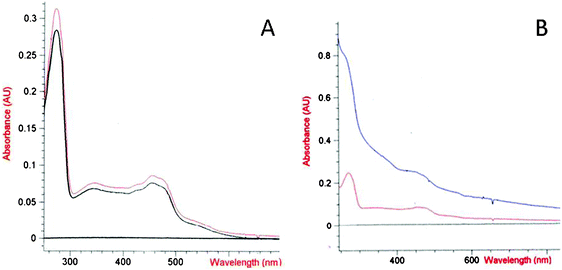 | ||
| Fig. 3 (A) Spectra of PHR sample before (red line) and after (black line) the CV experiment. (B) Spectra of PHR after DVP experiment without (red line) and with (blue line) treatment with 2M guanidinium chloride. | ||
Horse heart cytochrome c (hhc) was used as an artificial probe to assess the properties of the PHR surface involved in electron transfer and to confirm PHR electron transfer ability in the electrochemical cell set-up. Cytochrome c has been used in the past as a non-physiological redox partner for other reductases analogous to PHR. For example it has been demonstrated that in benzoate dioxygenase reductase hhc as an artificial electron acceptor involves the physiological redox transfer via FAD and iron–sulfur cluster, while other small molecules like potassium ferricyanide and 2,6 dichlorophenol indophenol, although good electron acceptors from this kind of reductases, might be reduced at good rates even in semi-apoforms lacking the 2Fe2S cluster.23 Moreover the well-known characteristics of hhc surface charges (highly positive and with a critical lysine ring surrounding the hot spot region for electron transfer) make it a good probe for charge–charge interactions.
Cyclic voltammetry was used to measure the pseudo-first order and second order rate constants for the reaction of PHR with hhc. In order to ensure that PHR could only interact with hhc and not directly with the electrode, a glassy carbon electrode was used after activation with nitric acid, a treatment which hinders any direct interaction of the electrode surface with PHR. As the interaction with PHR can only here occur via mediation by hhc, this method will be referred as mediated cyclic voltammetry (MCV).
The well defined voltammogram of hhc alone (75 μM), from which a diffusion controlled current value (id) was obtained at each tested ionic strength, was modified upon addition of increasing amount of PHR (0.5 to 4 μM) reduced by excess NADH (5 mM). A catalytic current ik was measured due to the regeneration of reduced hhc by PHR. The experimental ratio ik/id was related to the value kf/a by the Nicholson and Shain working curve. The plot of kf/a versus the scan rate gave the pseudo-first order constant kf.
The second order rate constants were calculated from the slope of the linear fittings of calculated kfversus the PHR concentration. As the nature of the interaction between charged molecules is studied by varying the ionic strength of buffer, the measurements were repeated over a range of ionic strengths from 0.01 to 0.08 M. Although the changes observed in the second order rate constant upon varying ionic strength in the tested range were within a narrow range of value (1.5 to 0.5 × 106 M−1 s−1) a significative trend of decrease of rate constants at higher ionic strengths can be observed (Fig. 4). The ionic strength dependence of PHR–hhc interaction further confirms that the protein remains in a folded and active form in the electrochemical cell set-up.
