Non-iminosugar glucocerebrosidase small molecule chaperones†
Juan Jose
Marugan
*a,
Wenwei
Huang
a,
Omid
Motabar
b,
Wei
Zheng
a,
Jingbo
Xiao
a,
Samarjit
Patnaik
a,
Noel
Southall
a,
Wendy
Westbroek
b,
Wendy A.
Lea
a,
Anton
Simeonov
a,
Ehud
Goldin
b,
Maria A.
DeBernardi
c and
Ellen
Sidransky
b
aNIH Chemical Genomics Center, National Human Genome Research Institute, National Institutes of Health, 9800 Medical Center Drive, Rockville, 20850, MD, USA. E-mail: maruganj@mail.nih.gov
bMedical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Building 35 Rm1A213, 35 Convent Drive, Bethesda, 20892, MD, USA
cJohns Hopkins University Microscopy Center, Montgomery County Campus, 9605 Medical Center Drive, Rockville, 20850, MD, USA
First published on 24th October 2011
Abstract
Small molecule chaperones are a promising therapeutic approach for the Lysosomal Storage Disorders (LSDs). Here, we report the discovery of a new series of non-iminosugar glucocerebrosidase inhibitors with chaperone capacity, and describe their structure–activity relationship (SAR), selectivity, cell activity and phamacokinetics.
Introduction
Gaucher disease is an autosomal recessive disorder that results from DNA mutations affecting the expression, function or translocation properties of the lysosomal hydrolase, glucocerebrosidase (EC 3.2.1.45, GCase).1 As a result of these changes, glucosylceramides, the enzyme's natural substrates, accumulate in lysosomes of macrophages, resulting in diverse disease manifestations.2–4Many mutant forms of GCase, including the prevalent Gaucher mutation N370S, are still functional. However, the altered amino acid sequence has a strong impact on the protein structure, preventing its proper folding to the conformation recognized by the corresponding lysosomal transporters in the endoplasmic reticulum (ER).4 As a consequence of misfolding, mutant GCase may accumulate in the ER, and is eventually tagged for proteasome-mediated breakdown. Small molecule chaperones can bind with these mutants, inducing or accelerating proper folding, and therefore are able to facilitate the tranlocation of the enzyme to the lysosome.5–7
Previous studies have shown that several iminosugar inhibitors of lysosomal hydrolases also display chaperone capacity.5,8–12 However, iminosugars tend to have poor selectivity, and their therapeutic window is very small.13–15 Therefore, alternative chaperone scaffolds are highly desirable. Herein, we present a new series of non-iminosugar small molecule chaperones.
Results and discussion
Previously, we described an approach for improving high-throughput screening techniques by executing assays for lysosomal hydrolases in a tissue-homogenate environment, which better emulated the cell's native environment.16,17 Using this tissue homogenate protocol,18 we identified compound ML156 (8i) with potent GCase inhibitory activity (Fig. 1). In addition, this compound was highly selective when tested with other hydrolases, including alpha-glucosidase and alpha-galactosidase. It did not exhibit auto-fluorescence, and it displayed inhibitory activity in isolated enzyme assays. Moreover, this compound effectively inhibited the hydrolysis of 4-methylumbelliferone β-D-glucopyranoside (blue substrate), resorufin-β-glucopyranoside (red substrate) and a fluorescent ceramide substrate.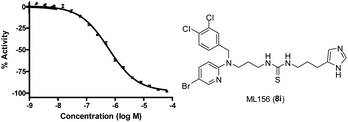 | ||
| Fig. 1 Concentration-response of lead compound ML156 (8i) in the primary screening assay for inhibition using tissue homogenate from a patient with genotype N370S N−1370S. The IC50 of this compound, measured in triplicate on three different days, was 580 ± 30 nM. | ||
As an initial evaluation of the potential chaperone capacity of ML156 (8i), we evaluated its effect on GCase function when the enzyme was incubated at 51 °C for 60 min (Fig. 2).19 Other known GCase chaperones, such as Isofagomine, have demostrated their capacity to thermostabilize this enzyme. In the absence of compound, the enzyme lost most of its hydrolytic capacity within 1 h. In contrast, in the presence of ML156 (8i) or Isofagomine as a control molecule, the enzyme was able to retain considerable function, indicating its ability to stabilize the functional conformation of GCase (Fig. 2). These data suggested that members of this chemical series had potential as chemical chaperones.
