Intrinsic protein disorder in human pathways†‡
Jessica H.
Fong
*,
Benjamin A.
Shoemaker
and
Anna R.
Panchenko
*
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, USA. E-mail: fongj@ncbi.nlm.nih.gov; panch@ncbi.nlm.nih.gov
First published on 20th October 2011
Abstract
We analyze human-specific KEGG pathways trying to understand the functional role of intrinsic disorder in proteins. Pathways provide a comprehensive picture of biological processes and allow better understanding of a protein's function within the specific context of its surroundings. Our study pinpoints a few specific pathways significantly enriched in disorder-containing proteins and identifies the role of these proteins within the framework of pathway relationships. Three major categories of relations are shown to be significantly enriched in disordered proteins: gene expression, protein binding and to a lesser degree, protein phosphorylation. Finally we find that relations involving protein activation and to some extent inhibition are characterized by low disorder content.
Introduction
A lack of well-defined structure is an important feature of many proteins, and approximately a third of eukaryotic proteins contain intrinsically disordered regions (IDRs).1,2 Although intrinsically disordered proteins (IDPs) have certain properties that distinguish them from proteins with well-defined structures, there are many different forms of disorder that are manifested as local flexible loops, flexible linkers and terminal regions, molten globules, or fully unstructured proteins.3–5 Disordered protein regions are characterized by low-complexity sequences and by amino acid compositions rich in hydrophilic and charged residues which inhibit formation of a hydrophobic core.6 Disorder is important to key cellular processes involving nucleic acid and protein binding such as transcription regulation7 and cell signaling,8–11 IDRs are also implicated in pathological conditions including cancer, diabetes, and heart disease,12,13 and can be potential drug targets.14,15Disorder has been implicated in a wide diversity of functional roles and mechanisms. A tremendous amount of attention has focused on the role of disorder in protein binding.4,16–20 Flexible loops and disordered regions can indeed fold upon binding to their interaction partners providing specific recognition elements in low affinity complexes.21,22 At the same time disorder can promote the formation of complexes without disorder-to-order transitions.17,23–26 In the human protein–protein interaction network, interactions between disordered proteins are preferred over interactions with ordered proteins, particularly among non-hub proteins.27 Disordered proteins are often enriched in post-translational modification sites28,29 and may degrade rapidly thereby enabling a rapid response to changes in protein concentration.13 Several programs have been developed to predict disordered regions from amino acid sequences30 with performance of the top method exceeding 83% average sensitivity and specificity, as demonstrated at CASP8.31 The applications of computational predictors to large protein datasets such as PDB,32protein complexes,17protein–protein interaction networks, and genetic interactions5 have presented a glimpse into the diverse role of disorder in biomolecular processes.33
Here, we investigate the extent to which disorder-mediated functions play a role in human biological pathways. Pathways provide a comprehensive picture of biological processes and allow better understanding of protein function within the specific context of its surroundings.34 For example, pathways in KEGG include a collection of manually curated biological pathways based on extensive surveys of the published literature.35KEGG pathways illustrate curated biomolecular relationships that have further been annotated with specific subtypes such as protein binding, activation and gene expression, and place these relations and the participating genes, complexes, and chemical compounds in the context of a particular biological process or disease. While much has been learned about the impact of IDPs on binding, complex formation and post-translational modifications, to our knowledge this is the first attempt to systematically analyze disorder-related functions within the framework of pathways.
Results
Disorder content in different pathways
First, we study the overall propensity for proteins and protein complexes from different pathways to be disordered. KEGG currently contains 148 non-metabolic and 86 metabolic human pathways. Nodes in each pathway denote genes (one or more), complexes (i.e.groups of gene nodes), chemical compounds, and orthologous gene groups. Binary relationships between entries, called “relations”, are labeled by type and subtype, for example, a protein–protein interaction of subtype “activation”. To study protein disorder, we focus on genes (including the “gene” and “complex” entries) and their relations. Each of the 5961 genes in the pathways has been assigned one protein isoform in KEGG. For each protein sequence, disordered regions were predicted using Disopred2.2 We calculate disorder content in a pathway as the average over unique nodes in the pathway, since repeated illustrations of a node in a pathway diagram may not indicate the importance of the node in the pathway. Fewer than 4% of nodes are repeated more than once in the same pathway. To reflect that nodes in KEGG may contain multiple genes or subunits that operate as a unit, we calculate the disorder content of each node as the number of residues predicted to be disordered in all proteins referenced by the node divided by the total length of all the proteins. We also consider disorder in all unique proteins in the pathways.Disorder contents in metabolic and non-metabolic pathways and proteins are presented in Fig. 1. As shown in this figure, proteins in non-metabolic pathways are much more disordered than in metabolic pathways (Wilcoxon rank test p-value ≪ 0.01). Namely, metabolic pathways contain on average 9.7% disorder while non-metabolic pathways contain on average 28.3% disorder in their nodes. According to the previous estimates using the same disorder prediction algorithm, the human proteome contains 21.6% disorder on average.2 Nearly all proteins in metabolic pathways are enzymes, explaining their highly structured nature. Only ∼2% of the proteins lack enzyme classification codes and many of these, on inspection, are in fact cytochrome C oxidase assembly proteins, or biosynthesis proteins. Nevertheless, 8.6% of metabolic proteins contain over 30% disorder (see Fig. 1). Among the most highly disordered enzymes are disease-related proteins (myeloid/lymphoid leukemia protein, Wolf-Hirschhorn syndrome), methyltransferases, polymerases, kinases, and receptor-binding proteins, consistent with our previous study which identified certain unstructured enzymes and their complexes.17 Many more enzymes with low disorder content are also disease associated.36
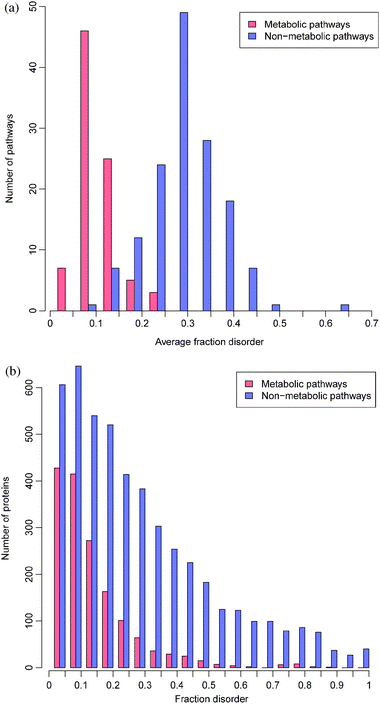 | ||
| Fig. 1 Distributions of disorder content among pathways (a) and proteins (b) for metabolic and non-metabolic pathways. | ||
Next, we checked individual pathways in terms of their disorder content. The pathways with highest disorder content are shown in Fig. 2 and all pathways are presented in Table S1 (ESI‡). Since the most disordered metabolic pathway has less average disorder content than the average non-metabolic pathway, we compared metabolic pathways to one another and non-metabolic pathways to one another to assess statistical significance of disorder enrichment. We identified a number of pathways for which interactions mediated by IDRs are widespread, spanning a spectrum of classifications from the KEGG pathway hierarchy. This list presented in Fig. 2 underscores the association between intrinsic disorder and human diseases such as diabetes, cancer, cardiovascular diseases and lupus. Previously, functional keyword analysis also showed that intrinsic disorder is associated with certain diseases.37 Among metabolic pathways, “Oxidative phosphorylation” and “Glycosaminoglycan biosynthesis” related pathways contain nodes and proteins significantly enriched with disorder (Fig. 2a). As can be seen from Fig. 2b, the non-metabolic pathway containing the most disorder is “Maturity onset diabetes of the young” (hsa04950) which includes on average 60% disordered proteins. Close examination of these proteins revealed that almost all of them are transcription factors with more than 80% disorder. Interestingly, the second most disorder-containing pathway, “SNARE interactions in vesicular transport” (hsa04130), involves SNARE proteins, which assemble into a four-helix, coiled-coil bundle in order to trigger the fusion of synaptic vesicles with the plasma membrane.38,39 Coupled disorder-to-order transition through coiled-coil formation and binding domain activation has been well characterized, in particular for DNA-binding domains such as the canonical GCN4 leucine zipper.12,40 Moreover we found that pathways directly related to gene expression and signal transduction are enriched with disordered proteins as well and the most prominent signal, as will be shown later, comes from transcription factors and proteins involved in transcription regulation (Fig. 2b).
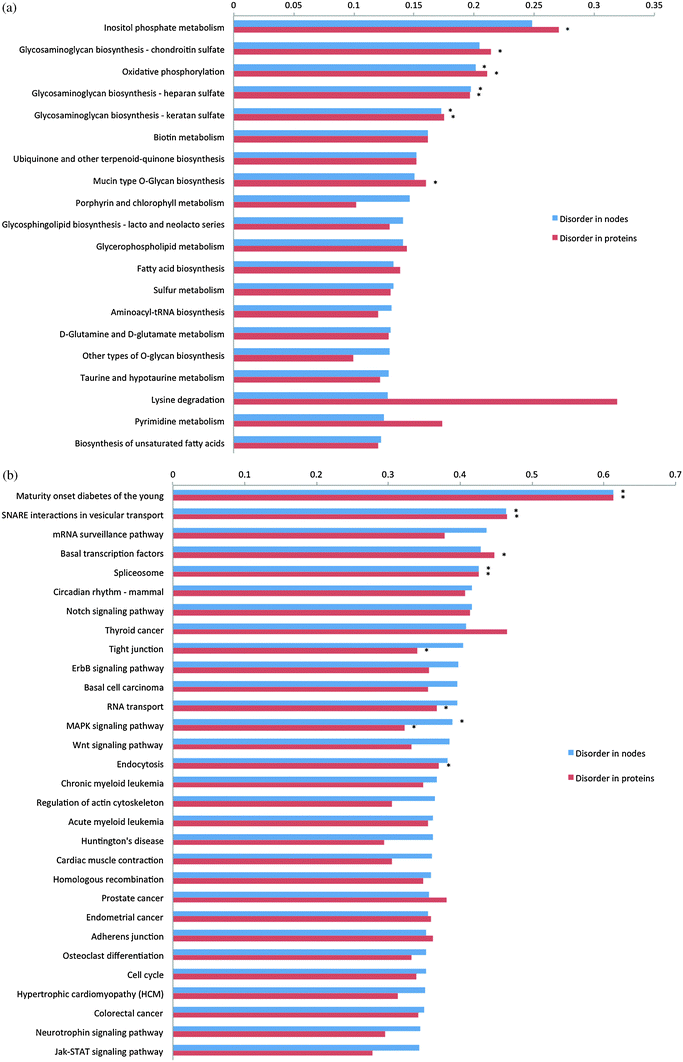 | ||
| Fig. 2 Bar plot showing an average disorder content in nodes and proteins for the top disorder containing metabolic (a) and non-metabolic pathways (b). Those pathways significantly enriched with disorder according to Fisher's exact test with Holm–Bonferroni correction for multiple testing are shown by asterisks. | ||
Fig. 3 shows disorder content in the KEGG pathway “Tight junction” (hsa04530), which includes relatively high disorder (40% in nodes on average) and at the same time is characterized by a variety of different relations (see next section). There are three major complexes, ZO-1 (TJP1, TJP2, TJP3), CRB3, and PARD6A, all of which contain disordered proteins colored in red and orange (disorder content above ∼75% and ∼50%, respectively). For example, a complex of membrane-associated tight junction proteins (TJP1-3) belongs to the class of so-called scaffolding proteins, which provide spatial and temporal coordination between different bound proteins. Scaffolding proteins of this particular pathway organize the transmembrane proteins and mediate coupling between membrane claudins and actin cytoskeleton. Other scaffolding proteins (Singulin, CGN, angiomotin, AMOTL1) and two transcription factors (CSDA and ASH1L) are also highly disordered. It has been suggested previously that disordered regions in scaffolding proteins provide the flexibility necessary for interactions between several bound partners and regulate the accessibility of binding sites depending on which other proteins are already bound to the scaffold.26
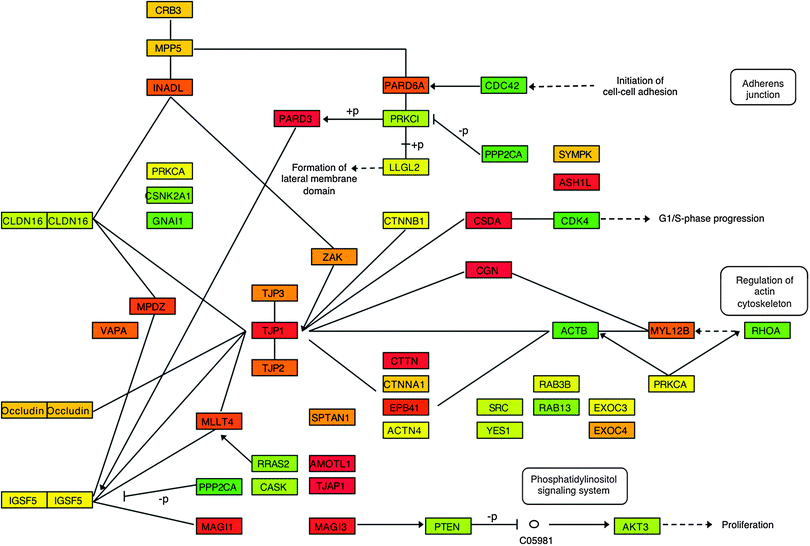 | ||
| Fig. 3 Diagram showing the disorder content of the nodes for the “Tight junction” (hsa04530) pathway, adapted from KEGG. Nodes are colored according to a gradient with 0% disorder colored green, 25% yellow, 50% orange, and 75% or higher red. | ||
Certainly, disorder can play an important role in pathways that do not exhibit overall enrichment in disorder, for example when IDPs represent a few key proteins within the pathway. Previous studies have addressed this case by characterizing disorder in hub proteins.4,16 Additionally, certain KEGG pathways including signaling and disease pathways have been shown to have significant preference for interactions between disordered proteins.27 Pathways may themselves be treated as a type of interaction network although they lack the information about binding interfaces, which has helped to show that date hubs might have greater disorder than non-hub or multi-interface proteins.18,20 We find limited overall correlation between fraction disorder and number of partners. We observe that nodes with a single partner are more structured (Wilcoxon signed rank test p-value < 0.003) and single-partner nodes have median disorder 0.24 compared to 0.31 for nodes with four partners. However, the correlation between disorder and number of partners does not extend above four partners (cor = 0.072). In fact the proteins with more than four partners are more structured than average, consistent with our previous findings.4 The few exceptions with both high degree and high disorder include p53, some kinases and transcription factors.