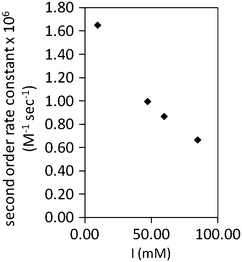 | ||
| Fig. 4 Plot of second order rate constants versus ionic strength (I) for the NADH dependent reduction of horse heart cytochrome c by PHR determined from kf values obtained by mediated cyclic voltammetry. Data points were obtained from triplicates and SD are below ±5% of the mean value. | ||
Direct electrochemistry of the PHO, the natural redox partner of PHR in phenol hydroxylase, was also studied by DPV on gold electrodes modified with Lys–Cys–Thr–Cys–Cys–Ala, a modification previously described for the catalytic moiety of methane monooxygenase from Methylococcus capsulatus (MMOX).55DPV showed two peaks at −74 and +396 mV (Fig. 5). The shape of the curve is nearly identical to that of MMOX, with two characteristic waves shifted towards more positive potentials, with differences of +66 and +146 mV for the lower and the higher potential waves respectively (MMOX: −142 and +248 mV).55
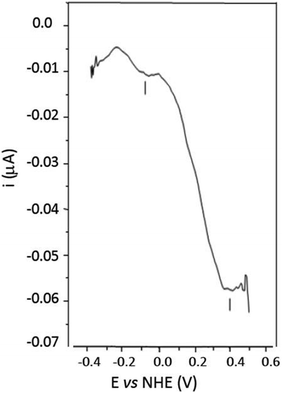 | ||
| Fig. 5 DPV scan of 13.35 μM PHO in MOPS 100 mM pH 7.0 on a gold electrode modified with Lys–Cys–Thr–Cys–Cys–Ala: scans were performed at 4.05 mV step potential and the background current due to the buffer has been subtracted. | ||
In conclusion, the results presented in this work support the hypothesis that the Em measured for PHR is compatible with electron transfer to PHO, and the entire phenol hydroxylase system from A. radioresistens functions at a high range of redox potentials when compared with other known multi-component systems with a similar structural and functional organisation. The 2Fe2S cluster of PHR, classified as thioredoxin-like or “third-kind”,28 shows unique redox properties with high midpoint potentials when compared to other ferredoxin-type 2Fe2S clusters. The DPV analysis performed on the PHO moiety on gold electrodes confirmed that the entire phenol hydroxylase system works at positive redox potentials and that the potential value measured for the PHO catalytic centre (+396 mV) is compatible with the electron supply by the PHR 2Fe2S redox centre.
Abbreviations
| PH | Phenol Hydroxylase from A. radioresistens |
| PHR | Phenol Hydroxylase Reductase |
| PHO | Phenol Hydroxylase Oxygenase |
| PHI | Phenol Hydroxylase Intermediate component |
| MMOX | Methane Monooxygenase |
| SCE | Saturated Calomel Electrode |
| NHE | Normal Hydrogen Electrode |
| CV | Cyclic Voltammetry |
| DVP | Differential Pulse Voltammetry |
| FAD | Flavin adenine Dinucleotide |
| Em | Midpoint potential |
References
- E. Pessione, F. Bosco, V. Specchia and C. Giunta, Microbios, 1996, 88, 213 Search PubMed.
- E. Pessione and C. Giunta, Microbios, 1997, 89, 105 Search PubMed.
- F. Briganti, E. Pessione, C. Giunta and A. Scozzafava, FEBS Lett., 1997, 416, 61 CrossRef CAS.
- E. Pessione, S. Divari, E. Griva, M. Cavaletto, G. L. Rossi, G. Gilardi and C. Giunta, Eur. J. Biochem., 1999, 265, 549 CrossRef CAS.
- S. Divari, F. Valetti, P. Caposio, E. Pessione, M. Cavaletto, E. Griva, G. Gribaudo, G. Gilardi and C. Giunta, Eur. J. Biochem., 2003, 270, 2244 CrossRef CAS.
- E. Griva, E. Pessione, S. Divari, F. Valetti, M. Cavaletto, G. L. Rossi and C. Giunta, Eur. J. Biochem., 2003, 270, 1434 CrossRef CAS.
- V. Shingler, F. C. Franklin, M. Tsuda, D. Holroyd and M. Bagdasarian, J. Gen. Microbiol., 1989, 135, 1083 CAS.
- I. Nordlund, J. Powlowski and V. Shingler, J. Bacteriol., 1990, 172, 6826 CAS.
- I. Nordlund, J. Powlowski, A. Hagstrom and V. Shingler, J. Gen. Microbiol., 1993, 139, 2695 CAS.
- J. Powlowski and V. Shingler, J. Bacteriol., 1990, 172, 6834 CAS.
- S. Ehrt, F. Schirmer and W. Hillen, Mol. Microbiol., 1995, 18, 13 CrossRef CAS.
- B. G. Fox, W. A. Froland, E. J. Dege and J. D. Lipscomb, J. Biol. Chem., 1989, 264, 10023 CAS.