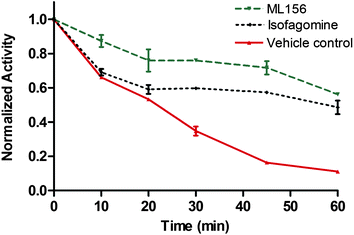 | ||
| Fig. 2 Thermofunctional stabilization studies with ML156 (8i) compared to the iminosugar Isofagomine. | ||
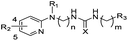
The synthesis of the selected compound ML156 (8i) and its analogues for SAR exploration is detailed in Schemes 1 to 3. As shown, reaction of 2-bromopyridines 1a–c with an excess of a diaminoalkane gave the corresponding 2-aminopyridines 3a–e. The primary amino group of 3a–e was then protected by a trityl group, yielding 4a–e. Subsequent selective alkylation of 4a–e with appropriate alkyl halides in dimethylformamide using lithium hexamethyldisilazide (LiHMDS) as a deprotonating agent afforded 5a–f and 5h–i, which were readily converted to amine 6a–i by trifluoroacetic acid mediated trityl group deprotection. The desired ureas 8j–k and thioureas 8a–i and 8l were prepared by a multicomponent one-pot reaction of 6, 7, and 1,1′-carbonyldiimidazole or 1,1′-thiocarbonyldiimidazole, followed by removal of the trityl protecting group (Scheme 1).
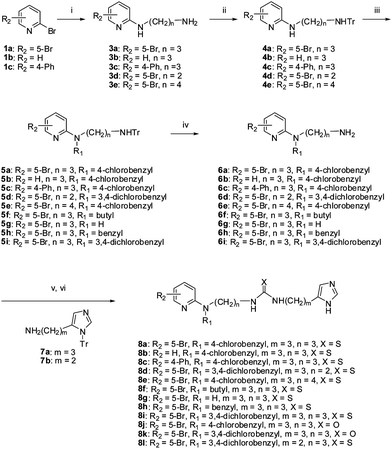 | ||
| Scheme 1 Reagents and conditions: (i) H2N(CH2)nNH2 (2), pyridine, reflux; (ii) TrCl, Et3N, THF, 0 °C-r.t.; (iii) LiHMDS, THF, R1X, r.t.; (iv) TFA, DCM; (v) 7, 1,1′-carbonyldiimidazole or 1,1′-thiocarbonyldiimidazole, iPrNEt2, DCM, μW, 130 °C, 10 min; (vi) TFA, DCM. | ||
Additional analogues were synthesized employing a Suzuki reaction of 5a with phenyl boronic acid, followed by trityl group deprotection to afford amine 9. Thiourea 10 was formed by a similar one-pot reaction of 9, 7a, and 1,1-thiocarbonyldiimidazole, followed by deprotection of the trityl group (Scheme 2).
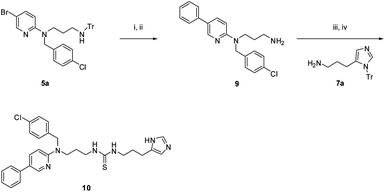 | ||
| Scheme 2 Reagents and conditions: (i) PhB(OH)2, Na2CO3, Pd(PPh3)4, 100 °C, 2 h; (ii) TFA, DCM; (iii) 7a, 1,1′-thiocarbonyldiimidazole, iPrNEt2, DCM, μW 150 °C, 10 min; (iv) TFA, DCM. | ||
Multicomponent reactions of 6a, 11, and 1,1-thiocarbonyldiimidazole yielded the corresponding thiourea 12. Trifluoroacetic acid mediated Boc-deprotection of 12 afforded the free amine 13a. Capping of the amine with an acetyl group gave the final compound 13b (Scheme 3).
