Structure–activity relationships of methyl-lysine reader antagonists†‡
J. Martin
Herold
a,
Lindsey Ingerman
James
a,
Victoria K.
Korboukh
a,
Cen
Gao
a,
Kaitlyn E.
Coil
b,
Dennis J.
Bua
b,
Jacqueline L.
Norris
a,
Dmitri B.
Kireev
a,
Peter J.
Brown
c,
Jian
Jin
a,
William P.
Janzen
a,
Or
Gozani
b and
Stephen V.
Frye
*a
aCenter for Integrated Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Division of Medicinal Chemistry and Natural Products, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States. E-mail: svfrye@email.unc.edu; Fax: +1 919-843-8465; Tel: +1 919-843-5486
bDepartment of Biology, Stanford University, Stanford, California 94305, United States. Fax: +1 650-725-8309; Tel: +1 650-736-7639
cStructural Genomics Consortium, University of Toronto, Toronto, M5G 1L7, Ontario, Canada
First published on 10th October 2011
Abstract
The interaction between methyl-lysine binding proteins and methylated histones plays a crucial role in the regulation of gene expression. Herein we describe the development of structure–activity relationships (SAR) surrounding UNC669, the first reported small molecule ligand for a methyl-lysine binding domain, using multiple assay formats. These studies revealed the key features required for successful inhibition of the L3MBTL1-methylated histoneprotein-protein interaction, while the selectivity of designed compounds against a panel of related methyl-lysine readers was also evaluated. Additionally, an optimized compound was demonstrated to successfully inhibit the recognition of H4K20me1 by L3MBTL1 in the context of an affinity pull down assay.
Introduction
A central biochemical mechanism for regulation of chromatin state is the post-translational modification of histones. Over the last decade, the enzymes and protein complexes that create, remove, and recognize these modifications have been characterized to varying degrees using the standard methodologies of molecular biology, cell biology, and biochemistry.1 However, there has been limited purposeful chemical biology or medicinal chemistry exploration of chromatin regulation.2–11 The recognition of the methylation-state of lysine residues in histones is a critical event in transcription. For example, different lysinemethylation marks (Kme) are associated with active (histone 3, lysine 4 dimethylation, i.e. - H3K4me2) and repressed transcriptional states (H3K9me2).12,13 The specific domains which recognize Kme have been described within several protein families: the plant homeodomain – PHD; the so-called ‘royal family’ – made up of Tudor, Agenet, chromo, PWWP and MBT domains; and the WD40 repeat proteins such as WDR5 and EED.When we initiated our program to discover antagonists of Kme binding domains, we focused on the MBT domain family because of its tractable size (9 MBT containing proteins in humans), unique binding mode, and abundant structural information.14,15 In addition to a distinct recognition motif and selectivity for Kme1–2 over Kme0 and Kme3, MBT-domain containing proteins have been functionally associated with important regulatory changes in the transcriptional state of chromatin regions and significant developmental biology.16–24 For example, the three MBT repeat fragment of human L3MBTL1 has been reported to compact nucleosomal arrays within the context of mono- and dimethylated states, but not the trimethylated state of H4K20 and H1bK26, leading to gene repression.25
We have recently reported the discovery of UNC669 (1) (Fig. 1) as a small molecule ligand of MBT domains, particularly L3MBTL1, for which a cocrystal structure was solved.14 To extend our exploration of this template we were particularly interested in how subtle changes in the structural features of nicotinamide1 could influence affinity and selectivity against Kme binding domains. Our initial Kme reader panel includes close homologues of L3MBTL1: L3MBTL3 and L3MBTL4, as well as SFMBT and MBTD1 in order to cover the other branches of the phylogenetic tree (Fig. 2). To gauge selectivity versus another methyl-lysine binding domain, the chromo domain CBX7 was chosen. While this panel of methyl-lysine binding domains represents only a fraction of this large target-class, inclusion of both closely related MBT domains and a less related binder that recognizes Kme3 provides an initial basis to assess both the selectivity of ligands and the tractability of another less similar binder for ligand discovery. We relied on an AlphaScreen® assay for initial protein profiling of all new scaffolds and isothermal titration calorimetry (ITC) as an orthogonal assay for confirmation of results for select compounds.26
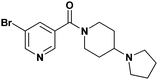 | ||
| Fig. 1 Structure of L3MBTL1inhibitor, UNC669 (1). | ||
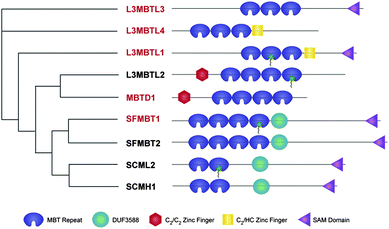 | ||
| Fig. 2 Phylogenetic tree of the human MBT domain protein family. The proteins in red are those included in our reader panel. | ||
Herein we detail our efforts to define the structure–activity relationships of small molecule methyl-lysine reader antagonists related to UNC669, with a focus on the MBT domain family. We also include confirmation of the activity of our most potent L3MBTL1antagonist in the context of an L3MBTL1 pull down assay.
