Order and disorder in large multi-site docking proteins of the Gab family—implications for signalling complex formation and inhibitor design strategies†
Philip C.
Simister
and
Stephan M.
Feller
Biological Systems Architecture Group, Department of Oncology, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, OX3 9DS, UK. E-mail: philip.simister@imm.ox.ac.uk; stephan.feller@imm.ox.ac.uk
First published on 20th September 2011
Abstract
Large multi-site docking (LMD) proteins of the Gab, IRS, FRS, DOK and Cas families consist of one or two folded N-terminal domains, followed by a predominantly disordered C-terminal extension. Their primary function is to provide a docking platform for signalling molecules (including PI3K, PLC, Grb2, Crk, RasGAP, SHP2) in intracellular signal transmission from activated cell-surface receptors, to which they become coupled. A detailed analysis of the structural nature and intrinsic disorder propensity of LMD proteins, with Gab proteins as specific examples, is presented. By primary sequence analysis and literature review the varying levels of disorder and hidden order are predicted, revealing properties and a physical architecture that help to explain their biological function and characteristics, common for network hub proteins. The virulence factor, CagA, from Helicobacter pylori is able to mimic Gab function once injected by this human pathogen into stomach epithelial cells. Its predicted differential structure is compared to Gab1 with respect to its functional mimicry. Lastly, we discuss how LMD proteins, in particular Gab1 and Gab2, and their protein partners, such as SH2 and SH3 domain-containing adaptors like Grb2, might qualify for future anti-cancer strategies in developing protein–protein interaction (PPI) inhibitors towards binary interactors consisting of an intrinsically disordered epitope and a structured domain surface.
 Philip C. Simister | Philip obtained his PhD in biochemistry from the University of Bristol, UK in 2004 studying the structural biology of signalling proteins. During a post-doctoral fellowship in France (CNRS, Gif-sur-Yvette) looking at uncharacterised domains of large Arf1 guanine-nucleotide exchange factors he became introduced to intrinsic protein disorder. Switching to molecular virology as a Junior Fellow of the French National Agency for AIDS and Hepatitis Research, he contributed to the molecular understanding of the RNA-dependent RNA polymerase of the hepatitis C virus by solving its structure from an HCV strain unique in its ability to replicate in cell culture without adaptive mutations. The structural insight enabled functional studies to identify a single polymorphism critical for this strain's replication fitness. Currently, Philip is a senior researcher at the University of Oxford, whose interests include understanding, and chemically targeting, the molecular interactions of proteins relevant to cancer, such as adaptors and large multi-site docking proteins. In his spare time recently he translated into English two specialist textbooks (Molecular and Cellular Enzymology; Chemogenomics and Chemical Genetics) from the original French. |
 Stephan M. Feller | Stephan studied biology and then joined the laboratory of renowned cancer virologist Hidesaburo Hanafusa at the Rockefeller University, New York. After some experimental detours, Stephan discovered that the SH3-N domains of Crk adaptor proteins serve as docking modules for signalling proteins like Abl kinase, SoS and C3G. He also proposed a model for conformational regulation of protein binding of c-Crk through phosphorylation by c-Abl. This work led to an offer to start his own junior research group at the University of Würzburg, Germany, where he continued his research into signalling by protein–protein interactions. Since moving in 2001 to the Weatherall Institute of Molecular Medicine in Oxford, Stephan's group investigates signal protein complex formation with biophysical and structural biology tools. He has also developed a strong interest in understanding the molecular heterogeneity of tumours on the signalling protein level. More recently the group has started to explore design principles in the molecular architecture of large signalling complexes believed to function as molecular signal computation machines that are aberrantly activated by oncogenic receptor proteins. |
Introduction
The binding of signalling factors to their cell-surface receptors is the first step in transducing many extracellular signals across the plasma membrane. These control numerous cellular processes in order to bring about a change of functional state within the cell. Once activated, a receptor transmits the signal to intracellular effector proteins, e.g. GTPases, kinases or phosphatases, in a process which is frequently achieved by coupling to large multi-site docking (LMD) proteins. These are believed to serve as signal integration platforms, enabling interaction with multiple protein partners to achieve effective coordination of the signalling responses.LMD proteins lack enzymatic activity and are involved in protein and plasma membrane binding. Well-studied examples include the Grb2-associated binder (Gab), fibroblast growth factor receptor substrate 2 (FRS2), insulin receptor substrate (IRS), downstream of kinase (DOK) proteins and Crk-associated substrate of 130 kDa (p130Cas; also known as breast cancer anti-oestrogen resistance protein 1, BCAR1) families. The one common feature of LMD proteins is their structural composition. They have folded N-termini, consisting of a small domain module i.e. pleckstrin homology, PH (Gab1–4), phosphotyrosine-binding, PTB (FRS-2α and 2β) or Src homology 3, SH3 (p130Cas family) domains. A slight variation to this is seen with IRS-1, -2 and -4, and DOK1–7, which have two adjacent folded N-terminal domains (PH and PTB). Beyond the folded N-terminus, there is a long C-terminal extension ostensibly devoid of major structural elements, and hence LMD proteins represent lesser-studied examples of intrinsically disordered (or unstructured) proteins (IDPs or IUPs). In all cases, the C-terminal tail comprises multiple phosphorylation sites for different tyrosine kinases. These sites serve as docking points for Src homology 2 (SH2) domain-containing adaptor proteins of the Crk (CT10 regulator of kinase), Grb2 (growth factor receptor-bound protein 2) and Nck (non-catalytic region of tyrosine kinase) families, which couple to a range of enzymes, and also for the direct binding of certain SH2-containing enzymes (e.g. the SH2 domain-containing phosphatase 2, SHP2 and phosphatidyl inositol-3 kinase, PI3K).