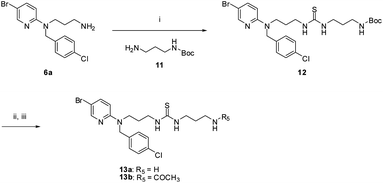 | ||
| Scheme 3 Reagents and conditions: Reagents and conditions: (i) 11, 1,1′-thiocarbonyldiimidazole, iPrNEt2, DCM, μW 130 °C, 10 min; (ii) TFA, DCM; (iii) for 13b only, CH3COCl, TEA, DCM. | ||
All analogues were tested in an assay using an N370S spleen homogenate assay (AID2590)18 to evaluate their inhibitory activity (Table 1). The resynthesized hit compound 8i displayed the same activity (0.6 μM) as the purchased sample (entries 1–2).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Cmpd. No. | R1 | R2 | R3 | n | m | X | IC50 (μM) |
| 1 | ML156,8i | 3,4-Dichlorobenzyl | 5-Br | 4-Imidazole | 3 | 3 | S | 0.6 ± 0.1 |
| 2 | 8a | 4-Chlorobenzyl | 5-Br | 4-Imidazole | 3 | 3 | S | 1.2 |
| 3 | 8f | Butyl | 5-Br | 4-Imidazole | 3 | 3 | S | 8.2 |
| 4 | 8h | Benzyl | 5-Br | 4-Imidazole | 3 | 3 | S | 5.8 |
| 5 | 8g | H | 5-Br | 4-Imidazole | 3 | 3 | S | 46.0 |
| 6 | 8b | 3,4-Dichlorobenzyl | H | 4-Imidazole | 3 | 3 | S | 7.3 |
| 7 | 10 | 4-Chlorobenzyl | 5-Ph | 4-Imidazole | 3 | 3 | S | 1.3 |
| 8 | 8c | 4-Chlorobenzyl | 4-Ph | 4-Imidazole | 3 | 3 | S | 4.0 |
| 9 | 8d | 3,4-Dichlorobenzyl | 5-Br | 4-Imidazole | 2 | 3 | S | 2.1 |
| 10 | 8e | 4-Chlorobenzyl | 5-Br | 4-Imidazole | 4 | 3 | S | 0.8 |
| 11 | 8l | 3,4-Dichlorobenzyl | 5-Br | 4-Imidazole | 3 | 2 | S | 18.3 |
| 12 | 8j | 3,4-Dichlorobenzyl | 5-Br | 4-Imidazole | 3 | 3 | 0.6 | |
| 13 | 8k | 4-Chlorobenzyl | 5-Br | 4-Imidazole | 3 | 3 | 1.0 | |
| 14 | 13a | 4-Chlorobenzyl | 5-Br | NH2 | 3 | 3 | S | 45.0 |
| 15 | 13b | 4-Chlorobenzyl | 5-Br | NHCOCH3 | 3 | 3 | S | inactive |
| 16 | 25 | 3,4-Dichlorobenzyl | 5-Br | CH3 | 3 | 3 | S | inactive |
| 17 | 8q | phenylethyl | 5-Br | 4-Imidazole | 3 | 3 | S | 2.8 |
| 18 | 8m | 4-chlorophenylethyl | 5-Br | 4-Imidazole | 3 | 3 | S | 2 |
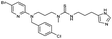
| 19 | 26 |
|
2.8 | |||||
| 20 | 27 |
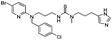
|
18 | |||||
| 21 | 24 |
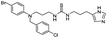
|
1.3 | |||||
Several positions of the hit compound ML156 (8i) were explored for SAR, yielding the following results: (a) Decreasing the size of the R1 substituent gradually weakened the activity (entries 1–5). (b) At the R2 position, placing a phenyl group at C5 resulted in better activity than placing the phenyl group at C4 (10vs.8c). (c) Ureas had activities similar to the thioureas (8ivs.8j and 8avs.8k). (d) Decreasing the distance between the thiourea group and the pyridyl moiety from 3C to 2C resulted in a small decrease in activity (8ivs.8d). On the other hand, increasing this carbon chain from 3C to 4C slightly increased the activity (8evs.8a). However, shortening the distance between the thiourea group and the imidazole group from 3C to 2C resulted in a 30-fold loss of activity (8ivs.8l). (e) The imidazole