Results and discussion
Having established nicotinamide1 as an effective ligand of the second MBT domain of L3MBTL1, multiple series of compounds were prepared in order to assess the effect of different substituents on the aromatic anchor, optimization of the (pyrrolidinyl)piperidine amine for the lysine pocket, and the effect of the linker between the aromatic anchor and the amine. AlphaScreen® data were then used to examine the potency of these variants in relation to compound 1. While this assay format is efficient in that it allows for the screening of a number of compounds in a high-throughput fashion, requiring minimal reagents, we also note that there can be variability in the results obtained from one day to the next. Consequently, compound 1, whose affinity for L3MBTL1 has been verified in multiple orthogonal assays, was included as a standard in each assay run.§ Furthermore, ITC was used as an orthogonal assay to confirm the affinity of key compounds.In the design of analogues of 1, it was apparent that the use of a benzoic acid anchor as opposed to a nicotinamide would provide greater synthetic opportunities, and as the X-ray co-crystal structure of 1 and L3MBTL1 revealed no specific favorable interaction between the pyridinenitrogen and the protein, we proceeded accordingly. In fact, the direct benzoic acid analogue of 1, compound UNC926 (2), showed a two-fold improvement in potency versusL3MBTL1. This improved affinity of 2 was confirmed by ITC, resulting in a Kd of 3.9 μM as opposed to 8.6 μM for 1 (see ESI).¶ Similarly to 1, compound 2 also exhibited a low micromolar affinity for the close homolog, L3MBTL3, with a decrease in affinity for the other MBT domains and no binding to CBX7.
Confident that the conversion to a benzoic acid core would not negatively affect the ligand's potency, we set out to investigate the effect of different bromine regioisomers of 2. Compounds 3 and 4 were prepared in an analogous fashion to 2, with bromine in the para- and ortho- positions, respectively, relative to the amide (Table 1). Both 3 and 4 were about 4-fold and 6-fold less potent against L3MBTL1, respectively, with little change in affinity for the remainder of the panel. The co-crystal structure of 1 bound to L3MBTL1 suggests that the bromine may have a favourable interaction with the surface of the protein, and this hypothesis appears to be supported by the decrease in affinity of compounds 3 and 4 for L3MBTL1. Furthermore, when the bromine is removed from the compound entirely, as seen in 5, the affinity for L3MBTL1 is comparable to that of 3 and 4, indicating that these alternate bromine regioisomers likely do not experience any unfavorable interactions.