- B. G. Fox, Y. Liu, J. E. Dege and J. D. Lipscomb, J. Biol. Chem., 1991, 266, 540 CAS.
- W. A. Froland, K. K. Andersson, S.-K. Lee, Y. Liu and J. D. Lipscomb, J. Biol. Chem., 1992, 267, 17588 CAS.
- K. E. Liu and S. J. Lippard, J. Biol. Chem., 1991, 266, 12836 CAS.
- A. C. Rosenzweig, G. A. Frederick, S. J. Lippard and P. Nordlund, Nature, 1993, 366, 537 CrossRef CAS.
- Y. Liu, J. C. Nesheim, S.-K. Lee and J. D. Lipscomb, J. Biol. Chem., 1995, 270, 24662 CrossRef CAS.
- Y. Liu, J. C. Nesheim, K. E. Paulsen, M. T. Stankovich and J. D. Lipscomb, Biochemistry, 1997, 36, 5223 CrossRef CAS.
- N. Elango, R. Radhakrishnan, W. A. Froland, B. J. Wallar, C. A. Earhart, J. D. Lipscomb and D. H. Ohlendorf, Protein Sci., 1997, 6, 556 CrossRef CAS.
- D. E. Coufal, P. Tavares, A. S. Pereira, B. H. Hyunh and S. J. Lippard, Biochemistry, 1999, 38, 4504 CrossRef CAS.
- R. Davydov, A. M. Valentine, S. K. Panicucci, B. M. Hoffman and S. J. Lippard, Biochemistry, 1999, 38, 4188 CrossRef CAS.
- J. Green and H. Dalton, J. Biol. Chem., 1988, 263, 17561 CAS.
- M. Yamaguchi and H. Fujisawa, J. Biol. Chem., 1981, 256, 6783 CAS.
- J. Muller, A. A. Lugovskoy, G. Wagner and S. J. Lippard, Biochemistry, 2002, 41, 42 CrossRef.
- L. L. Chatwood, J. D. Gross, G. Wagner and S. J. Lippard, Biochemistry, 2004, 43, 11983 CrossRef CAS.
- A. Karlsson, Z. M. Beharry, D. Matthew Eby, E. D. Coulter, E. L. Neidle, D. M. Kurtz Jr., H. Eklund and S. Ramaswamy, J. Mol. Biol., 2002, 318, 261 CrossRef CAS.
- A. Marchler-Bauer, J. B. Anderson, M. K. Derbyshire, C. DeWeese-Scott, N. R. Gonzales, M. Gwadz, L. Hao, S. He, D. I. Hurwitz, J. D. Jackson, Z. Ke, D. Krylov, C. J. Lanczycki, C. A. Liebert, C. Liu, F. Lu, S. Lu, G. H. Marchler, M. Mullokandov, J. S. Song, N. Thanki, R. A. Yamashita, J. J. Yin, D. Zhang and S. H. Bryant, Nucleic Acids Res., 2009, 37, 205.
- J. Meyer, FEBS Lett., 2001, 509, 1 CrossRef CAS.
- J. Meyer, J. Biol. Inorg. Chem, 2008, 13, 157 CrossRef CAS.
- H. Mo, S. S. Pochapsky and T. C. Pochapsky, Biochemistry, 1999, 38, 5666 CrossRef CAS.
- M. Kostic, S. S. Pochapsky, J. Obenauer, H. Mo, G. M. Pagani, R. Pejchal and T. C. Pochapsky, Biochemistry, 2002, 41, 5978 CrossRef CAS.
- F. Capozzi, S. Ciurli and C. Luchinat, Struct. Bonding, 1998, 90, 127 CAS.
- R. Langen, G. M. Jensen, U. Jacob, P. J. Stephens and A. Warshel, J. Biol. Chem., 1992, 267, 25625 CAS.
- P. J. Stephens, D. R. Jollie and A. Warshel, Chem. Rev., 1996, 96, 2491 CrossRef CAS.