group at the R3 position was found to be very important for optimal activity. Replacing the imidazole with a free amino group (13a), an acetamide (13b), or a methyl group (25) resulted in significant reduction or complete loss of activity. (f) Selective methylation of the thiourea NH between the thiourea and pyridine resulted in a 2-fold loss of activity (26vs.8a). Methylation of the other thiourea NH between the thiourea and imidazole resulted in a 10-fold loss of activity (27vs.8a). (g) Replacing the pyridine ring with a benzene ring (24vs.8a), as shown in entry 21, yielded a similar level of activity.
Our lead compound (ML156, 8i) has been previously described in the literature as an agonist of the Somatostatin receptor SST4.20 For this reason, we decided to evaluate several analogues of the series in a human SST4 receptor binding affinity assay (Table 2). When comparing data from the receptor binding assay and the GCase assay, it was clear that the SARs did not correlate with each other. For example, compound 24 was about 10-fold more potent than compound 27 in the N370S spleen homogenate GCase assay, but 24 was about 5-fold less potent than 27 in the SST4 binding affinity assay. These results suggest that the SST4 binding affinity of this series could be further diluted without diminishing the GCase activity through additional structure modifications.
| Entry | Compound # | GCase IC50 (nM) | SST4 Ki (nM) |
|---|---|---|---|
| 1 | 8i | 600 | 17 |
| 2 | 8g | 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
690 |
| 3 | 26 | 2800 | 19 |
| 4 | 24 | 1300 | 160 |
| 5 | 27 | 18000 |
26 |
We then evaluated the GCase chaperone capacity of our best compounds, as well as the known chaperone of Isofagomine with laser scanning confocal microscopy in both wild type fibroblasts and fibroblasts from patients with genotype N370S N−1370S.21 The cells were treated with 1 μM of the selected compounds, let rest for five days, and then stained both with an Alexa555-labeled antibody for GCase (red) and an Alexa488-labeled antibody for the lysosomal marker LAMP-1 (green). Fig. 3 shows high magnification images resulting from these translocation studies. Increased co-localization between LAMP-1 (green), representing the lysosomes, and the N370S GCase enzyme (red) is clearly observed after treatment with 1 μM of 24 (Fig. 3C) or 8i (Fig. 3D) compared to vehicle-only treatment (Fig. 3A). These compounds show translocation capacities that are similar to the iminosugar GCase chaperone Isofagomine (Fig. 3B), which has been advanced to clinical trials. In addition, these compounds display a lower GCase inhibitory activity when compared to Isofagomine (IC50 = 80 nM, using the N370S tissue homogenate assay with the blue substrate), and therefore, they may offer a wider therapeutic window.
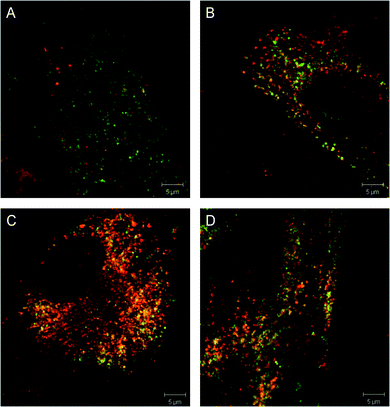 | ||
| Fig. 3 Translocation studies of N370S GCase in Gaucher fibroblasts (genotype N370S N−1370S). Gaucher fibroblasts were stained for LAMP-1 (green) and GCase (red) in the presence of (A) DMSO (vehicle), (B) 1 μM Isofagomine, (C) 1 μM of compound 24, (D) 1 μM of compound ML156 (8i). Scale bar is 5 μm. | ||
We further evaluated the stability of our chemical series under in vitro conditions. Compounds were incubated in mouse liver microsomes for 60 min in the presence of NADPH. After incubation with our lead compound (ML156, 8i), 58% of the parent molecule remained, suggesting that this compound is metabolically stable. This promising result prompted us to evaluate the in vivo pharmacokinetic profile of ML156 (8i). Table 3 shows the levels and pharmacokinetic parameters of ML156 (8i) in plasma following intraperitoneal administration of the compound to mice at a dose of 30 mg kg−1.
| Dose (mg kg−1) | Dose rose | Sampling time (h) | Concentration (ng mL−1) | Mean (ng mL−1) | Mean (μM) | SD | CV (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| Individual | |||||||||
| 30 | IP | 0 | BQL | BQL | BQL | BQL | BQL | N/A | N/A |
| 0.083 | 1820 | 1820 | 1410 | 1683 | 3.03 | 237 | 14.1 | ||
| 0.25 | 2420 | 3010 | 1640 | 2357 | 4.24 | 687 | 29.2 | ||
| 0.5 | 2530 | 2270 | 3270 | 2690 | 4.84 | 519 | 19.3 | ||
| 1 | 3740 | 3770 | 3150 | 3553 | 6.39 | 350 | 9.84 | ||
| 2 | 2870 | 3590 | 3840 | 3433 | 6.17 | 504 | 14.7 | ||
| 4 | 2560 | 2030 | 2570 | 2387 | 4.29 | 309 | 12.9 | ||
| 8 | 1190 | 357 | 714 | 754 | 1.35 | 418 | 55.5 | ||
| 12 | 118 | 127 | 80.7 | 109 | 0.20 | 24.5 | 22.6 | ||
| 24 | 8.04 | 12.6 | 9.98 | 10.2 | 0.02 | 2.29 | 22.4 | ||
| PK parameters | Unit | Estimate | |||||||
| Tmax | h | 1.00 | |||||||
| Cmax | ng mL−1 | 3553 | |||||||
| T1/2 | h | 2.57 | |||||||
| AUClast | h*ng mL−1 | 20600 |
|||||||
| AUCINF | h*ng mL−1 | 20700 |
|||||||
The high stability of ML156 (8i) is shown in Table 3. This compound maintained plasma levels above the GCase IC50 for more than 8 h. Additional pharmacokinetic studies disclosed that reasonable levels of the compound also reached the brain (please see the supporting information†). This finding is very encouraging, suggesting that this compound could have utility in the treatment of the neuronopathic forms (Type 2 and 3) of Gaucher disease.
Conclusions
We have reported the discovery and initial SAR study of a novel non-iminosugar small molecule chemical series with encouraging GCase chaperone activity. Initial pharmacokinetic studies of IP administered ML156 (8i) show good penetration of this molecule in plasma, brain and liver. The level of the compound in brain is especially important, as the current standard-of-care, enzyme replacement therapy with recombinant GCase, has no effect on the neurological manifestations of the disease due to its poor CNS distribution. Therefore, this series provides an excellent lead candidate for further development and testing in animal models of Gaucher disease. Further in vivo evaluation is currently in progress.Acknowledgements
We thank Xianlong Chu, Xin Chen, Ruihong Tao (Alputon Inc.) and the Alputon chemistry team for their participation in this project. We also thank Ms Allison Mandich for critical review of the manuscript. This research was supported by the Molecular Libraries Initiative of the NIH Roadmap for Medical Research (U54MH084681) and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.Notes and references
- G. A. Grabowski, Lancet, 2008, 372, 1263–1271 CrossRef CAS.