| Compound ID | R | AlphaScreen®IC50 (μM) | |||||
|---|---|---|---|---|---|---|---|
| L3MBTL1 | L3MBTL3 | L3MBTL4 | MBTD1 | SFMBT1 | CBX7 | ||
| a IC50 values are the average of at least 5 values from at least 2 independent trials ± the standard deviation. b The screens reported herein are performed up to 30 μM. For those compounds with an IC50 greater than 30 μM, the percent inhibition at 30 μM is reported and compounds reported as not active (n/a) showed no inhibition at 30 μM. | |||||||
| 1 |
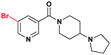
|
7.9 ± 1.1 | 4.8 ± 1.1 | >30 (41%) | >30 (23%) | >30 (n/a) | >30 (9%) |
| 2 |
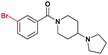
|
3.9 ± 1.1 | 3.2 ± 1.0 | 15.6 ± 1.1 | >30 (44%) | >30 (12%) | >30 (11%) |
| 3 |
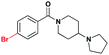
|
14.7 ± 1.0 | 4.1 ± 1.0 | >30 (30%) | >30 (24%) | >30 (n/a) | >30 (n/a) |
| 4 |
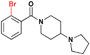
|
22.7 ± 1.0 | 6.8 ± 1.0 | >30 (29%) | >30 (31%) | >30 (n/a) | >30 (17%) |
| 5 |
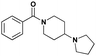
|
17.8 ± 1.1 | 6.2 ± 1.0 | >30 (40%) | >30 (22%) | >30 (n/a) | >30 (n/a) |
| 6 |
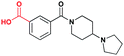
|
> 30 (31%) | 6.9 ± 1.0 | >30 (26%) | >30 (4%) | >30 (n/a) | >30 (4%) |
| 7 |
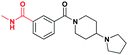
|
13.3 ± 1.0 | 6.4 ± 1.0 | >30 (32%) | >30 (21%) | >30 (n/a) | >30 (n/a) |
While positional variation of the bromine was tolerated to a certain extent, replacement of the bromine in 2 with an acidic functional group as seen in 6 results in a significantly less potent inhibitor against L3MBTL1 (Table 1). As there are multiple acidic residues surrounding the lysine binding pocket, it is reasonable to suggest that this diminished binding is due to electrostatic repulsion between the acidic functionality of 6 and a nearby acidic residue. Interestingly, binding of benzoic acid6 to L3MBTL3 is not diminished to the same extent despite the fact that the majority of these acidic residues are conserved, and in general, L3MBTL3 is less sensitive to structural variations in this scaffold. Affinity for L3MBTL1 can be partially rescued by converting the carboxylic acid of 6 to a methylbenzamide (7), however the potency remains about 3-fold weaker than 2.
The crystal structure of 1 bound to L3MBTL1 (PDB 3P8H) revealed that the (pyrrolidinyl)piperidine amine effectively filled the lysine binding cavity, maximizing the number of van der Waals contacts in the pocket, while also positioning the basic amine in close proximity to Asp355 in order to make a key hydrogen bond. With the 3-bromobenzamide scaffold as an anchor, we set out to further investigate the optimum amine for the L3MBTL1 binding pocket for both improvement in binding affinity as well as selectivity. Therefore, analogues of 2 were prepared in order to vary the size and disposition of the basic amine containing substituent. For example, expanding the pyrrolidine ring of 2 to a piperidine in 8 resulted in no activity against L3MBTL1 and only modest affinity for MBTD1 within the panel (Table 2), consistent with there being a similar steric requirement for all of these MBT domains.15ITC was used to successfully confirm that compound 8 does not bind L3MBTL1 (see supporting information).
| Compound ID | R | AlphaScreen®IC50 (μM) | |||||
|---|---|---|---|---|---|---|---|
| L3MBTL1 | L3MBTL3 | L3MBTL4 | MBTD1 | SFMBT1 | CBX7 | ||
| a IC50 values are the average of at least 5 values from at least 2 independent trials ± the standard deviation. b The screens reported herein are performed up to 30 μM. For those compounds with an IC50 greater than 30 μM, the percent inhibition at 30 μM is reported and compounds reported as not active (n/a) showed no inhibition at 30 μM. | |||||||
| 2 |
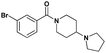
|
3.9 ± 1.1 | 3.2 ± 1.0 | 15.6 ± 1.1 | >30 (44%) | >30 (12%) | >30 (11%) |
| 8 |
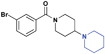
|
>30 (5%) | >30 (21%) | >30 (n/a) | 29.7 ± 1.1 | >30 (n/a) | >30 (3%) |
| 9 |
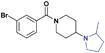
|
>30 (6%) | >30 (25%) | >30 (n/a) | >30 (16%) | >30 (n/a) | >30 (n/a) |
| 10 |
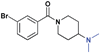
|
>30 (5%) | >30 (18%) | >30 (n/a) | >30 (1%) | >30 (n/a) | >30 (n/a) |
| 11 |
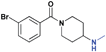
|
>30 (n/a) | >30 (9%) | >30 (n/a) | >30 (3%) | >30 (n/a) | >30 (3%) |
| 12 |
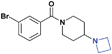
|
>30 (32%) | 23.8 ± 1.1 | >30 (4%) | >30 (20%) | >30 (n/a) | >30 (11%) |
| 13 |
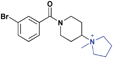
|
>30 (n/a) | >30 (13%) | >30 (n/a) | >30 (3%) | >30 (n/a) | >30 (17%) |
| 14 |
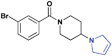
|
8.6 ± 1.0 | 8.7 ± 1.1 | >30 (28%) | >30 (24%) | >30 (n/a) | >30 (18%) |
| 15 |
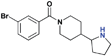
|
>30 (6%) | >30 (52%) | >30 (n/a) | 9.2 ± 1.1 | >30 (n/a) | >30 (n/a) |
| 16 |
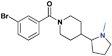
|
>30 (3%) | 21.0 ± 1.2 | >30 (n/a) | >30 (32%) | >30 (n/a) | >30 (n/a) |
| 17 |
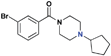
|
>30 (n/a) | >30 (5%) | >30 (n/a) | >30 (n/a) | >30 (n/a) | >30 (1%) |
| 18 |
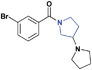
|
>30 (18%) | >30 (40%) | >30 (n/a) | >30 (13%) | >30 (n/a) | >30 (4%) |
| 19 |
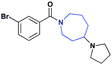
|
28.9 ± 1.0 | 6.7 ± 1.1 | >30 (28%) | 15.2 ± 1.0 | >30 (n/a) | >30 (13%) |
| 20 |
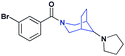
|
23.6 ± 1.0 | 3.6 ± 1.1 | >30 (16%) | >30 (31%) | >30 (n/a) | >30 (n/a) |
While we had previously shown that 2-methyl-pyrrolidines could bind to L3MBTL1 in the context of a flexible aliphatic linker,14 this substitution was not tolerated in 9 with the more rigid and bulky piperidine linker. We were surprised to find that the sterically more compact dimethylamine analogue 10 also showed no activity for L3MBTL1, a result which was consistent across the entire panel of Kme binders and further confirmed by ITC (see supporting information). Similarly, the monomethylated derivative 11 also demonstrated no affinity for any of the reader proteins, highlighting the distinction between the SAR in this series and the canonical recognition of Kme1 and Kme2 in the context of native histones. Converting the pyrrolidine to a smaller azetidine ring as in 12 similarly results in a decrease in binding affinity for both L3MBTL1 and L3MBTL3 relative to 2. The reversal in affinity of these compounds is most likely due to a decrease in the number of favorable van der Waals contacts in the pocket or small changes in the position of the positive charge. The protonated amine must be appropriately positioned to form a hydrogen bond with a nearby aspartic acid residue, as this interaction is imperative for binding. This key hydrogen bond has been identified previously by site-directed mutagenesis, and we have further confirmed this here, as methylating the amine renders 13 inactive. Interestingly, the quaternary ammonium analogue, 13, also does not interact with the Kme3 reader, CBX7, which was an outcome we might have anticipated. One final modification made to the pyrrolidine moiety was conversion to a pyrroline ring as shown in 14, resulting in a 2 to 3-fold decrease in affinity both for L3MBTL1 and L3MBTL3 relative to 2.
The next series of compounds were designed in order to investigate the position of the nitrogen to potentially further optimize hydrogen bonding with residue D355 of L3MBTL1. When comparing the co-crystal structure of L3MBTL1 with a H4K20me2peptideversusnicotinamide1, it appeared that the lysinenitrogen was positioned slightly deeper in the binding cavity versus that of the small molecule. Consequently, 15 was prepared to evaluate whether displacing the nitrogen in this fashion would have a positive or negative impact on binding. While it appeared in overlays of the crystal structures containing L3MBTL1 with both H4K20Me2 and 1 that the basic nitrogen in 15 would more closely mimic that of the peptide, this did not translate into a more potent binder than 2 against L3MBTL1 and L3MBTL3. Compound 15 was also methylated in order to increase the basicity of the amine. Not surprisingly based on the inactivity of 9, compound 16 was inactive against L3MBTL1, but a slight improvement in potency for L3MBTL3 relative to 15 was observed. It is worth noting that 15 is one of the few compounds that binds MBTD1, indicating that positioning the nitrogen in this fashion may aid in achieving selectivity for other readers; however, 16 was determined to be inactive against MBTD1. A more shallow placement of the nitrogen in the (cyclopentyl)piperazine analogue 17 resulted in inactivity against all proteins, indicating that the position of the nitrogen in 2 is optimal for binding to L3MBTL1.
Previous work on this scaffold demonstrated an improvement in affinity in moving from a flexible alkyl-pyrrolidine to the more rigidified amine seen in 1. While an enhanced affinity for L3MBTL1 was established through the introduction of the piperidine ring, it had yet to be determined if other ring systems would be equally potent at this position. The synthesis and evaluation of 18 and 19, where the piperidine was replaced with a smaller pyrrolidine and larger azepane respectively, demonstrated that the (pyrrolidinyl)azepane modification was better tolerated as a Kme mimic than the (pyrrolidinyl)pyrrolidine. To further increase the hydrophobicity, rigidity, and size of the piperidine ring, an azabicyclooctane scaffold was introduced in the synthesis of compound 20. While 20 showed about a 6-fold decrease in affinity for L3MBTL1, similar to that of compound 19, it remained equipotent against L3MBTL3 with a low micromolar IC50. This again shows the ability to modulate the selectivity between reader proteins based on minimal structural changes in the ring system.
Finally, to complete our initial investigation of the structure activity relationships of analogues of 1, the nature of the linker between the aromatic anchor and the amine was varied (Table 3). While the amide moiety connects the amine-containing ring in a fairly rigid fashion, anchoring the amine in the lysine pocket with little flexibility, we felt it was worth exploring linkers with different conformational preferences and electrostatic properties with the hopes that a higher affinity interaction would result.
| Compound ID | R | AlphaScreen®IC50 (μM) | |||||
|---|---|---|---|---|---|---|---|
| L3MBTL1 | L3MBTL3 | L3MBTL4 | MBTD1 | SFMBT1 | CBX7 | ||
| a IC50 values are the average of at least 5 values from at least 2 independent trials ± the standard deviation. b The screens reported herein are performed up to 30 μM. For those compounds with an IC50 greater than 30 μM, the percent inhibition at 30 μM is reported and compounds reported as not active (n/a) showed no inhibition at 30 μM. | |||||||
| 2 |
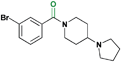
|
3.9 ± 1.1 | 3.2 ± 1.0 | 15.6 ± 1.1 | >30 (44%) | >30 (12%) | >30 (11%) |
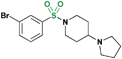
| 21 |
|
>30 (48%) | 3.7 ± 1.1 | >30 (6%) | 21.9 ± 1.1 | >30 (n/a) | >30 (n/a) |
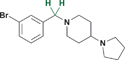
| 22 |
|
>30 (20%) | 13.3 ± 1.1 | >30 (10%) | 18.6 ± 1.1 | >30 (n/a) | >30 (n/a) |
| 23 |

|
>30 (25%) | 16.0 ± 1.1 | >30 (13%) | 9.0 ± 1.0 | >30 (n/a) | >30 (n/a) |
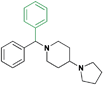
| 24 |
|
20.3 ± 1.0 | 2.4 ± 1.0 | >30 (17%) | >30 (36%) | >30 (n/a) | >30 (n/a) |
It has been shown previously that amides and sulfonamides display relatively equal potency against MBT domains in the context of small molecule binders containing aliphatic pyrrolidines. In contrast, compound 21 shows a decrease in potency against L3MBTL1 relative to 2, while maintaining equipotency against L3MBTL3 and gaining affinity for MBTD1. When the carbonyl is removed entirely as in the benzylated amine (22), a more significant change is observed. This compound is inactive against L3MBTL1 and L3MBTL4, with about a 4-fold decrease in potency against L3MBTL3. However in the case of MBTD1, an improvement in potency is observed.
In moving from the benzyl substituted analogue to a benzylmethylamine (rac-23) the affinity for MBTD1 is slightly greater, while remaining relatively unchanged against the other MBT domains. Interestingly, preparation of the dibenzyl substituted amine revealed that the affinity for L3MBTL3 could be regained entirely, while simultaneously reverting back to an inactive inhibitor of MBTD1. As the structural and physical properties of 24 and 2 are quite different, it is likely that there are multiple factors at play in dictating the potency of 24. For example, it is possible that while still maintaining a level of flexibility, the dibenzylgroup also picks up some unfavorable steric interactions with surrounding residues in the case of MBTD1. Structural studies with MBT domains and the key compounds from this SAR series will hopefully lead to a greater understanding of the basis for design of more potent and selective ligands.