Disorder in different types of biomolecular interactions
Pathway data provide an invaluable source of manually curated relationships between proteins, nucleic acids, small molecules and other cell components. We utilize these relations and analyze them with respect to the propensity of one or both participating entries to be disordered. Metabolic pathways contain only two types of relations (between enzymes and compounds or between other pathways and compounds), notably including no explicit protein–protein binding relations, and the proteins involved in these have average fraction disorder at most 0.1.For non-metabolic pathways there are different types of relations between proteins and compounds that are described in detail in the KEGG documentation (http://www.genome.jp/kegg/xml/docs/). Table 1 lists relations with significantly greater/less disorder content (column 4). Gene expression, phosphorylation and protein–protein binding/association relations are consistently enriched in disorder. Interestingly, nodes participating in protein phosphorylation are over-represented in disorder yet the proteins for these relations are under-represented in disorder. To explain this effect, we identified a single node “R” in pathway hsa04740 that contains many GPCR-like olfactory receptors (altogether 356 proteins) with limited disorder content of about 5%. Olfactory receptors constitute one of the largest families in the human genome and this node participates in protein phosphorylation, inhibition, and activation, contributing a large fraction of proteins from these relation types. Previous studies indeed showed that flexible regions and intrinsically disordered regions have a tendency to contain phosphorylation sites and phosphorylation might induce disorder-to-order as well as order-to-disorder transitions.7,41 This is consistent with the regulatory role of phosphorylation in disordered regions and the requirement of phosphorylation sites to be accessible for interactions with kinases and phosphatases. We also showed that proteins and nodes participating in “activation” relations are consistently depleted in disorder content (Table 1). These categories include, for example, activation of G-proteins by GPCR (G-protein coupled receptors), adenylyl cyclases (relation type “PPrel, activation”) or activation of receptor proteins by different ligands (“PCrel, activation”).