- H. Beinert, R. H. Holm and E. Munck, Science, 1997, 277, 653 CrossRef CAS; I. Bertini, C. Luchinat, A. Provenzani, A. Rosato and P. R. Vasos, Proteins, 2002, 46, 110 CrossRef CAS.
- B. S. Perrin Jr. and T. Ichiye, Proteins, 2010, 78, 2798 Search PubMed; E. Denke, T. Merbitz-Zahradnik, O. M. Hatzfeld, C. H. Snyder, T. A. Link and B. L. Trumpower, J. Biol. Chem., 1998, 273, 9085 CrossRef CAS.
- H. A. Heering, B. M. Bulsink, W. R. Hagen and T. E. Meyer, Biochemistry, 1995, 34, 14675 CrossRef CAS.
- T. A. Link, W. R. Hagen, A. J. Pierik, C. Assmann and G. van Jagow, Eur. J. Biochem., 1992, 208, 685 CrossRef CAS.
- T. A. Link, O. M. Hatzfeld, P. Unalkat, J. K. Shergill, R. Cammack and J. R. Mason, Biochemistry, 1996, 35, 7546 CrossRef CAS.
- A. Riedel, S. Fetzner, M. Rampp, F. Lingens, U. Liebl, J. L. Zimmermann and W. Nitschke, J. Biol. Chem., 1995, 270, 30869 CrossRef CAS.
- L. Banci, I. Bertini, G. G. Savellini and C. Luchinat, Inorg. Chem., 1996, 35, 4248 CrossRef CAS.
- S. G. Sligar and I. C. Gunsalus, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1078 CAS.
- L. Avila, M. Wirtz, R. A. Bunce and M. Rivera, JBIC, J. Biol. Inorg. Chem., 1999, 4, 664 CrossRef CAS.
- C. Lei, U. Wollenberger, C. Jung and F. W. Scheller, Biochem. Biophys. Res. Commun., 2000, 268, 740 CrossRef CAS.
- M. Wirtz, V. Oganesyan, X. Zhang, J. Studer and M. Rivera, Faraday Discuss., 2000, 116, 221 RSC , discussion 257.
- M. Rivera, R. Seetharaman, D. Girdhar, M. Wirtz, X. Zhang, X. Wang and S. White, Biochemistry, 1998, 37, 1485 CrossRef CAS.
- S. Bagby, P. D. Barker, L. H. Guo and H. A. Hill, Biochemistry, 1990, 29, 3213 CrossRef CAS.
- S. Mitra and S. J. Elliott, Biochemistry, 2009, 48, 3813 CrossRef CAS.
- W. Sokol and J. A. Howell, Biotechnol. Bioeng., 1981, 23, 2039 CrossRef CAS.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706 CrossRef CAS.
- M. F. J. M. Verhagen, R. B. G. Wolbert and W. R. Hagen, Eur. J. Biochem., 1994, 221, 821 CAS.
- W. R. Hagen, Eur. J. Biochem., 1989, 182, 523 CAS.
- H. A. Heering and W. R. Hagen, J. Electroanal. Chem., 1996, 404, 249 CrossRef CAS.
- M. F. J. M. Verhagen, T. A. Link and W. R. Hagen, FEBS Lett., 1995, 361, 75 CrossRef CAS.
- J. Kazlauskaite, H. A. Hill, P. C. Wilkins and H. Dalton, Eur. J. Biochem., 1996, 241, 552 CrossRef CAS.
- A. Aliverti, W. R. Hagen and G. Zanetti, FEBS Lett., 1995, 368, 220 CrossRef CAS.
- J. Lund and H. Dalton, Eur. J. Biochem., 1985, 147, 291 CAS.
- I. C. Gunsalus and G. C. Wagner, In Methods in Enzymology, ed. S. Fleisher and L. Packer, Academic Press, New York, 1978 Search PubMed.
- J. K. Shergill and R. Cammack, Biochim. Biophys. Acta, 1994, 1185, 43 CAS.
- G. Battistuzzi, M. D'Onofrio, M. Borsari, M. Sola, A. L. Macedo, J. J. Moura and P. Rodrigues, JBIC, J. Biol. Inorg. Chem., 2000, 5, 748 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |