Synthesis of hybrid 4-anilinoquinoline triazines as potent antimalarial agents, their in silico modeling and bioevaluation as Plasmodium falciparumtransketolase and β-hematin inhibitors†
Moni
Sharma
a,
Kuldeep
Chauhan
a,
Shikha S.
Chauhan
a,
Ashok
Kumar
a,
Shiv Vardan
Singh
b,
Jitendra K.
Saxena
b,
Pooja
Agarwal
c,
Kumkum
Srivastava
c,
S.
Raja Kumar
c,
Sunil K.
Puri
c,
Priyanka
Shah
d,
M. I.
Siddiqi
d and
Prem M. S.
Chauhan
*a
aMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute Chattar Manzil, P.O. Box 173, Mahatma Gandhi Marg, Lucknow 226001, India. E-mail: prem_chauhan_2000@yahoo.com; Fax: +91 522 2623405; Tel: +91 522 2612411premsc58@hotmail.com
bDivision of Biochemistry, CSIR-Central Drug Research Institute, Lucknow 226001, India
cDivision of Parasitology, CSIR-Central Drug Research Institute, Lucknow 226001, India
dMolecular and Structural Biology Division, CSIR-Central Drug Research Institute, Lucknow 226001, India
First published on 3rd November 2011
Abstract
Analogues of a novel class of hybrid 4-anilinoquinoline triazines have been synthesized with the aim of identifying the compounds with improved antimalarial activity preserving the potency of parent drug chloroquine (CQ). All the synthesized molecules were evaluated in vitro for their antimalarial activity against chloroquine-sensitive 3D7 and chloroquine-resistant K1 strains of P. falciparum. Molecules were also screened for their cytotoxicity towards VERO cell line. Sixteen compounds (17, 19, 26, 27, 29, 31, 32, 33, 35, 36, 37, 39, 40, 49, 50, and 52) exhibited excellent antimalarial activity with IC50 values ranging from 1.36–4.63 ng ml−1 and were also found to be nontoxic with good selectivity index. In silico activity prediction as well as enzyme inhibitory activity against P. falciparumtransketolase reveals that the molecules are also good inhibitors of the enzymeP. falciparumtransketolase. The compound 52 showed good in vivo activity by oral route and resulted in survival of 3 out of 5 mice till day 28.
Introduction
Since the discovery (1932) and development (1946) of CQ at the end of world war II,1CQ (1) has been used extensively for the treatment of malaria and become the foundation of malaria therapy.2,3 After chloroquine, an impressive number of compounds (aminoquinoline derivatives, sulphadoxine, pyrimethamine, cycloguanil 2 and others) (Fig. 1) have also been developed for the treatment of malaria.4,5 The global campaign to eradicate malaria, launched in 1955 and phased out by the end of the 1960s, has been dubbed a misguided failure and as a consequence the current malaria situation has become worrisome.6,7 Furthermore, efforts to control the disease are hampered by increased resistance of parasites,8–14 and vectors to drugs and insecticides.15–17 With 243 million cases and 863![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 attributed deaths reported globally,18,19 nearly half of the world population is at risk of contracting malaria.20 The disease is caused by protozoan parasites of the genus Plasmodium. There are four major species of the malaria parasite P. falciparum, P. vivax, P. malariae and P. ovale that are responsible for the spread of the disease in humans. In addition to these four major species, P. knowlesi is a recently discovered species that also infects humans.21 Among these, P. falciparum remains the most problematic.22 Currently, artemisinin-based combination therapies (ACTs) have been adopted globally as the first line of treatment.23 The three artemisinin derivatives (artemether 3, dihydroartemisinine 4 and artemotil 5) (Fig. 1) currently used in ACTs are so closely related chemically that parasites resistant to one will probably be resistant to all.24 Therefore, we desperately need new drugs, and toward that end, new drug targets for the treatment of resistant malaria.