| Relation | All nodes | “From” and “to” nodes | ||||||
|---|---|---|---|---|---|---|---|---|
| # Nodes | DO | Significance | # A | # B | DO A | DO B | Significance | |
| GErel, repression | 17 | 0.438 | 10 | 7 | 0.460 | 0.405 | ||
| PPrel, phosphorylation | 376 | 0.377 | >N, <P | 116 | 191 | 0.282 | 0.466 | A < B |
| PPrel, expression | 28 | 0.359 | 12 | 15 | 0.511 | 0.209 | A > B | |
| GErel, expression | 255 | 0.350 | >N, >P | 58 | 179 | 0.536 | 0.261 | A > B |
| PPrel, binding_association | 646 | 0.342 | >N, >P | 199 | 258 | 0.330 | 0.314 | |
| PPrel, dissociation | 52 | 0.340 | 22 | 20 | 0.367 | 0.359 | ||
| PPrel, inhibition | 429 | 0.320 | <P | 188 | 178 | 0.310 | 0.323 | |
| PCrel, binding_association | 170 | 0.318 | 43 | 77 | 0.289 | 0.289 | ||
| PPrel, dephosphorylation | 78 | 0.317 | 30 | 47 | 0.303 | 0.318 | ||
| PPrel, missing_interaction | 58 | 0.312 | 28 | 19 | 0.323 | 0.265 | ||
| PPrel, indirect_effect | 283 | 0.302 | 127 | 109 | 0.296 | 0.320 | ||
| PCrel, phosphorylation | 12 | 0.301 | 7 | 5 | 0.275 | 0.336 | ||
| PPrel, ubiquination | 52 | 0.298 | 22 | 30 | 0.280 | 0.312 | ||
| PPrel, activation | 1245 | 0.287 | <P | 428 | 458 | 0.278 | 0.292 | |
| GErel, indirect_effect | 21 | 0.282 | 6 | 15 | 0.216 | 0.309 | ||
| ECrel, activation | 20 | 0.278 | 7 | 8 | 0.242 | 0.275 | ||
| PCrel, indirect_effect | 23 | 0.278 | 3 | 20 | 0.100 | 0.304 | ||
| PCrel, inhibition | 26 | 0.272 | 11 | 15 | 0.206 | 0.320 | ||
| ECrel, compound | 46 | 0.270 | 11 | 15 | 0.234 | 0.312 | ||
| PPrel, compound | 83 | 0.246 | 28 | 41 | 0.246 | 0.239 | ||
| PCrel, activation | 139 | 0.233 | <N, <P | 31 | 81 | 0.208 | 0.262 | |
Some of the relations have directionality from node A to node B, including protein activation, phosphorylation, and gene expression. To investigate differences in disorder between “from” and “to” nodes, we perform the Wilcoxon signed rank test to determine if the disorder content is significantly greater or less in A over B (Table 1). We found two significant trends. First is that the disorder content of the transcription factors and proteins regulating gene expression is significantly higher than their target genes/proteins (p-value ≪ 0.0001). Second, the disorder content is significantly less in kinases than their target proteins. We do not observe any significant differences in disorder for phosphatases and their relations with target proteins. Among the particular nodes linked by these relations, the average difference between disorder in “from” and “to” nodes is −0.16 for protein phosphorylation and 0.21 for gene expression.
Discussion
We analyze human-specific KEGG pathways trying to understand the functional role of intrinsic disorder in proteins within the specific context of their surroundings. Even though disorder has been shown previously to be abundant among proteins with certain functions, we pinpoint a few specific pathways significantly enriched in disorder containing proteins, and identify their role within the framework of pathway relationships. Pathway analysis showed that metabolic pathways contain much less disordered proteins and regions than non-metabolic pathways. Moreover, KEGG pathways provide manually curated annotations of different relations between interacting biomolecules. The analysis of this high-quality data showed three major categories of relations which contain disordered proteins: gene expression, protein binding and, to a lesser degree, protein phosphorylation. Importantly, we show that relations involving activation and to some extent inhibition are characterized by low disorder content. Indeed, protein activity is generally modulated through reversible transitions between different discrete active/inactive conformations or oligomeric states. According to the “conformational selection” hypothesis, all conformations preexist and binding of the ligand selects only the relevant conformations out of the entire ensemble.42 The scaffold that supports such changes should provide a rigid frame and at the same time, relatively short switch regions might be flexible or partially disordered to facilitate transitions between active and inactive conformations. The flexibility of disordered protein regions may offer an advantage for effective search through the large number of conformations and might be advantageous in molecular recognition events. For some reactions, particularly enzymatic reactions, however, conformational selection time realized in disordered proteins can be much longer than protein turnover time.43 In such cases the well-structured environment is required to perform specific function. In contrast to activation/inhibition relations, we found the most disorder content in transcription factors and relations involving their interactions with the target genes. Indeed, it has been previously observed that about half of the sequences of transcription factors are disordered regions and certain DNA/RNA-binding domains are either totally disordered (AT-hooks, basic domains) or contain structured modules connected by flexible linkers.2,44–47 Moreover, transcription factors folding upon binding might occur through an induced fit mechanism which might not require the preexistence of structured conformations before binding.43,48 Therefore disordered and structured protein regions might maintain various biological functions through these mechanisms. These mechanisms may also play a key role in molecular adaptation to different environments; highly unstructured, rapidly evolving viral proteins on one hand and highly structured proteins from thermophilic organisms on the other hand provide such extreme examples.47The influence of disorder on protein function and binding is difficult to overestimate. The same is true for the role of a protein's surroundings and pathways in which it participates. Our study is only the beginning of pathway-oriented analysis of intrinsic disorder, which ultimately might help to understand the mechanisms of biomolecular recognition and regulation through flexible regions and disordered proteins.
Experimental
Exploring pathway data
Human-specific pathways in KEGG were obtained in KGML (Kegg XML) format from the KEGG web site. To study disorder, we will consider proteins indicated by entry (aka node) types “gene” which indicates one or more human genes and “group” which indicates complexes of nodes. The pathways in this study contain 5961 genes. One protein isoform for each gene has been provided by KEGG (ftp://ftp.genome.jp/pub/kegg/genes/organisms/hsa/h.sapiens.pep). “Relations” specify relationships between genes and complexes as well as chemical compounds, and other pathways. Some types of relations are clearly undirected (e.g. “binding/association”, “dissociation”) while others are directed (e.g. “activation”, “phosphorylation”). We annotate the genes in each node with all relations of that node and the complexes of which it is a member. The KGML encoding of human-specific pathways does not provide relations for entries that represent orthologous groups without known homologs in human (entry type “ortholog”). While inclusion of these relations, which may be obtained from the reference pathways, may disputably be valid to increase completeness in annotation, we ascertain that a relatively small number of relations are missing and do not include these relations in our analysis.Disorder prediction
Disorder predictions were made for all proteins in our dataset using Disopred v.2.4.2 We used the default settings, which aim to assign 5% of residues as disordered, with only one change. To speed up the computation of sequence profiles, we executed PSI-BLAST with the uniref 90 database instead of the default non-redundant (nr) database.Acknowledgements
This work was supported by the Intramural Research Program of the NIH, National Library of Medicine.References
- A. K. Dunker, Z. Obradovic, P. Romero, E. C. Garner and C. J. Brown, Genome Inf. Ser., 2000, 11, 161–171 CAS.