In order to further confirm the ability of our most potent L3MBTL1 ligand (2) to antagonize the interaction between the three MBT repeats of L3MBTL1 (L3MBTL13xMBT) and H4K20me1, we performed a series of pull down assays (Fig. 3). It should be noted that the H4K20me0peptide serves as a control for the specificity of the MBT-methyllysine interaction (compare lanes 2 and 3 of Fig. 3A). In addition to active compounds nicotinamide1 and benzamide2, compound 8 was also evaluated as an inactive control in this assay. Consistent with AlphaScreen® and ITC results, compound 8 had little effect on the binding of the 3xMBT repeats of L3MBTL1 to H4K20me1. In contrast, compounds 1 and 2 both inhibited binding of the 3xMBT domain to H4K20me1. In agreement with the reported binding affinites (Table 1), 2 demonstrated stronger inhibition than 1 (compare lanes 3 and 6 of Fig. 3A). Thus, 1 and 2 inhibit the association of L3MBTL13xMBT with the appropriate histonepeptides in a dose-dependent manner.
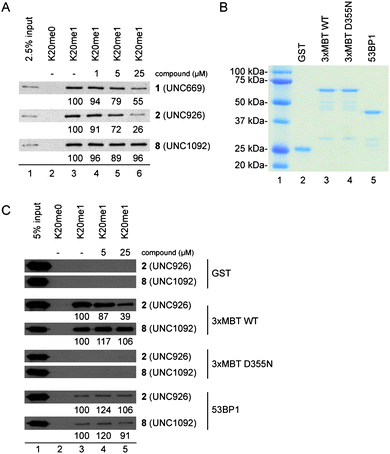 | ||
| Fig. 3 Compound 2 selectively inhibits the L3MBTL13xMBT-H4K20me1 interaction in a dose-dependent manner. A) Dose-dependent inhibition of the L3MBTL13xMBT-H4K20me1 interaction by 2 in peptide pull down assays. Western analysis of peptide pull down pellets with GST-fused MBT repeats of L3MBTL1 and biotinylated peptides. B) Coomassie stained gel of GST-fused domains used in Figure 3C. 3xMBT-D355N is a point mutant in the second MBT domain of L3MBTL1 (see text). C) Compound 2 selectively interferes with the binding of L3MBTL13xMBT to H4K20me1. Western analysis of peptide pull down pellets with indicated GST-fusion domain and biotinylated peptides. ImageJ software (NIH) was used for quantitative densitometric analysis of the gel band intensities. The signals for binding assays with compound are reported as a percentage of the H4K20me1 pellet without compound. | ||
Next, the specificity of 2 for the MBT repeats of L3MBTL1 was evaluated. The ability of compound 2 to bind the Tudor domain of 53BP1, another H4K20me1-interaction domain, was evaluated (Fig. 3B and 3C). Due to their structural similarities, both MBT and Tudor domains are members of the royal family of proteins. Binding assays were also conducted with GST alone or with a GST-fused binding-null mutant of L3MBTL13xMBT, L3MBTL1D355N, as negative controls. Neither GST alone nor L3MBTL1D355N bound to the peptides, regardless of peptidemethylation or small molecule concentration. Consistent with the results of Fig. 3A, the addition of 2 inhibits the binding of L3MBTL13xMBT to H4K20me1, whereas treatment with 8 has no effect on binding. The specificity of 2 for L3MBTL1 was further demonstrated, as neither 2 nor 8 had an effect on the binding of 53BP1 to H4K20me1, even at 25 μM. The results of the pull down binding assays support both AlphaScreen® and ITC results, and demonstrate specificity of compound 2 for L3MBTL1 over 53BP1.
Conclusions
Methyl-lysine binding domains make up a large and diverse family of epigenetic regulators which read the post-translational methylation of lysine residues to modulate critical protein-protein interactions involved in gene expression. We report here SAR studies on small molecule antagonists of this reading event which expand our understanding of inhibitor design and the potential for selectivity within a small subset of domains. In addition to activity in a high-throughput AlphaScreen® assay and direct binding studies by ITC, activity of an optimized compound (2, UNC926) in an affinity pull down assay confirms functional antagonism in this format and supports further development of this series toward cellularly active chemical probes.27 Given the emerging validation of some methyl-lysine binding domains as drug targets,28 our efforts to establish the druggability of this target-class will progress with an emphasis on improved potency, documented selectivity versus a more extensive panel of readers, and activity in cellular assays.Acknowledgements
This work is supported by NIH grant number RCIGM090732 to S.F. and the Structural Genomics Consortium, a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. Postdoctoral fellowships for L.A.I. and J.M.H. from the Carolina Partnership are gratefully acknowledged. This work was also supported in part by grants from the NIH to O.G. (R01 GM079641). K.E.C. is supported by a Hubert Shaw and Sandra Lui Stanford Graduate Fellowship. D.J.B. is supported by the Stanford DARE Fellowship. We thank Professor Cheryl Arrowsmith for helpful discussions, Dr Krzysztof Krajewski and Professor Brian Strahl for support with peptide synthesis, and Dr Ashutosh Tripathy for helpful discussions concerning ITC. We also thank the SGC for providing the constructs and/or plasmids for L3MBTL1, L3MBTL3, L3MBTL4, MBTD1, CBX7, and SFMBT1.Notes and references
- K. A. Gelato and W. Fischle, Biol. Chem., 2008, 389, 353–363 CrossRef CAS.