Comprehensive reviews have been recently published covering the functional aspects of LMD proteins to which the reader is referred.1–5 In this insight review we focus on a detailed understanding of the structural nature of LMDs, in particular mammalian members of the Gab family. The structural architecture of the CagA protein from Helicobacter pylori also comes under scrutiny, as it can hijack the same pathways usually associated with normal Gab protein signalling. The details of these important physical aspects of LMDs have been largely overlooked. Furthermore, the reach of IDP research has not really covered these key proteins, and thus we provide a closer investigation of what can be discerned about their structural properties from analyses of their sequences and the literature.
LMD proteins of the Gab family
Gab family proteins are key mediators of signalling processes during normal growth and development. They are found in the genomes of higher eukaryotes from mammals, fish and flies down to the level of more primitive metazoans, such as the starlet sea anemone, Nematostella vectensis.6 Upon activation, they become tethered to the plasma membrane through interaction of their N-terminal PH domains with the membrane lipid phosphatidyl inositol-3,4,5-trisphosphate (PIP3). PIP3 is converted from phosphatidylinositol-(4,5)-bisphosphate (PIP2) in the phosphorylation reaction catalysed by PI3K.A wide range of receptors has been reported to transmit signals via the Gab proteins, including receptor tyrosine kinases (RTKs), G protein-coupled receptors (GPCRs), cytokine receptors, multi-chain immune recognition receptors and integrins.1 Their normal mode of interaction with the cytoplasmic portion of the receptor is achieved by indirect coupling to adaptor proteins, the most predominant being Grb2, which is a 25 kDa protein composed of a central SH2 domain flanked by terminal SH3 domains (SH3-N and SH3-C). Its SH2 domain can bind directly to phosphorylated tyrosine residues within the receptor. The SH3-C domain interacts with the Gab tail by docking onto proline-rich segments containing an atypical SH3 domain binding motif, RxxK. Interestingly, this interaction may be transient, at least in some cases.7 Gab1 is unique in having an additional means to couple to the hepatocyte growth factor receptor c-Met, an RTK implicated in a plethora of human cancers; it can bind directly with a 13-amino-acid segment present in its unstructured tail region.8
Other adaptors and docking proteins can act as accessory proteins for Gab1 and 2 in concert with Grb2 in coupling to receptors. These include Src homology domain-containing transforming protein 1 (Shc1), linker for activation of T-cells (LAT) and Nck1. Gab3 can bind to the Grb2-related adaptor, Mona/Gads, in cells of haematopoietic lineage.9 Furthermore, Gab1 has been shown to form a tripartite docking complex with Grb2 and the LMD protein FRS2α in fibroblast growth factor receptor (FGFR) signalling.10
Effector proteins involved in signal transduction downstream of Gab proteins
In addition to the above-mentioned adaptor and docking proteins that partner with Gab proteins, these also act as platforms on to which a range of effector enzymes can dock. Multiple sites of tyrosine phosphorylation allow SH2 domain-containing proteins to couple to Gab. These include the SHP2 phosphatase, the p85 subunit of PI3K, phospholipase C gamma (PLCγ), Ras GTPase accelerating protein (RasGAP), and the ubiquitin ligase casitas B-lineage lymphoma proto-oncogene (c-Cbl).1,11,12 Additionally, Gab2 was reported to be phosphorylated on specific serine residues, which provide docking sites for 14-3-3 proteins.13Functions of Gab proteins in normal physiology
In cells, signalling pathways requiring Gab proteins promote various outcomes, including cell proliferation and survival, but also cell scattering/migration and invasiveness. These studies are not the focus of this article, but are discussed in a recent, excellent review1 (and references therein). It is instructive, nevertheless, to describe here the non-redundant roles of Gab proteins during normal development, since the effects of individual gab gene deletions in mouse models have been reported. Gab1 is essential for embryo survival, since gab1 gene knockout mice (Gab1−/−) are not viable and only reach day 14 to 18 of gestation. They display various morphological defects in the heart, skin and muscles of the diaphragm and limbs, as well as smaller livers, open eyelids and problems with placental development.14,15On the other hand, Gab2 and Gab3 knockout mice live to a normal age. Whereas Gab3-deficient mice show no obvious phenotype,16 Gab2-deficient mice, while generally healthy, have distinct impairments. The high affinity immunoglobulin-epsilon receptor, FcεRI, in mast cells requires Gab2 to activate PI3K in the allergic response. Thus, Gab2 deficiency results in impaired allergic reactions such as passive cutaneous and systemic anaphylaxis.17 Also, bone marrow-derived mast cell growth was reported to be reduced in Gab2−/− mice due to defective signalling by the c-Kit receptor.18 Other bone-related deficiencies have been observed in Gab2−/− mice including osteopetrosis (an increase in bone density) and reduced bone resorption. These result from defective osteoclast differentiation involving the RANK (Receptor Activator of Nuclear Factor κ B) receptor, which requires association to Gab2.19 Additionally, normal haematopoesis is perturbed in Gab2 knockout mice as revealed by a poor haematopoietic cell response to early-acting cytokines.20 Somewhat surprisingly, Gab4 has not yet been analysed at all.
Several studies have been carried out with organ-specific knockouts in mice. For instance, liver-specific Gab1 knockouts have shown that Gab1 participates in the regeneration of the liver in conjunction with SHP2.21 Interestingly, Gab1 can also negatively modulate the effects of hepatic insulin signalling amplified by the docking proteins IRS1 and 2, in spite of its structural similarity.22 Both of these physiological effects are elicited by activation of the ERK pathway. Very recently, three independent studies have simultaneously reported the crucial role of Gab1 in promoting postnatal angiogenesis using Gab1 endothelial cell-specific knockout (ecKO) mice and hindlimb ischaemia models.23–25 Both the VEGF23 and c-Met24 receptors were implicated in mediating blood vessel development. The Gab1-ecKO mice were viable with no obvious vascular defects, which indicates that Gab1 in the endothelium plays no crucial role during developmental vasculogenesis. Effects on angiogenesis were not observed for conventional Gab2 knockout mice.24
The particular contributions of distinct protein binding sites, and hence downstream pathway effects, were unraveled for Gab1.26 In this important study, knock-in mutant mice were generated lacking the SHP2, Grb2 and c-Met binding sites. The most severe phenotype, embryonic lethality, occurred with Gab1ΔShp2/ΔShp2 mice, possibly by inactivating Ras signalling. The unusual, direct binding and Grb2 adaptor-linked recruitment mechanisms to the c-Met receptor are non-redundant. While either appears to be usable for normal limb muscle, placenta and liver development, as well as palatal closure, both the Grb2 and c-Met binding modes are jointly required. Thus, the need for c-Met-binding sites in Gab1 is tissue specific.26
The structural architecture of Gab proteins
Secondary structure prediction
Domain architecture analysis of the Gab proteins with Pfam27 or SMART28 describes the known PH domain at the N-terminus, along with reported proline-rich segments, phosphorylation and protein-binding sites i.e., eukaryotic linear motifs (ELMs) (Fig. 1). Apart from these, the rest of the sequence is of unrecognised composition, typical for IDPs. This particular arrangement, i.e. a folded N-terminal domain followed by an extended, disordered tail, is interesting; it turns out not to be unique to Gab proteins and the related LMD proteins mentioned above, but can be found within at least 50 human proteins, many of which are potentially involved in cell signalling.29 This raises the question of whether there is a particular need for the folded region to be at the N-terminus in these proteins, which will be discussed below. The unstructured tail of Gab proteins is enriched in ELMs, which are commonly observed in disordered protein regions.30,31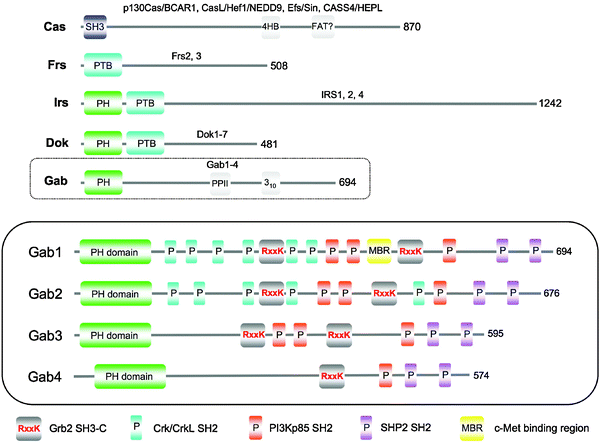 | ||
| Fig. 1 Comparison of the structural architecture of human large multi-site docking (LMD) proteins. Representative members of each family (p130Cas, Frs2, IRS1, Dok1, and Gab1) are illustrated with their corresponding SMART domain structure and previously described secondary structure elements. Protein lengths are also indicated. The human Gab family is expanded in the bottom (enclosed) schematic, with the sites of protein binding and phosphorylation displayed, as described in the key below the panel. | ||
Sequence alignment of all four human Gab proteins illustrates the high sequence conservation in the PH domain (Fig. 2). However, it is clear that beyond the PH domain the Gab members bear less resemblance to one another. Curiously, the extreme C-terminus regains significant sequence homology, even when comparing across all family members. Other regions of higher identity correspond to conserved protein interaction sites, the first Grb2 SH3-C binding motif being notably absent in Gab4, which, disregarding its short length, otherwise resembles Gab2 the most closely.