000 attributed deaths reported globally,18,19 nearly half of the world population is at risk of contracting malaria.20 The disease is caused by protozoan parasites of the genus Plasmodium. There are four major species of the malaria parasite P. falciparum, P. vivax, P. malariae and P. ovale that are responsible for the spread of the disease in humans. In addition to these four major species, P. knowlesi is a recently discovered species that also infects humans.21 Among these, P. falciparum remains the most problematic.22 Currently, artemisinin-based combination therapies (ACTs) have been adopted globally as the first line of treatment.23 The three artemisinin derivatives (artemether 3, dihydroartemisinine 4 and artemotil 5) (Fig. 1) currently used in ACTs are so closely related chemically that parasites resistant to one will probably be resistant to all.24 Therefore, we desperately need new drugs, and toward that end, new drug targets for the treatment of resistant malaria.
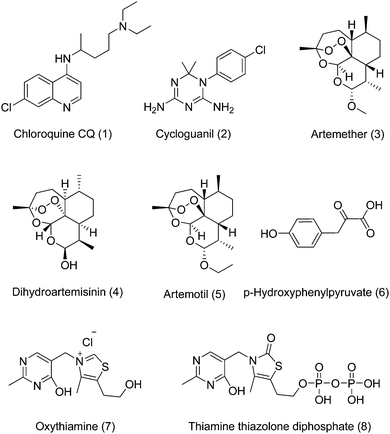 | ||
| Fig. 1 Important antimalarials (1–5) and transketolase inhibitors (6–8). | ||
In 1987 Peters et al. first introduced the bitherapy strategy for the treatment of malaria, but this bitherapy strategy in malaria failed to prevent the emergence of resistance.25 On the above basis, another strategy i.e. hybridization approach has recently been developed.26 Molecular hybridization involves the rational design of new chemical entities by the fusion (usually via a covalent linker) of two drugs, both active compounds and/or pharmacophoric units recognized and derived from known bioactive molecules.27 Furthermore, to circumvent antimalarial drug resistance, hybridization is quite an attractive strategy, particularly when the pharmacophores/active molecules being merged possess independent modes of antimalarial action. Due to its advanced mode of action and high selectivity, hybrid molecule-based chemotherapy appears as a beneficial tool in the current trend of drug discovery.
Among old and new targets of malaria chemotherapy, the host heme molecule attacked by chloroquine is still one of the most attractive targets for drug development.28–30 The biology, mode of action and resistance of chloroquine has been extensively studied in the literature.31–36 Moreover in retrospect, CQ is a wonder drug cheap, safe, easily affordable and most effective and therefore the use of the haloquinoline core of CQ is simply too strong to abandon. On another hand Plasmodium falciparumtransketolase, a vital enzyme of the non-oxidative branch of pentose phosphate pathway has been reported as a novel target in Plasmodium falciparum.37 The selectivity of the target arises due to adequate difference between the human and parasite's transketolase. The enzyme catalyzes the cleavage of a carbon–carbon bond adjacent to a carbonyl group in keto sugars and transfers a ketol moiety to aldosugar.38 Recently the P. falciparumtransketolase has been cloned, expressed and characterized.39 The mechanism of action of transketolase has been mediated by its cofactor thiamine pyrophosphate (ThPP), which is coordinated to divalent metal ions.40,41p-Hydroxyphenyl pyruvate (6), a natural analogue of transketolase substrate is proved to be a reversible and competitive inhibitor of transketolase with respect to substrate.42 Molecules such as oxythiamine (7) and thiamine thiazolone diphosphate (8) also acting on this target have poor activity and low selectivity and therefore clinical applicability of this class of compound is inadequate.43,44
In light of all the above observations, several hybrid molecules (9–12) (Fig. 2) were recently developed by our group as well as by others in malaria chemotherapy.45–55 As a part of our continuing efforts in malaria chemotherapy and to address the urgent need for new antimalarial agents, here we further optimize the derivatives of hybrid 4-anilinoquinoline-triazine (16–52) (Fig. 2, Table 1) as antimalarial agents.
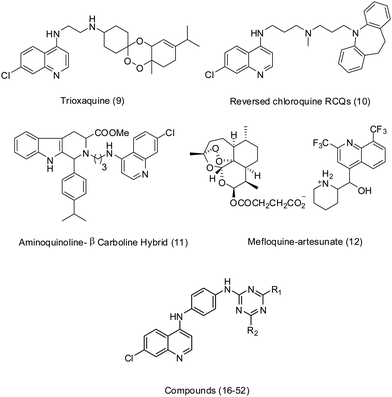 | ||
| Fig. 2 Hybrid antimalarials. | ||
| Compd. No. | R1 | R2 | a IC50 (ng mL−1) | b SI | |
|---|---|---|---|---|---|
| 3D7 | K1 | ||||
| a IC50 (ng mL−1): concentration corresponding to 50% growth inhibition of the parasite. b SI: Selectivity index (IC50 values of cytotoxic activity/IC50 values of antimalarial activity). | |||||
| 16 |