- Y. Chang, X. Zhang, J. R. Horton, A. K. Upadhyay, A. Spannhoff, J. Liu, J. P. Snyder, M. T. Bedford and X. Cheng, Nat. Struct. Mol. Biol., 2009, 16, 312–317 CAS.
- S. Chen, M. Borowiak, J. L. Fox, R. Maehr, K. Osafune, L. Davidow, K. Lam, L. F. Peng, S. L. Schreiber, L. L. Rubin and D. Melton, Nat. Chem. Biol., 2009, 5, 258–265 CrossRef CAS.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067–1073 CrossRef CAS.
- D. Huangfu, R. Maehr, W. Guo, A. Eijkelenboom, M. Snitow, A. E. Chen and D. A. Melton, Nat. Biotechnol., 2008, 26, 795–797 CrossRef CAS.
- E. Nicodeme, K. L. Jeffrey, U. Schaefer, S. Beinke, S. Dewell, C.-w. Chung, R. Chandwani, I. Marazzi, P. Wilson, H. Coste, J. White, J. Kirilovsky, C. M. Rice, J. M. Lora, R. K. Prinjha, K. Lee and A. Tarakhovsky, Nature, 2010, 468, 1119–1123 CrossRef CAS.
- M. Ott and E. Verdin, Chem. Biol., 2010, 17, 417–418 CrossRef CAS.
- N. R. Rose, S. S. Ng, J. Mecinovic, B. M. Lienard, S. H. Bello, Z. Sun, M. A. McDonough, U. Oppermann and C. J. Schofield, J. Med. Chem., 2008, 51, 7053–7056 CrossRef CAS.
- Y. Shi, J. T. Do, C. Desponts, H. S. Hahm, H. R. Scholer and S. Ding, Cell Stem Cell, 2008, 2, 525–528 CrossRef CAS.
- Y. Xu, Y. Shi and S. Ding, Nature, 2008, 453, 338–344 CrossRef CAS.
- M. Vedadi, D. Barsyte-Lovejoy, F. Liu, S. Rival-Gervier, A. Allali-Hassani, V. Labrie, T. J. Wigle, P. A. Dimaggio, G. A. Wasney, A. Siarheyeva, A. Dong, W. Tempel, S. C. Wang, X. Chen, I. Chau, T. J. Mangano, X. P. Huang, C. D. Simpson, S. G. Pattenden, J. L. Norris, D. B. Kireev, A. Tripathy, A. Edwards, B. L. Roth, W. P. Janzen, B. A. Garcia, A. Petronis, J. Ellis, P. J. Brown, S. V. Frye, C. H. Arrowsmith and J. Jin, Nat. Chem. Biol., 2011, 7, 566–574 CrossRef CAS.
- M. A. Adams-Cioaba and J. Min, Biochem. Cell Biol., 2009, 87, 93–105 CrossRef CAS.
- J. Ernst, P. Kheradpour, T. S. Mikkelsen, N. Shoresh, L. D. Ward, C. B. Epstein, X. Zhang, L. Wang, R. Issner, M. Coyne, M. Ku, T. Durham, M. Kellis and B. E. Bernstein, Nature, 2011, 473, 43–49 CrossRef CAS.
- J. M. Herold, T. J. Wigle, J. L. Norris, R. Lam, V. K. Korboukh, C. Gao, L. A. Ingerman, D. B. Kireev, G. Senisterra, M. Vedadi, A. Tripathy, P. J. Brown, C. H. Arrowsmith, J. Jin, W. P. Janzen and S. V. Frye, J. Med. Chem., 2011, 54, 2504–2511 CrossRef CAS.
- D. Kireev, T. J. Wigle, J. Norris-Drouin, J. M. Herold, W. P. Janzen and S. V. Frye, J. Med. Chem., 2010, 53, 7625–7631 CrossRef CAS.