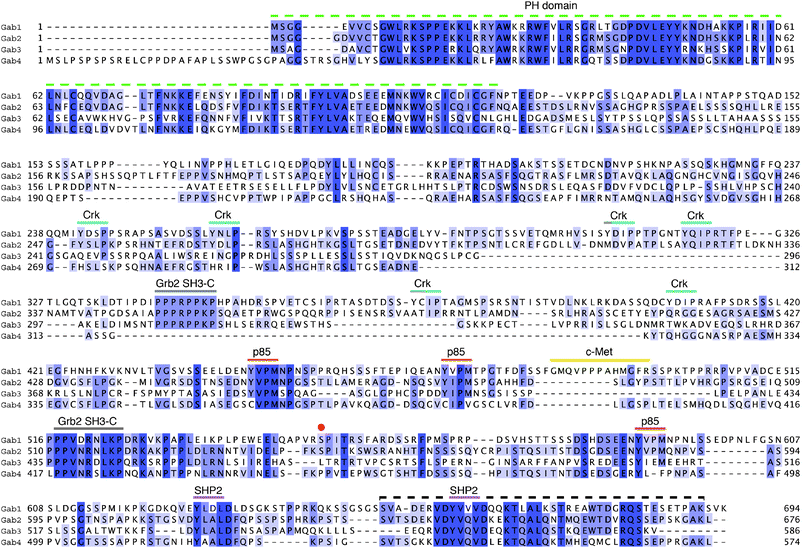 | ||
| Fig. 2 Sequence alignment of human Gab family members. Gab1–4 are shown coloured by percentage identity using JalView.128 The PH domain and protein interaction regions on Gab1 as shown in Fig. 1 are here mapped more precisely using the same colour code. Note: Crk refers to the binding epitopes for the SH2 domains Crk and CrkL proteins; p85 is the subunit from PI3 kinase; the red point indicates the site of serine phosphorylation involved in PH domain autoregulation.44 The dashed black line at the C-terminus indicates a hitherto unrecognised region of homology, which is also predicted to contain secondary structure. | ||
Exploring Gab1's amino-acid sequence further using secondary structure prediction (PSIPRED server32) reveals that secondary structure is correctly predicted for the N-terminal PH domain segment, but the majority of the remaining chain towards the C-terminus lacks predicted structural elements. However, a small region near to the C-terminus is predicted with medium-to-high confidence to contain α and β structure (Fig. 3, top panel). What is more, this region maps to the C-terminal section of closer homology observed in the alignment of Gab protein sequences (Fig. 2, black dashed line). This sequence homology is observed in all human Gab proteins and in lower metazoans, including the frog species Xenopus laevis, the zebrafish Danio rerio, and to a lesser extent in the fruit fly Drosophila melanogaster (not shown). This is interesting, as the total lengths and composition of the Gab sequences are varied, yet they begin and end in a similar structural manner. This stretch of high homology is an unexpected finding, as we know of no known reports defining a structural requirement for the extreme C-terminus in the functional roles of all Gabs elucidated to date. We note, however, that this region harbours the sites of SHP2 phosphatase binding. High conservation of sequence across family members in structured proteins is generally linked to their related function and therefore it is tempting to speculate that, if not fully explained by the presence of SHP2 sites, the C-terminus may serve an additional and as yet unidentified functional role in Gab-family proteins.
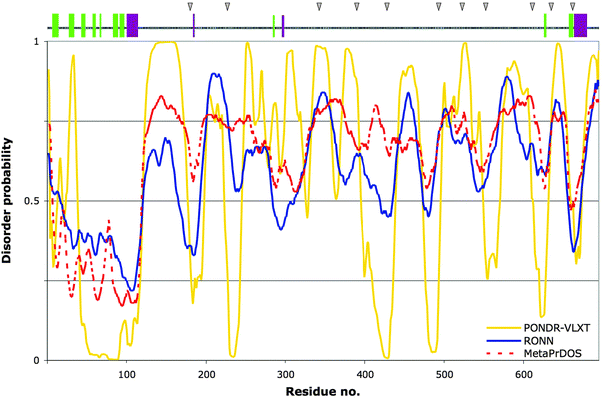 | ||
| Fig. 3 Prediction of structural order and disorder for human Gab1. Bottom plot: Three predictors of disorder were used to analyse the protein sequence of human Gab1 for comparison: PONDR-VLXT (yellow), RONN (blue) and MetaPrDOS (red, dashed line). Values above or below the 0.5 tendency level represent disorder and order, respectively. Top panel: PSIPRED secondary structure prediction for Gab1, represented as coloured bars along the sequence: α-helices in dark magenta, β-strands in light green. The grey triangles above indicate the positions of the Gab1-PH domain-binding sites along Gab1's tail, as determined by peptide array (see Table 1 and ref. 29). | ||
Disorder prediction
Currently there have been over 60 predictors reported for disorder alone.33 We analysed the Gab1 sequence using a selection of programs that are predictors of protein disorder (PONDR VL-XT, RONN and MetaPrDOS) to obtain a more informed view.34 The PONDR VL-XT algorithm35 uses feedforward neural networks incorporating information about disordered and ordered sequences from structural studies (X-ray crystallography, NMR and CD spectroscopy) in the training set. The disordered segments are based on long internal regions and sequences five or more residues in length residing at the termini. We found that the PH domain is correctly predicted to be ordered, whereas the remainder of the protein has a tendency to be mostly disordered with shorter, regularly spaced segments displaying a greater likelihood of order (Fig. 3). This was largely confirmed by the program RONN,36 a regional order neural network-based approach, although some segments having a predicted tendency towards order judged by PONDR-VLXT were less marked in this case. Thirdly, the MetaPrDOS program was compared, which in the version used is an amalgamation of seven distinct predictors (PrDOS, DISOPRED2, DisEMBL, DISPROT [VSL2P], DISpro, IUPred and POODLE-S).37 The meta approach effectively averages the individual scores and such methods have been shown to give an improved prediction accuracy (see also PONDR-FIT,38 and MD39). The order/disorder profile with this method overlays more closely on that of RONN's output, but in general less order was predicted C-terminal to the PH domain. A quite similar profile has been reported for Gab240 using the single predictor, IUPred.41 The differences in the results from various methods are not surprising as they are trained on different datasets, which may include different types of disorder.42 This makes it harder to state with confidence exactly where order might occur for an uncharacterised, generally disordered protein. Nevertheless it would appear that disorder is a general characteristic of the C-terminal extension, possibly punctuated by more ordered segments. These low disorder probability segments are indicative of regions that may undergo disorder-to-order transitions and thus be involved in functional interactions.43Self-association and autoregulation of Gab1
One purpose for the extended, intrinsic disorder within Gab1 was implied from the insight that a site far from the PH domain can autoregulate its membrane binding. This emerged, unexpectedly, from studies with HEK293T cells transfected with a chimeric cytokine receptor protein.44 Those cells have an endogenously high level of PI3K activity. Since the resulting PIP3 is the ligand for the Gab1 PH domain, one might expect that it would reside permanently at the cell membrane. However, only upon stimulation of a chimeric EpoR/IL-6 receptor by Epo, did Gab1, which in unstimulated cells appeared diffusely located in the cytoplasm, become recruited to the plasma membrane. Studies with serial Gab1 truncations and subsequent mutational analysis pinpointed serine 552 (S552) as being a key phosphorylation site within the disordered protein tail. A S552E mutation resulted in constitutive membrane localisation regardless of the activation state of the chimeric receptor. By contrast, a S552A mutant protein remained in the cytoplasm, even after Epo treatment. Thus, Gab1, when not phosphorylated on S552, is prevented from docking on to the membrane with its PH domain. Phosphorylation appears to remove the block and implicates the S552 region in an indirect or direct autoregulatory membrane recruitment mechanism. An indirect effect could, for example, involve regulation by already characterised partner proteins such as SHP2, Grb2 and PI3K. However, deletion in turn of their respective binding sites on Gab1 did not prevent membrane targeting after chimeric receptor stimulation with Epo.44 An alternative plausible mechanism could be the folding-back of the unfolded tail, which either directly or indirectly interacts with the PH domain, thereby occluding the PIP3 binding site. Thus, in a recently published study, we probed a peptide array of overlapping peptides covering the entire mouse Gab1 (mGab1) sequence with purified mGab1 PH domain expressed as a GST fusion (GST–mGab1-PH).29 The GST–mGab1-PH domain (but not GST alone) did indeed bind directly to the region encompassing S552. Furthermore, an interaction was seen with several other distinct regions; the overlapping peptide consensus sequences from the corresponding binding regions are listed in Table 1. While this single technique requires validation by other methods, several associated observations are worthy of mention.| No. | Overlapping binding sequence | Interaction partner | Local structure |
|---|---|---|---|
| a ID = intrinsic disorder. | |||
| 1 | 179DPQDYLLLI187 | — | Low IDa predicted |
| 2 | 219SHQTPASSQSK229 | — | Low ID predicted |
| 3 | 335 DTIPDIPPPRPPK347 | Grb2 SH3-C | PP-II helix (by experiment6) |
| 4 | 383PSRSNTISTVDLNKL397 | — | Low ID predicted |
| 5 | 415SDRSSSLEGFHSQYK429 | — | Low ID predicted |
| 6 | 491 PPPAHMGFR 499 | c-Met | Low ID predicted |
| 7 | 517 PPPVDRNLK 525 | Grb2 SH3-C | 310 helix (by experiment6) |
| 8 | 549PVRSPITRS555 | Gab1 PH | Low ID predicted |
| 9 | 607SNSLDGGSSPMNK619 | — | Low ID predicted |
| 10 | 631LDLDSGKSTPPRK643 | — | Low ID predicted |
| 11 | 655DERVDYVVVDQQK667 | SHP2 SH2 | Low ID predicted |
Firstly, it is interesting that all three YxxM (PI3K p85 subunit SH2 binding motifs) and all six YxxP tyrosine phosphorylation sites (putative Crk/CrkL SH2 binding sites) in Gab1 fall in regions outside of these binding sequences, which may represent specific disordered ‘domains’.45 However, the two SHP2 phosphatase SH2 interaction sites are located within or close to two of these peptide stretches; the significance, if any, of these differences remains to be understood. Another feature of these data is that a further three putative self-binding sequences overlap wholly or nearly completely (the corresponding amino-acid residues are written in bold text, Table 1) with specific known protein-binding regions, i.e. the c-Met binding motif, as well as the two Grb2 SH3-C binding segments encompassing RxxK motifs, in addition to the SHP2 site mentioned.
It is also of note that natural variants reported in Uniprot for the human Gab1 protein (accession code: Q13480; 90% sequence identity to mGab1), assuming they are not sequencing errors, occur in equivalent sequence regions in mGab1 that displayed no binding in our peptide array analysis.29 In contrast, a T387N somatic mutation discovered a few years ago during sequence analysis of 11 breast cancer samples46 lies in the centre of binding sequence 4 (Table 1), and being a non-conservative side-chain alteration could conceivably affect self-binding of the Gab1 tail. Whether these changes are driver mutations or simply passenger mutations, however, is not yet known.
Lastly, all but two of these PH-interacting regions appear to lie in areas of the Gab1 sequence that in the human sequence (shown as grey triangles in Fig. 3, top panel) are predominantly predicted to anticorrelate with disorder according to PONDR-VLXT and to some extent, RONN. Although Table 1 lists the sequences of the mouse Gab1 homologue, the PONDR-VLXT prediction for mGab1 (not shown) yields a profile that is nearly perfectly superimposable on that of the human protein; for instance, 8 of the 11 peptides are 100% identical (three are between 80 and 92% conserved: peptides 2, 5 and 9 in Table 1), thus the argument most likely holds. The two sequences lying significantly outside the more ordered regions are, incidentally, sequences 3 and 7. Ironically, these are the only two sequence regions of a Gab protein for which experimental data exist, having been crystallised in complex with the SH3-C domain from Grb2.6 In the co-complexes reported, these sequences harbouring RxxK motifs are not disordered but adopt distinct structures: a polyproline type II (PP-II) helix and a 310 helix6 (see also a related study47). Recent work indicates that Gab1–Grb2 SH3-C complexes may be transient.7 Therefore, the Gab1 interaction epitopes may well be available for intramolecular binding to the PH domain when Grb2 is absent. If the additional and as yet uncharacterised sites are genuine protein-binding sequences, their being more ordered is consistent with the notion that sequences with a low disorder probability within otherwise disordered protein regions may be involved in disorder-to-order transitions in the context of protein–protein interactions (PPIs).48 This has been shown for RNase E, for instance, where protein-binding segments including one involved in self-association, corresponds to low predicted disorder scores.49
Theoretical prediction methods are useful for predicting α and β structures. However, despite their assumed increased abundance in intrinsically unstructured protein regions,50 PP-II helices have always proved difficult to predict with great accuracy. The same applies for 310 helices. Recent attempts have reported PP-II helix prediction accuracies of about 60% based on information derived from a global analysis of tetrapeptides within the PDB,51 or 70% using a support vector machine (SVM) learning approach.52 However, these methods have not been incorporated into commonly used secondary structure prediction software. It becomes clear that only through focused experimental studies can these important structures be identified, as seen with Gab2, when peptides representing the two Grb2 binding sites bound with distinct helical backbone conformations, discussed above.6 Again, the importance of experiment is underlined here in order to elucidate the exact structural nature of intrinsically disordered regions (IDRs). Quite how the remainder of the polypeptide chain beyond the PH domain arranges itself is therefore open to study by biophysical methods, e.g. small-angle X-ray scattering (SAXS), cryo-electron microscopy and nuclear magnetic resonance (NMR) spectroscopy.
The multiple binding sites in Gab1 discovered by peptide array led to the proposal that the long tail region might actually loop back several times on to the folded PH domain as a matter of course, after ribosomal exit (N-terminal folding nucleation [NFN] hypothesis29). A key consequence of this model is that the protein's overall shape would be more compact, and specific segments of the unfolded polypeptide chain capable of moving into closer spatial proximity to other more remote sequence sites in a defined way. The principal benefit of this spatial form, aside from any advantage in terms of preventing unwanted associations and resisting degradation,53 is with respect to Gab1's critical role as a signal integration platform. Related to this, the locations of the tyrosine phosphorylation sites are not random, they are clustered in specific portions of Gab1's polypeptide chain, suggestive of intrinsic order to their positions. The interaction of specific effector molecules in particular regions of Gab may thus be facilitated by the geometry dictated by the potential anchoring sites along the chain. This could be complemented by the oligomerisation of some Gab interacting proteins, e.g. as reported for the CrkL protein.54
Regulation by distal parts of a protein becomes possible by having a highly flexible, extended linker that can fold back over a long range. A few examples of IDPs participating in such regulatory processes, each with their own unique characteristics, include the following: the cell cycle regulator p27Kip1 when in complex with Cdk2 and cyclin A55 (see also ref. 56). The N-terminus of p27Kip1 is attached to this complex and its C-terminus can fold back onto the complex, in a phosphorylation-dependent manner. Intramolecular autoregulation is observed within other proteins regulating microtubule dynamics, most notably the cytoplasmic linker protein of 170 kDa, CLIP170.57 CLIP170 contains many structured segments, but employs a flexible linker to allow long-range auto-inhibition, in this case through the folding back of a coiled-coil domain by a phosphorylation-independent mechanism.58 The disordered translocation domain of the bacterial antibiotic colicin N, from E. coli, can self-associate to its folded domain, thereby possibly affording it some protection from mammalian host proteases.59
Gab mimicry by a bacterial virulence factor
The case of CagA protein from Helicobacter pylori
An intriguing case that possibly does not fit the self-association model is provided by the CagA protein from the bacterial pathogen Helicobacter pylori. CagA has been described as being capable of functioning as a Gab-like adaptor.60,61H. pylori is a rod-shaped bacterium that infects the stomach epithelial cell layer. Infection takes place in several steps. In the early stages, H. pylori secretes proteases, which digest the cell-cell junctions. Very recently, HtrA (high-temperature requirement A), a presumed dual-function chaperone/protease, was identified as being a key virulence factor fulfilling this role. HtrA is secreted from the periplasm and able to cleave E-cadherin, the principal component of adherens junctions, as its substrate.62 This leads to the breakdown of intracellular junctions, allowing H. pylori to invade the interstitial space and to attach to cell surfaces. Briefly, attachment involves the assembly of H. pylori's type IV secretion system (T4SS). This is encoded by the cytotoxin-associated gene (cag) pathogenicity island, a region of approximately 32 genes, including cagA. The T4SS includes an extended tubular pilus, which protrudes through the bacterium's outer membrane and docks onto transmembrane α5β1 integrin receptors of the host cell.63,64 The CagL, CagY, CagI and CagA proteins are necessary for the interaction with the β1 integrin subunit,63,64 implying that they are situated on the exterior face of the pilus. CagA is additionally injected down the pilus into the cytoplasm of the epithelial cell, whereupon it undergoes phosphorylation on E-P-I-Y-A sequence motifs located towards its C-terminus by Src family kinases65 and c-Abl.66 Subsequently, CagA recruits various SH2 domain-containing proteins,65e.g. SHP2 and Grb2,67 enabling CagA effectively to hijack signalling pathways that are normally managed by Gab proteins. This process brings about rearrangements of the actin cytoskeleton, cell scattering and elongation and hence a migratory behaviour, termed the ‘hummingbird’ phenotype (covered in a recent mini-review68), reminiscent of the cellular response to Gab activation.