- J. J. Ward, J. S. Sodhi, L. J. McGuffin, B. F. Buxton and D. T. Jones, J. Mol. Biol., 2004, 337, 635–645 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS.
- J. H. Fong and A. R. Panchenko, Mol. BioSyst., 2010, 6, 1821–1828 RSC.
- J. Bellay, S. Han, M. Michaut, T. Kim, M. Costanzo, B. J. Andrews, C. Boone, G. D. Bader, C. L. Myers and P. M. Kim, Genome Biol., 2011, 12, R14 CrossRef CAS.
- P. Romero, Z. Obradovic, X. Li, E. C. Garner, C. J. Brown and A. K. Dunker, Proteins, 2001, 42, 38–48 CrossRef CAS.
- J. Gsponer, M. E. Futschik, S. A. Teichmann and M. M. Babu, Science, 2008, 322, 1365–1368 CrossRef CAS.
- H. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, V. N. Uversky and Z. Obradovic, J. Proteome Res., 2007, 6, 1882–1898 CrossRef CAS.
- L. M. Iakoucheva, C. J. Brown, J. D. Lawson, Z. Obradovic and A. K. Dunker, J. Mol. Biol., 2002, 323, 573–584 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Curr. Opin. Struct. Biol., 2002, 12, 54–60 CrossRef CAS.
- Y. Levy, P. G. Wolynes and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 511–516 CrossRef CAS.
- A. K. Dunker, C. J. Oldfield, J. Meng, P. Romero, J. Y. Yang, J. W. Chen, V. Vacic, Z. Obradovic and V. N. Uversky, BMC Genomics, 2008, 9(suppl 2), S1 CrossRef.
- M. M. Babu, R. van der Lee, N. S. de Groot and J. Gsponer, Curr. Opin. Struct. Biol., 2011, 21, 432–440 CrossRef CAS.
- A. Fernandez, S. Bazan and J. Chen, Trends Pharmacol. Sci., 2009, 30, 66–71 CrossRef CAS.
- S. J. Metallo, Curr. Opin. Chem. Biol., 2010, 14, 481–488 CrossRef CAS.
- A. K. Dunker, M. S. Cortese, P. Romero, L. M. Iakoucheva and V. N. Uversky, FEBS J., 2005, 272, 5129–5148 CrossRef CAS.
- J. Fong, B. A. Shoemaker, S. O. Garbuzynskiy, M. Y. Lobanov, O. V. Galzitskaya and A. R. Panchenko, PLoS Comput. Biol., 2009, 5(3), e1000316 Search PubMed.
- P. M. Kim, A. Sboner, Y. Xia and M. Gerstein, Mol. Syst. Biol., 2008, 4, 179 CrossRef.
- P. M. Kim, L. J. Lu, Y. Xia and M. B. Gerstein, Science, 2006, 314, 1938–1941 CrossRef CAS.
- M. Higurashi, T. Ishida and K. Kinoshita, Protein Sci., 2008, 17, 72–78 CrossRef CAS.
- P. E. Wright and H. J. Dyson, J. Mol. Biol., 1999, 293, 321–331 CrossRef CAS.
- K. Sugase, H. J. Dyson and P. E. Wright, Nature, 2007, 447, 1021–1025 CrossRef CAS.
- H. Hegyi, E. Schad and P. Tompa, BMC Struct. Biol., 2007, 7, 65 CrossRef.
- M. Y. Lobanov, B. A. Shoemaker, S. O. Garbuzynskiy, J. H. Fong, A. R. Panchenko and O. V. Galzitskaya, Nucleic Acids Res., 2010, 38, D283–D287 CrossRef CAS.
- B. Meszaros, I. Simon and Z. Dosztanyi, Phys. Biol., 2011, 8, 035003 CrossRef.
- M. S. Cortese, V. N. Uversky and A. K. Dunker, Prog. Biophys. Mol. Biol., 2008, 98, 85–106 CrossRef CAS.