|

|
22.80 | 1.54 | 55.26 |
| 17 |

|
|
2.47 | ND | 149.49 |
| 18 |
|

|
40.02 | 78.07 | 122.19 |
| 19 |

|

|
1.36 | 5.71 | 551.47 |
| 20 |

|

|
128.74 | 106.53 | 75.13 |
| 21 |
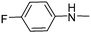
|
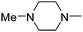
|
8.97 | 2.06 | 121.51 |
| 22 |
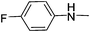
|
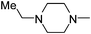
|
11.11 | 19.79 | 88.21 |
| 23 |
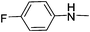
|

|
9.81 | 95.41 | 1325.18 |
| 24 |
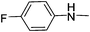
|
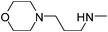
|
15.82 | ND | 682.05 |
| 25 |

|

|
41.75 | >500 | 520.00 |
| 26 |

|
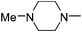
|
2.41 | 16.87 | 360.99 |
| 27 |

|

|
2.11 | 4.96 | 317.53 |
| 28 |

|

|
30.60 | 6.49 | 101.30 |
| 29 |
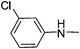
|

|
3.82 | 8.82 | 180.15 |
| 30 |

|

|
7.80 | 35.15 | 66.66 |
| 31 |
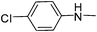
|

|
4.14 | 9.81 | 67.63 |
| 32 |

|
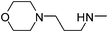
|
4.63 | 8.58 | 481.64 |
| 33 |

|

|
4.26 | ND | 117.37 |
| 34 |

|

|
8.90 | ND | 72.95 |
| 35 |

|

|
2.98 | ND | 2496.64 |
| 36 |

|
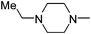
|
4.26 | 3.08 | 75.11 |
| 37 |
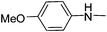
|

|
3.81 | 155.06 | 1307.08 |
| 38 |

|

|
5.04 | 4.86 | 168.65 |
| 39 |
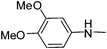
|

|
2.58 | 2.30 | 624.03 |
| 40 |

|

|
4.58 | 2.60 | 412.66 |
| 41 |

|

|
12.64 | 68.23 | 74.36 |
| 42 |
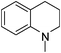
|

|
20.84 | 11.64 | 35.98 |
| 43 |
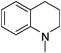
|
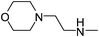
|
17.35 | 274.95 | 588.47 |
| 44 |
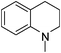
|
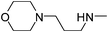
|
14.70 | >500 | 439.54 |
| 45 |

|
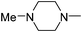
|
28.12 | ND | 48.36 |
| 46 |
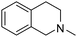
|

|
12.12 | 116.20 | 75.90 |
| 47 |

|
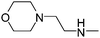
|
45.21 | ND | 1160.00 |
| 48 |
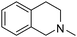
|
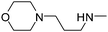
|
20.32 | 55.48 | 435.53 |
| 49 |

|

|
3.22 | 2.86 | 121.18 |
| 50 |
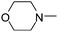
|

|
2.16 | 14.68 | 430.55 |
| 51 |
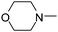
|
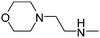
|
5.87 | 53.17 | 41.56 |
| 52 |

|

|
2.76 | 2.66 | 3318.84 |
| CQ (mean ± S.D.) | 2.45 ± 1.08 | 255.86 ± 65.58 | 8983.00 | ||
Results and discussion
Chemistry
Synthesis of the designed inhibitors was achieved in an economical way using inexpensive starting materials. A relatively straightforward synthetic approach (Scheme 1) was followed for the synthesis of target N2-(4-(7-chloroquinolin-4-ylamino)phenyl)-N4-(4-haloaryl)-N6-(aminoalkyl)-1,3,5-triazine-2,4,6-triamines (16–52). Syntheses of mono aryl/alkyl amino substituted analogues of triazines (13a–13j) were carried out by the simple ipso nucleophilic substitution reaction of amines with trichlorotriazine at 0 °C in dry tetrahydrofuran (THF). Reaction of commercially available 4,7-dichloroquinoline with p-phenylenediamine again via ipso nucleophilic substitution reaction led to the synthesis of N1-(7-chloroquinolin-4-yl) benzene-1,4-diamine (14). The so formed N1-(7-chloroquinolin-4-yl) benzene-1,4-diamine (14), further underwent the ipsosubstitution reaction with mono aryl/alkyl amino substituted analogues of triazines (13a–13j) to afford corresponding disubstituted triazines (15a–15j). Finally, compounds (15a–15j) were subjected to nucleophilic substitution reaction with various aliphatic amines to furnish the title compounds 16–52 in excellent yield. During the synthesis of all the intermediates and final hybrid molecules, excellent chemoselectivity was observed which might be due to the nucleophilic potency of the different amines.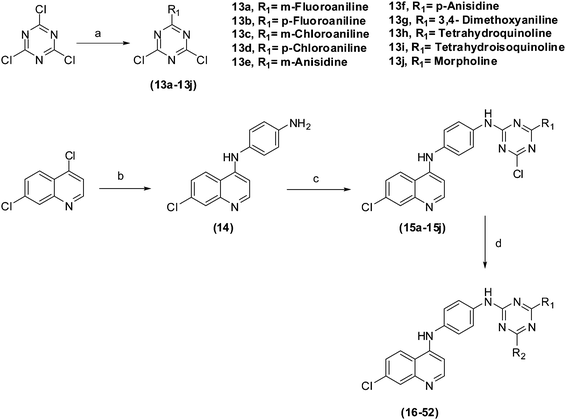 | ||
| Scheme 1 Reagents and conditions: (a) 0 °C, THF, 2 h; (b) p-phenylenediamine, p-TSA, EtOH, 3 h; (c) mono amino aryl/alkyl substituted triazines, THF, reflux, 8 h; (d) various amines, THF, reflux, 5 h. | ||
Biological activity
All the synthesized hybrid molecules (16–52) were evaluated in vitro and in vivo for their antimalarial activity. In vitroantimalarial activity was evaluated against CQ-sensitive 3D7 strain and CQ-resistant K1 strain (MRA-159, MR4, ATCC Manassas Virginia) of P. falciparum using a standardized inexpensive assay based on SYBER Green I.56 The IC50 values were calculated from experiments carried out in triplicate. The cytotoxic evaluation of all the hybrid molecules was determined against VERO cell line using MTT assay.57 The in vivodrug responses of selected compounds were evaluated in swiss mice infected with N-67 strain of P. yoelii which is innately resistant to CQ.58 Moreover, to find out the mode of action of these molecules, effects of all the compounds upon the inhibition of β-hematin formation were also investigated using the β-hematin inhibitory (BHIA) assay.59 The in silico studies of some selected molecules were modeled with the SYBYL 7.1 molecular modeling program (Tripos Associates, Saint Louis, MO)60 using the FlexX module. The eight compounds selected on the basis of maximum FlexX61 score were further biologically screened against Plasmodium falciparumtransketolase enzyme.37 All the results are discussed in their respective sections.Among the 37 hybrid prototypes prepared (16–52) and tested, 16 compounds (17, 19, 26, 27, 29, 31, 32, 33, 35, 36, 37, 39, 40, 49, 50, and 52), exhibited excellent IC50 values ranging from 1.36–4.63 ng mL−1. Five compounds (17, 19, 26, 27, and 50), showed IC50 values in the range of 1.36–2.47 ng mL−1 which were more potent than CQ. Four compounds displayed an IC50 value ranging from 2.58–3.22 ng mL−1 which were comparable to CQ. Other molecules were found to be less active than CQ, among which nine compounds showed IC50 values ranging from 3.81–5.87 ng mL−1, ten compounds exhibited IC50 values ranging from 7.80–17.35 ng mL−1 and eight compounds displayed IC50 in the range of 20.32–45.21 ng mL−1. During the synthesis of the desired molecules, p-phenylene diamine was selected as linker between the 4-aminoquinoline moiety and triazine nucleus. The selection of this linker was optimized by considering the analogy between the side chain of CQ and p-phenylene diamine as demonstrated in Fig. 3.
 | ||
| Fig. 3 Optimization of linker and lipophilicity. | ||
In the previous literature, several side chain modified derivatives of CQ and their significance are also described.62 Furthermore, to enhance the lipophilicity and acquire rigidity in these molecules, the flexible linker of CQ was replaced by a rigid and more lipophilic linker i.e. p-phenylene diamine and as a consequence, the synthesized molecules were found to be more active as compared to the parent drug (CQ).
Further, the activity of these molecules depends upon the substitution pattern around the triazine nucleus and thus great strides were taken to establish the complete SAR in these molecules. During the study of the substitution pattern of aryl/alkyl amines around the triazine nucleus it was observed that the meta substituted arylamines have more impact on antimalarial activity than the para substituted arylamines. The m-fluoroanilino substituted compounds having N-ethyl piperazine (17) and 4-(3-aminopropyl)morpholine (19) unit as substituents and p-fluoroaniline analogues with the same substituents (22, 24) at the triazine nucleus showed IC50 values of 2.47 (17), 1.36 (19) and 11.11 (22), 15.82 (24) respectively. Similarly, m-chloroanilino substituted derivatives with amino substituents such as N-methyl piperazine, N-ethyl piperazine and 4-(3-aminopropyl)morpholine showed superior activity with IC50 values of 2.41 (26), 2.11 (27) and 3.82 ng mL−1 (29) respectively in comparison to p-substituted derivatives with similar substitution having IC50 values of 7.80 (30), 4.14 (31) and 4.63 ng mL−1 (32) respectively. The m-anisidine substituted derivative with 4-(3-aminopropyl)morpholine substitution has an IC50 value of 2.98 ng mL−1 (35) while p-anisidine with the same substitution showed an IC50 value of 3.18 ng mL−1 (37). These results clearly indicate the preference for m-substituted aryl amines over the p-substituted aryl amines. In the case of dimethoxy substituted derivatives (38–40) a combined effect of substituents was observed with IC50 value 5.04, 2.58, 4.58 ng mL−1.
Regardless of the cyclic/acyclic amino substitution at the triazine nucleus, arylamino substituents have more impact on antimalarial activity compared to non aromatic or reduced aromatic systems such as tetrahydroquinoline (41–44) and tetrahydroisoquinoline (45–48). These results indicate that aromatic system is necessary to maintain the favorable interactions that are responsible for antimalarial activity. While in place of aromatic amines, morpholino substituted derivatives (49–52) also showed very good activity with IC50 values of 3.22, 2.16, 5.87, 2.76 ng mL−1. Here the heteroatom might be playing an important role in maintaining the favorable interactions.
The effect of different acyclic and cyclic amines was also established and it was observed that N-methyl piperazine, N-ethyl piperazine and 4-(3-aminopropyl)morpholine are significant for antimalarial activity. The activity results of the hybrid molecules with these amines in combination with various anilines were found to be consistent. Compounds 26, 33, 38 and 49, where N-methyl piperazine substituent is present, have shown promising antimalarial activity ranging from 2.41–5.04 ng mL−1. While in the case of N-ethyl piperazine (17, 27, 31, 36, 39, and 50) the antimalarial activity ranges from 2.11–4.26 ng mL−1. Among 4-(3-aminopropyl) morpholine and 4-(2-aminoethyl) morpholine analogues, the latter have shown dramatically reduced activity however 4-(3-aminopropyl) morpholine substituted compounds (19, 29, 32, 35, 37, 40, and 52) have an excellent activity profile ranging from 1.36–4.63 ng mL−1. In the case of piperidine substituted derivatives the antimalarial activity was very disappointing. The activity of N-methyl piperazine, N-ethyl piperazine and 4-(3-aminopropyl)morpholine is mainly attributed to the basic character of the side chain which is absent in the case of piperidine.
Among the 37 molecules synthesized, 30 molecules were further screened against CQ-resistant (K1) strain of P. falciparum. Several molecules showed very good in vitroantimalarial activity (Table 1). Ten molecules (17, 19, 21, 27, 36, 38, 39, 40, 49, and 52) have shown promising antimalarial activity with IC50 values ranging from 1.54–5.71 ng mL−1. In several compounds the activity against resistant parasite is even better as compared to sensitive strains. In malaria chemotherapy, the mechanism of resistance mainly depends upon the active efflux of the drug (CQ) inside the parasite food vacuole and an increase in the basic character of the molecule leads to influx of that molecule at their active site. Therefore, it could be illustrated that the activity of our synthesized molecules against resistant strain depends upon the basic character of the side chain. Finally the complete SAR study concerning the various substitution patterns around the triazine nucleus suggested that meta as well as parasubstituted anilines with halosubstitution and substitution with electron donating groups in combination with N-methyl piperazine, N-ethyl piperazine and 4-(3-aminopropyl)morpholine are well tolerated for antimalarial activity.
| Compound No. | IC50 (μg mL−1) | Compound No. | IC50 (μg mL−1) | Compound No. | IC50 (μg mL−1) |
|---|---|---|---|---|---|
| a The IC50 represents the concentration of compound that inhibits β-hematin formation by50%. | |||||
| 16 | 3.03 | 29 | 4.56 | 42 | 6.55 |
| 17 | 2.06 | 30 | 2.17 | 43 | 2.31 |
| 18 | 2.70 | 31 | 2.05 | 44 | 2.40 |
| 19 | 2.06 | 32 | 2.25 | 45 | 2.81 |
| 20 | 3.10 | 33 | 2.05 | 46 | 2.25 |
| 21 | 2.08 | 34 | 2.25 | 47 | 2.08 |
| 22 | 2.64 | 35 | 3.49 | 48 | 2.09 |
| 23 | 2.38 | 36 | 2.08 | 49 | 6.50 |
| 24 | 2.17 | 37 | 2.34 | 50 | 4.17 |
| 25 | 2.43 | 38 | 3.05 | 51 | 9.82 |
| 26 | 2.42 | 39 | 2.45 | 52 | 2.98 |
| 27 | 2.06 | 40 | 2.30 | CQ | 3.65 |
| 28 | 2.01 | 41 | 2.48 |
| Compound No. | FlexX Score Energy(Kcal mol−1) | IC50(μM) |
|---|---|---|
| 17 | −31.71 | 75.0 ± 1.88 |
| 19 | −34.29 | 76.7 ± 1.17 |
| 26 | −34.56 | 80.2 ± 0.94 |
| 27 | −31.96 | 74.5 ± 1.17 |
| 29 | −35.60 | 76.4 ± 2.12 |
| 37 | −33.31 | 72.7 ± 1.64 |
| 39 | −30.90 | 85.5 ± 0.42 |
| p -hydroxyphenylpyruvate (control) | −16.0 | 103.0 ± 1.41 |
As obvious from the visual inspection of the bound ligands in the active site of P. falciparumtransketolase shown in Fig. 4a, it was observed that top ranking docked poses of the seven compounds fitted very well into the transketolase active site. To illustrate the probable mechanism of the protein–ligand interaction, docking of compound 19 with P. falciparumtransketolase is described in detail in Fig. 4b. Tight interaction between ligand and protein was observed as evident from several hydrogen bonds between polar atoms of compound 19 and those of active site residues Ala269, Gly30, Asp473 and Arg361.
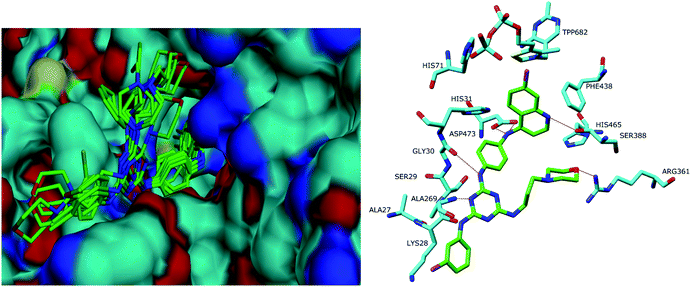 | ||
| Fig. 4 (left) All the docked poses into the binding cleft of P. falciparumtransketolase. (right) Docked pose of 19 into the active site of P. falciparumtransketolase. | ||
The binding site for TPP (a prosthetic group of the enzyme) is characterized by a number of hydrophobic interactions including the π–π stacking interactions with Phe441, Phe444 and the phenyl ring of TPP which are important for ligand binding. Besides, several hydrophobic interactions have also been observed with residues TPP 682, Val129, Ala132, Ile133, Ala135, His136, Ala171, Leu174, Ala175, Leu178, Leu180, Arg182, Ile194, His31, Ile421 and Phe438. Some π–π interactions have also been observed between aromatic rings of 19 with those of residues His31 and Phe438. These interactions further confirm our earlier finding for the necessity of aromatic substitution at triazine nucleus.
| Compound | Dose (mg kg−1 × 4 days) | % suppression on day 4 | Mice alive on day 28 |
|---|---|---|---|
| 52 | 100 mg kg−1 × 4 days | 99.99 | 3/5 |
| 16 | 100 mg kg−1 × 4 days | 99.99 | 0/5 |
| 19 | 100 mg kg−1 × 4 days | 99.99 | 0/5 |
| 21 | 100 mg kg−1 × 4 days | 99.99 | 0/5 |
| 23 | 100 mg kg−1 × 4 days | 99.99 | 0/5 |
| 30 | 100 mg kg−1 × 4 days | 99.99 | 0/5 |
| 42 | 100 mg kg−1 × 4 days | 99.00 | 0/5 |
| 50 | 100 mg kg−1 × 4 days | 99.99 | 0/5 |
| 16 | 50 mg kg−1 × 4 days | 99.82 | 0/5 |
| 42 | 50 mg kg−1 × 4 days | 77.82 | 0/5 |
| 50 | 50 mg kg−1 × 4 days | 93.54 | 0/5 |
| 52 | 50 mg kg−1 × 4 days | 98.39 | 0/5 |
Conclusion
In summary, a series of novel and highly potent hybrid antimalarial agents were designed and synthesized. The synthesized prototypes exhibited promising in vitro antiplasmodial activity where several compounds showed remarkable antimalarial potency in vitro, especially against CQ-R Pf (K1) strain. Inhibition of hemozoin formation was found to be a possible mechanism by which the tested analogues exert their anti plasmodial activity and several analogues were found to be more potent than chloroquine in inhibiting β-hematin formation. The in silico modeling results and the in vitro enzymatic activity against Plasmodium falciparumtransketolase could also contribute to the observed antiplasmodial activities. The introduction of aromatic lipophilic side chain led to a significant improvement in the antimalarial activity as compared to parent drug (CQ). Compound 52 showed a promising antiplasmodial activity in vivo against P. yoelli. Further pharmacological and pharmacokinetic development of these hybrid molecules is currently in progress. As malaria is a disease of developing nations, notably, simple and economically cheap starting materials are used for the development of these novel antimalarials.Acknowledgements
M.S., K.C., and S.S.C thank the Indian Council of Medical Research and the Council of Scientific and Industrial Research, India, for the award of Senior Research Fellowship. We are also thankful to S.A.I.F. Division, CDRI, Lucknow, for providing spectroscopic data. We thank MR4 for providing us with malaria parasites contributed by Dr Dennis Kyle.References
- J. Wiesner, R. Ortmann, H. Jomaa and M. Schlitzer, Angew. Chem., Int. Ed., 2003, 42, 5274 CrossRef CAS.
- R. G. Ridley, Nature, 2002, 415, 686 CrossRef CAS.
- I. M. Hastings, P. G. Bray and S. A. Ward, Science, 2002, 298, 74 CrossRef CAS.
- M. Schlitzer, ChemMedChem, 2007, 2, 944 CrossRef CAS.
- D. A. Fidock, Nature, 2010, 465, 297 CrossRef CAS.
- M. Enserink, Science, 2010, 330, 1736 CrossRef CAS.
- J. D. Sachs, Science, 2002, 298, 122 CrossRef CAS.
- T. E. Wellems, Science, 2002, 298, 124 CrossRef CAS.
- P. A. Winstanley, Parasitol. Today, 2000, 16, 146 CrossRef CAS.
- K. Wengelnik, V. Vidal, M. L. Ancelin, A. M. Cathiard, J. L. Morgat, C. H. Kocken, M. Calas, S. Herrera, A. W. Thomas and H. J. Vial1, Science, 2002, 295, 1311 CrossRef CAS.
- C. Wongsrichanalai and S. R. Meshnick, Emerg. Infect. Dis., 2008, 14, 716 CrossRef.
- A. M. Dondorp, et al. , N. Engl. J. Med., 2009, 361, 455 CrossRef CAS.
- W. A. Guiguemde, et al. , Nature, 2010, 465, 311 CrossRef CAS.
- D. E. Goldberg, Science, 2002, 296, 482 CrossRef CAS.
- World Health Organisation, World Malaria Report (World Health Organisation, Geneva, 2009) Search PubMed.
- P. Müller, M. J. Donnelly and H. Ranson, BMC Genomics, 2007, 8, 36 CrossRef.
- B. G. Knols, T. Bukhari and M. Farenhorst, Future Microbiol., 2010, 5, 339 CrossRef.
- World Health Organization. World malaria report. http://www.who.int/malaria/publications/atoz/9789241563901/en/index.html ( 2009).
- F. J. Gamo, et al. , Nature, 2010, 465, 305 CrossRef CAS.
- W. Fang, J. V. Rodríguez, A. K. Ghosh, M. J. Lorena, A. Kang and R. J. St. Leger, Science, 2011, 331, 1074 CrossRef CAS.
- T. T. Ta, A. Salas, M. Ali-Tammam, C. M. Martínez, M. Lanza, E. Arroyo and J. M. Rubio, Malaria J., 2010, 9, 219 CrossRef.
- P. M. S. Chauhan and S. K. Srivastava, Curr. Med. Chem., 2001, 8, 1535 CAS.
- M. Enserink, Science, 2005, 307, 33 CrossRef CAS.
- M. Enserink, Science, 2010, 328, 844 CrossRef CAS.
- W. Peters and B. L. Robinson, Ann. Trop. Med. Parasitol., 1987, 81, 459 Search PubMed.
- B. Meunier, Acc. Chem. Res., 2008, 41, 69 CrossRef CAS.
- S. S. Chauhan, M. Sharma and P. M. S. Chauhan, Drug News Perspect., 2010, 23, 632 CrossRef CAS.
- D. J. Sullivan Jr., H. Matile, R. G. Ridley and D. E. Goldberg, J. Biol. Chem., 1998, 273, 31103 CrossRef CAS.
- S. Pagola, P. W. Stephens, D. S. Bohle, A. D. Kosar and S. K. Madsen, Nature, 2000, 404, 307 CrossRef CAS.
- H. Ginsburg, S. A. Ward and P. G. Bray, Parasitol. Today, 1999, 15, 357 CrossRef CAS.
- A. B. S. Sidhu, D. V. Pinard and D. A. Fidock, Science, 2002, 298, 210 CrossRef CAS.
- H. S. Banyal and C. D. Fitch, Life Sciences, 1982, 31, 1141 CrossRef CAS.
- H. Ginsburg, O. Famin, J. Zhang and M. Krugliak, Biochemical Pharmacology, 1998, 56, 1305 CrossRef CAS.
- A. F. Slater, Pharmacol. Ther., 1993, 57, 203 CrossRef CAS.
- M. Foley and L. Tilley, Pharmacol. Ther., 1998, 79, 55 CrossRef CAS.
- J. E. Hyde, FEBS J., 2007, 274, 4688 CrossRef CAS.
- S. Joshi, A. R. Singh, U. Saqib, P. C. Misra, M. I. Siddiqi and J. K. Saxena, J. Biophy. Chem., 2010, 1, 96 CrossRef CAS.
- B. Comín-Anduix, J. Boren, S. Martinez, C. Moro, J. J. Ce- ntelles, R. Trebukhina, N. Petushok, W. N. Lee, L. G. Boros and M. Cascante, Eur. J. Biochem., 2001, 268, 4177 CrossRef.
- S. Joshi, A. R. Singh, A. Kumar, P. C. Misra, M. I. Siddiqi and J. K. Saxena, Mol. Biochem. Parasitol., 2008, 160, 32 CrossRef CAS.
- D. S. Shreve, M. P. Holloway, J. C. Haggerty and H. Z. Sable, J. Biol. Chem., 1993, 258, 12405 Search PubMed.
- A. Schellenberger, J. Nutr. Sci. Vitaminol (Tokyo), 1992, 392 CrossRef CAS.
- O. N. Solovjeva and G. A. Kochetov, FEBS Letters, 1999, 462, 246 CrossRef CAS.
- L. G. Boros, J. Puigjaner, M. Cascante, W. N. Lee, J. L. Brandes, S. Bassilian, F. I. Yusuf, R. D. Williams, P. Muscarella, W. S. Melvin and W. J. Schirmer, Cancer Res., 1999, 57, 4242 Search PubMed.
- U. Nilsson, Y. Lindqvist, R. Kluger and G. Schneider, FEBS Lett., 1993, 326, 145 CrossRef CAS.
- O. D. Cabaret, F. B. A. Robert and B. Meunier, Chem. Bio. Chem., 2000, 4, 281 CrossRef.
- L. K. Basco, O. D. Cabaret, M. Ndounga, F. S. Meche, A. Robert and B. Meunier, Antimicrob. Agents Chemother., 2001, 45, 1886 CrossRef CAS.
- I. Opsenica, D. Opsenica, C. A. Lanteri, L. Anova, W. K. Milhous, K. S. Smith and B. A. Solaja, J. Med. Chem., 2008, 51, 6216 CrossRef CAS.
- S. J. Burgess, A. Selzer, J. X. Kelly, M. J. Smilkstein, M. K. Riscoe and D. H. A. Peyton, J. Med. Chem., 2006, 49, 5623 CrossRef CAS.
- J. X. Kelly, M. J. Smilkstein, R. Brun, S. Wittlin and R. A. Cooper, Nature, 2010, 459, 270 CrossRef.
- Kumar, K. Srivastava, S. Raja Kumar, S. K. Puri and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2010, 20, 7059 CrossRef CAS.
- N. Sunduru, K. Srivastava, S. Rajakumar, S. K. Puri, J. K. Saxena and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2009, 19, 2570 CrossRef CAS.
- N. Sunduru, M. Sharma, K. Srivastava, S. Rajakumar, S. K. Puri, J. K. Saxena and P. M. S. Chauhan., Bioorg. Med. Chem., 2009, 17, 6451 CrossRef CAS.
- A. Kumar, K. Srivastava, S. R. Kumar, M. I. Siddiqi, S. K. Puri, J. K. Sexana and P. M. S. Chauhan, Eur. J. Med. Chem., 2011, 46, 676 CrossRef CAS.
- L. Gupta, K. Srivastava, S. Singh, S. K. Puri and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 2008, 18, 3306 CrossRef CAS.
- F.de P. Varotti, A. C. C. Botelho, A. A. Andrade, R. C. de Paula, E. M. S. Fagundes, A. Valverde, L. M. U. Mayer, J. S. Mendonc, M. V. N. de Souza, N. Boechat and A. U. Krettlil, Antimicrob. Agents Chemother., 2008, 52, 3868 CrossRef CAS.
- S. Singh, R. K. Srivastava, M. Srivastava, S. K. Puri and K. Srivastava, Experimental Parasitology., 2011, 127, 318 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS.
- S. K. Puri and N. Singh, Exp. Parasitol., 2000, 94, 8 CrossRef CAS.
- A. V. Pandey, N. Singh, B. L. Tekwani, S. K. Puri and V. S. Chauhan, J. Pharm. Biomed. Anal., 1999, 20, 203 CrossRef CAS.
- SYBYL Ver 7.1, St. Louis, MO, Tripos Inc., 2005.
- M. Rarey, B. Kramer, T. Lengauer and G. Klebe, J. Mol. Biol., 1996, 261, 470 CrossRef CAS.
- D. De, F. M. Krogstad, L. D. Byers and D. J. Krogstad, J. Med. Chem., 1998, 41, 4918 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, protocols of biological assays. See DOI: 10.1039/c1md00188d |
| This journal is © The Royal Society of Chemistry 2012 |