CagA has been categorised as a Gab mimic based on its capability to interact with partners of Gab and elicit similar functional effects in human cells.60 A remarkable demonstration of CagA's mimicry was provided by transgenic studies in the fruit fly Drosophila melanogaster containing loss-of-function Dos (Daughter of Sevenless) mutants.61 Dos is a Gab-related docking protein, which signals downstream of different receptors, including the Sevenless receptor (Sev).69 Sev is a tyrosine kinase receptor essential for R7 UV photoreceptor cell development in the compound eye of Drosophila. Dos is coupled to Sev via Drk, the Drosophila homologue of mammalian Grb2 family adaptors. Drk binding to Dos requires two RxxK motifs and deletion of these leads to a loss of all R7 cells.70 Dos inactivation is lethal during development; few pupae are formed and flies do not develop to adulthood.69 When the CagA protein was overexpressed in flies lacking wild-type Dos, more than double the number of pupae were generated. Secondly, in homozygous dos mutants, which produce few cells and photoreceptors, the overexpression of either Dos itself or CagA is able to restore cell growth to similar levels along with the generation of equivalent numbers of eye photoreceptors.61 These experiments neatly revealed how CagA can functionally mimic Dos.
In humans, H. pylori infection is a major risk factor for gastric inflammation and cancers, largely due to this capability of promoting Gab-like signalling, but this is not all that is required. CagA can also associate with the apoptosis-stimulating protein of p53 (ASPP2) in order to subvert its tumour suppressor function. Following this interaction, the cell's balance of the pro-apoptotic transcription factor, p53, becomes perturbed, whereupon it undergoes increased proteasomal degradation in an ASPP2-dependent manner.71
Curiously, CagA lacks an N-terminal PH domain or indeed any other known folded domain, apart from three short, putative coiled-coil domains predicted by SMART. Furthermore, its sequence length is nearly twice that of Gab1 and 2 and thus CagA appears structurally unrelated to the Gab family. Nevertheless, the N-terminal region has been ascribed a role in localising CagA to the membrane.72 In fact, it would seem that CagA has a more complex mechanism of membrane attachment with selective binding to distinct membrane substructures, requiring the interplay of regions within both the N- and C-termini.73 These regions remain to be characterised in more detail.
Predominant structural order predicted for CagA
We subjected a CagA sequence (Uniprot code: P55980; strain 26695 [ATCC 700392]; note: many other H. pylori variants exist) to disorder prediction and secondary structural analysis. Both of these approaches generated consistent results. The three disorder predictors (PONDR-VLXT, RONN and MetaPrDOS) in general all predicted a mixed profile, with rather more residues having a propensity towards order along the entire length of the protein sequence (Fig. 4). As was the case for hGab1, the programs RONN and MetaPrDOS yield per-residue probability profiles that are quite closely matched. Also, analogous to the comparison with the hGab1 sequence, PONDR-VLXT predicts regions of order/disorder that are largely overlapping with these programs, but distinctly more structural order is defined for certain sections of the polypeptide chain that are more ambiguously predicted by RONN/MetaPrDOS (e.g. residues 800–900).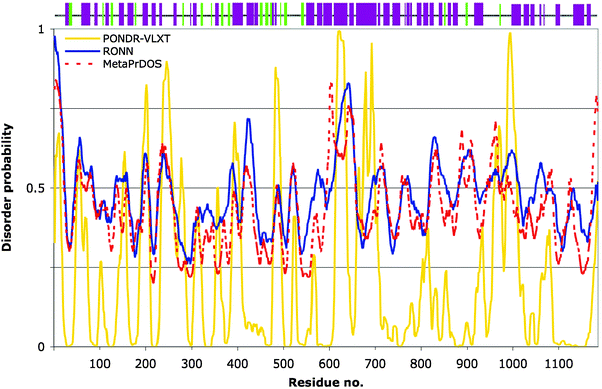 | ||
| Fig. 4 Prediction of structural order and disorder for Helicobacter pylori CagA protein. Bottom plot: Three predictors of disorder were used to analyse the protein sequence of CagA for comparison: PONDR-VLXT (yellow), RONN (blue) and MetaPrDOS (red, dashed line). Values above or below the 0.5 tendency level represent disorder and order, respectively. Top panel: The PSIPRED secondary structure prediction for CagA, represented as coloured bars along the sequence: α-helices in dark magenta, β-strands in light green. | ||
The secondary structural prediction for CagA by the PSIPRED server32 is represented in Fig. 4 (top panel). It is evident from this output that CagA has a predicted, high tendency to form mainly α-helices throughout the sequence with some β-strands in the N-terminal half, albeit with variable confidence levels (not shown). This is in stark contrast to the Gab proteins. The widespread distribution of secondary structural elements corroborates the mixed order/disorder prediction. Importantly, these combined analyses strongly support the notion that CagA is structurally unrelated to the Gab proteins. Therefore, it is remarkable that H. pylori has found a means to mimic the cellular response promoted by Gab signalling, using a protein of dissimilar architecture. Clearly, there are other constraints on H. pylori, such as (i) the need to inject this virulence factor across the host cell membrane, a process that may require specific structural handles alongside a degree of plasticity, and (ii) to interact with the pilus,64,74 that may dictate why CagA adopts its particular form. An additional difference with respect to CagA's structure, in contrast to eukaryotic Gab proteins, is that CagA can undergo multimerisation once injected, which is necessary for its ability to cause the hummingbird phenotype.75 The functional mimicry would therefore seem to reside in the presence of the correct short linear motifs, i.e. c-Src phosphorylation sites, SHP2 and Grb2 docking sites etc. This is supported by the fact that in transgenic fruit flies, CagA mediates its effects by way of the Corkscrew (CSW) protein, the equivalent of SHP2 phosphatase in Drosophila.76 In csw-null flies, CagA failed to increase the number of R7 photoreceptors.61 It is also conceivable that the structural context of these motifs is important and CagA may recreate a similar local geometry to the Gab proteins at sites of protein–protein interactions. It would be interesting to investigate experimentally whether predicted helical regions in CagA do indeed form defined tertiary structure. To date, only a short fragment has been structurally analysed: a 14-residue CagA peptide possessing no regular secondary structure was crystallised in complex with the human kinase PAR1b/MARK2.77 This was unexpected since the reported co-crystallisation experiment was set up with a 120-residue portion of CagA (amino acids 885 to 1005) encompassing the E-P-I-Y-A motifs, but only the 14 amino acids showed visible electron density, implying flexible disorder for the remaining 104 residues.77 This is quite consistent with the output from our disorder analysis: all three predictors used indicate that disorder predominates in this region (Fig. 4). Furthermore, secondary structure is predicted to be the most sparse in part of this region (Fig. 4, top panel).