- L. Addou-Klouche, J. Adelaide, P. Finetti, N. Cervera, A. Ferrari, I. Bekhouche, F. Sircoulomb, C. Sotiriou, P. Viens, S. Moulessehoul, F. Bertucci, D. Birnbaum and M. Chaffanet, Mol. Cancer, 2010, 9, 213 CrossRef.
- N. Gurvich, F. Perna, A. Farina, F. Voza, S. Menendez, J. Hurwitz and S. D. Nimer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22552–22557 CrossRef CAS.
- H. Honda, K. Takubo, H. Oda, K. Kosaki, T. Tazaki, N. Yamasaki, K. Miyazaki, K. A. Moore, Z.-i. Honda, T. Suda and I. R. Lemischka, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2468–2473 CrossRef CAS.
- N. Kalakonda, W. Fischle, P. Boccuni, N. Gurvich, R. Hoya-Arias, X. Zhao, Y. Miyata, D. Macgrogan, J. Zhang, J. K. Sims, J. C. Rice and S. D. Nimer, Oncogene, 2008, 27, 4293–4304 CrossRef CAS.
- P. A. Northcott, Y. Nakahara, X. Wu, L. Feuk, D. W. Ellison, S. Croul, S. Mack, P. N. Kongkham, J. Peacock, A. Dubuc, Y. S. Ra, K. Zilberberg, J. McLeod, S. W. Scherer, J. Sunil Rao, C. G. Eberhart, W. Grajkowska, Y. Gillespie, B. Lach, R. Grundy, I. F. Pollack, R. L. Hamilton, T. Van Meter, C. G. Carlotti, F. Boop, D. Bigner, R. J. Gilbertson, J. T. Rutka and M. D. Taylor, Nat. Genet., 2009, 41, 465–472 CrossRef CAS.
- F. Perna, N. Gurvich, R. Hoya-Arias, O. Abdel-Wahab, R. L. Levine, T. Asai, F. Voza, S. Menendez, L. Wang, F. Liu, X. Zhao and S. D. Nimer, Blood, 2010, 116, 2812–2821 CrossRef CAS.
- L. A. Saddic, L. E. West, A. Aslanian, J. R. Yates, S. M. Rubin, O. Gozani and J. Sage, J. Biol. Chem., 2010, 285, 37733–37740 CrossRef CAS.
- L. E. West, S. Roy, K. Lachmi-Weiner, R. Hayashi, X. Shi, E. Appella, T. G. Kutateladze and O. Gozani, J. Biol. Chem., 2010, 285, 37725–37732 CrossRef CAS.
- S. Wu, R. C. Trievel and J. C. Rice, FEBS Lett., 2007, 581, 3289–3296 CrossRef CAS.
- P. Trojer, G. Li, R. J. Sims, 3rd, A. Vaquero, N. Kalakonda, P. Boccuni, D. Lee, H. Erdjument-Bromage, P. Tempst, S. D. Nimer, Y. H. Wang and D. Reinberg, Cell, 2007, 129, 915–928 CrossRef CAS.
- T. J. Wigle, J. M. Herold, G. A. Senisterra, M. Vedadi, D. B. Kireev, C. H. Arrowsmith, S. V. Frye and W. P. Janzen, J. Biomol. Screening, 2009, 15, 62–71 CrossRef.
- S. V. Frye, Nat. Chem. Biol., 2010, 6, 159–161 CrossRef CAS.
- G. G. Wang, J. Song, Z. Wang, H. L. Dormann, F. Casadio, H. Li, J. L. Luo, D. J. Patel and C. D. Allis, Nature, 2009, 459, 847–851 CrossRef CAS.
Footnotes |
| † This article is part of a MedChemComm web theme issue on epigenetics. |
| ‡ Electronic supplementary information (ESI) available: In vitro assay conditions, representative AlphaScreen® and ITC binding curves, experimental procedures including spectra for all final compounds. See DOI: 10.1039/c1md00195g |
| § The discrepancy between the data for compound 1 reported herein and that published previously is the result of a change in the specific histonepeptide used in the L3MBTL3 AlphaScreen® assay. |
| ¶ The reported ITC values are an average of 3 or more repeat experiments. The small difference in potency of 1 for L3MBTL1 reported herein as measured by ITC in comparison to previously published results is due to the fact that the experimental conditions were optimized to improve protein stability. The concentration of NaCl in the experiments was increased to 75 mM from 25 mM previously. |
| This journal is © The Royal Society of Chemistry 2012 |