- A. R. Sawkar, W. D'Haeze and J. W. Kelly, Cell. Mol. Life Sci., 2006, 63, 1179–1192 CrossRef CAS.
- K. S. Hruska, M. E. LaMarca, C. R. Scott and E. Sidransky, Hum. Mutat., 2008, 29, 567–583 CrossRef CAS.
- A. H. Futerman and M. G. van, Nat. Rev. Mol. Cell Biol., 2004, 5, 554–565 CrossRef CAS.
- T. D. Butters, R. A. Dwek and F. M. Platt, Glycobiology, 2005, 15, 43R–52R CrossRef CAS.
- A. R. Sawkar, W.-C. Cheng, E. Beutler, C.-H. Wong, W. E. Balch and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15428–15433 CrossRef CAS.
- T. D. Butters, Expert Opin. Pharmacother., 2007, 8, 427–435 CrossRef CAS.
- Z. Yu, A. R. Sawkar, L. J. Whalen, C.-H. Wong and J. W. Kelly, J. Med. Chem., 2007, 50, 94–100 CrossRef CAS.
- H.-H. Chang, N. Asano, S. Ishii, Y. Ichikawa and J.-Q. Fan, FEBS J., 2006, 273, 4082–4092 CrossRef CAS.
- P. Compain, O. R. Martin, C. Boucheron, G. Godin, L. Yu, K. Ikeda and N. Asano, ChemBioChem, 2006, 7, 1356–1359 CrossRef CAS.
- L. Yu, K. Ikeda, A. Kato, I. Adachi, G. Godin, P. Compain, O. Martin and N. Asano, Bioorg. Med. Chem., 2006, 14, 7736–7744 CrossRef CAS.
- M. Egido-Gabas, D. Canals, J. Casas, A. Llebaria and A. Delgado, ChemMedChem, 2007, 2, 992–994 CrossRef CAS.
- J. Diot, M. I. Garcia-Moreno, S. G. Gouin, M. C. Ortiz, K. Haupt and J. Kovensky, Org. Biomol. Chem., 2009, 7, 357–363 CAS.
- G. Horne, F. X. Wilson, J. Tinsley, D. H. Williams and R. Storer, Drug Discovery Today, 2011, 16, 107–118 CrossRef CAS.
- J.-Q. Fan, Trends Pharmacol. Sci., 2003, 24, 355–360 CrossRef CAS.
- J. J. Marugan, W. Zheng, O. Motabar, N. Southall, E. Goldin, E. Sidransky, R. A. Aungst, K. Liu, S. K. Sadhukhan and C. P. Austin, Eur. J. Med. Chem., 2010, 45, 1880–1897 CrossRef CAS.
- J. J. Marugan, W. Zheng, O. Motabar, N. Southall, E. Goldin, W. Westbroek, B. K. Stubblefield, E. Sidransky, R. A. Aungst, W. A. Lea, A. Simeonov, W. Leister and C. P. Austin, J. Med. Chem., 2011, 54, 1033–1058 CrossRef CAS.
- The results of the GC screens and follow up studies can be found in PubChem (AID's: 2101, 2613, 2592, 2588, 2595, 2597, 2596, 2577, 2578, 2587, 2589, and 2593).
- For details, please check the supporting information†.
- M. Ankersen, M. Crider, S. Liu, B. Ho, H. S. Andersen and C. Stidsen, J. Am. Chem. Soc., 1998, 120, 1368–1373 CrossRef CAS.
- O. Goker-Alpan, E. A. Wiggs, M. J. Eblan, W. Benko, S. G. Ziegler, E. Sidransky and R. Schiffmann, J. Pediatr., 2008, 153, 89–94 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1md00200g |
| This journal is © The Royal Society of Chemistry 2012 |