- K. Shimizu and H. Toh, J. Mol. Biol., 2009, 392, 1253–1265 CrossRef CAS.
- L. M. Iakoucheva, P. Radivojac, C. J. Brown, T. R. O'Connor, J. G. Sikes, Z. Obradovic and A. K. Dunker, Nucleic Acids Res., 2004, 32, 1037–1049 CrossRef CAS.
- M. O. Collins, L. Yu, I. Campuzano, S. G. Grant and J. S. Choudhary, Mol. Cell. Proteomics, 2008, 7, 1331–1348 CAS.
- F. Ferron, S. Longhi, B. Canard and D. Karlin, Proteins, 2006, 65, 1–14 CrossRef CAS.
- O. Noivirt-Brik, J. Prilusky and J. L. Sussman, Proteins, 2009, 77(suppl 9), 210–216 CrossRef CAS.
- M. Y. Lobanov, E. I. Furletova, N. S. Bogatyreva, M. A. Roytberg and O. V. Galzitskaya, PLoS Comput. Biol., 2010, 6, e1000958 Search PubMed.
- J. Gsponer and M. M. Babu, Prog. Biophys. Mol. Biol., 2009, 99, 94–103 CrossRef CAS.
- L. Y. Geer, A. Marchler-Bauer, R. C. Geer, L. Han, J. He, S. He, C. Liu, W. Shi and S. H. Bryant, Nucleic Acids Res., 2010, 38, D492–D496 CrossRef CAS.
- M. Kanehisa, S. Goto, M. Furumichi, M. Tanabe and M. Hirakawa, Nucleic Acids Res., 2010, 38, D355–D360 CrossRef CAS.
- U. Midic, C. J. Oldfield, A. K. Dunker, Z. Obradovic and V. N. Uversky, Protein Pept. Lett., 2009, 16, 1533–1547 CrossRef CAS.
- V. N. Uversky, C. J. Oldfield, U. Midic, H. Xie, B. Xue, S. Vucetic, L. M. Iakoucheva, Z. Obradovic and A. K. Dunker, BMC Genomics, 2009, 10(suppl 1), S7 CrossRef.
- A. T. Brunger, Q. Rev. Biophys., 2005, 38, 1–47 CrossRef CAS.
- D. E. Gordon, M. Mirza, D. A. Sahlender, J. Jakovleska and A. A. Peden, EMBO Rep., 2009, 10, 851–856 CrossRef CAS.
- E. K. O'Shea, J. D. Klemm, P. S. Kim and T. Alber, Science, 1991, 254, 539–544 CAS.
- I. Radhakrishnan, G. C. Perez-Alvarado, D. Parker, H. J. Dyson, M. R. Montminy and P. E. Wright, Cell, 1997, 91, 741–752 CrossRef CAS.
- D. D. Boehr and P. E. Wright, Science, 2008, 320, 1429–1430 CrossRef CAS.
- A. G. Turjanski, J. S. Gutkind, R. B. Best and G. Hummer, PLoS Comput. Biol., 2008, 4, e1000060 Search PubMed.
- P. B. Sigler, Nature, 1988, 333, 210–212 CrossRef CAS.
- J. Liu, N. B. Perumal, C. J. Oldfield, E. W. Su, V. N. Uversky and A. K. Dunker, Biochemistry, 2006, 45, 6873–6888 CrossRef CAS.
- Y. Minezaki, K. Homma, A. R. Kinjo and K. Nishikawa, J. Mol. Biol., 2006, 359, 1137–1149 CrossRef CAS.
- I. N. Berezovsky, Phys. Biol., 2011, 8, 035002 CrossRef.
- C. J. Oldfield, J. Meng, J. Y. Yang, M. Q. Yang, V. N. Uversky and A. K. Dunker, BMC Genomics, 2008, 9(suppl 1), S1 CrossRef.
Footnotes |
| † Published as part of a Molecular BioSystems themed issue on intrinsically disordered proteins: Guest Editor M. Madan Babu. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c1mb05274h |
| This journal is © The Royal Society of Chemistry 2012 |