Gab proteins in disease processes and cellular networks
Gab proteins in human cancers
Gab proteins have been implicated in several cancers of blood and tissues. The chromosomal 11q13 locus comprising the Gab2 gene is commonly amplified in breast, ovarian, head and neck, and oesophageal cancer.78 Gab2 is overexpressed in oestrogen receptor (ER)-positive cells79 and a subset of breast cancers is driven by Gab2 overexpression coupled to Neu/ErbB2/HER receptor signalling.80 Increased Gab2 expression is seen in gastric carcinomas, although it appeared not to correlate with late-stage disease progression.81 Gab2 overexpression has also been shown to potentiate metastatic melanoma,82 and very recently was observed in malignant lung cancer tissue.83 Furthermore, Gab2 is instrumental in sustaining the oncogenic properties of the Bcr-Abl fusion protein in chronic myelogenous leukaemia (CML).84 Gab2 also features in a range of other haematological neoplasias, such as juvenile myelomonocytic leukaemia (JMML),85 acute leukaemia (AML)86 and acute lymphoblastic leukaemia (ALL) amongst others (see Wöhrle et al., 20091 and references therein for a fuller treatment).Gab1's role in tumourigenesis has been implied through its essential connection with c-Met receptor signalling,87 which is activated (mutated or overexpressed) in a vast array of cancers.88 Other receptor-driven tumourigenesis pathways implicate Gab1, for example, in EGFR signalling within intestinal adenomas,89 and glioblastoma cells,90 as well as in hyaluronan-mediated CD44 signalling in metastatic breast tumours.91
Therefore, the direct link between Gab1 and 2 in numerous oncogenic mechanisms raises the possibility that they may be useful target areas for the development of novel therapeutics.
Gab proteins as network hubs
Hub proteins occupy central and critical points in biological networks and while their definition is inexact, several characteristics of hubs frequently present themselves,92–95 including: (i) they interact with multiple protein partners (different benchmarks are used: either a minimum of 5, or above 10 partners) (ii) they are often more disordered than non-hub proteins; (iii) they may be more evolutionarily conserved than non-hub proteins; (iv) they have lower connectivity to other hubs; (v) removal of a hub can be detrimental to the network, as opposed to removal of a non-hub protein; (vi) they tend to correspond to essential genes. Hub proteins have been proposed to fall into one of two types, date or party hubs, depending on their coexpression profile with respect to partner proteins.95 However, this dichotomy is disputed.96While not nearly as promiscuous in their interactions as other well-characterised hubs, such as the DNA binding proteins and tumour suppressors BRCA1 and p53, the list of Gab-interacting partners as well as the structural and functional features of Gab proteins leads to the notion that these may indeed be network hubs. As described earlier, a gab1 gene deletion gives a lethal phenotype in mice. However, deletion of gab2 and 3 produces viable mice. This may reflect their more restricted expression profile during development, since Gab2 can interact with the same effector proteins as Gab1, thus its network promiscuity is essentially equivalent.
Hub proteins are a very frequent point of dysregulation contributing to human pathologies. Their pivotal role in coordinating various inputs and outputs requires fine control and thus their integrity within the cell has multiple adverse repercussions when compromised. Given the central role docking proteins such as Gab1 and 2 play in mediating cellular signalling responses after activation of growth factor receptors, it is perhaps surprising that they are not more frequently mutated in human cancers. This seems to be contrary to the typical scenario with BRCA1 and p53, which are found to be severely mutated in many cancers. One possible explanation could be that it is practically impossible to mimic tyrosine phosphorylation by mutations in disordered tails of proteins in order to generate oncogenic signals, while it is much easier to disrupt many anti-oncogenic signals that are based on protein–protein binding events, or to activate enzymatic proteins by mutation.
In line with this, mutations in Gab-associated proteins are more frequently reported. Some examples include the D61G gain-of-function mutation in SHP2, which is linked to JMML.97,98 Deletion of Gab2 in D61G mutant mice alleviates the aberrant activity, indicating once again the important role it plays in propagating signals via the SHP2 phosphatase. Presumably, the mere presence of sufficient expressed Gab2 in this context is required to sustain the cancerous phenotype. PI3K is mutated in most breast cancer subtypes,98,99 many of which harness Gab signalling functions. PLCγ can also adopt gain-of-function mutations relevant to cancer.100
In any case, it is possible that additional disease-associated mutations in Gab proteins await discovery. To be effective in driving disease processes, we hypothesise that these mutations might: (i) enhance their interaction with the membrane and/or receptors, perhaps residing in the PH domain, or by mimicking Gab1 Ser552 phosphorylation; (ii) decrease affinity for negative regulators, e.g. 14-3-3 in the case of Gab2, or certain tyrosine phosphatases, or (iii) promote structural organisation favourable to signalling-complex assembly e.g. by modifying the potential anchor points listed in Table 1.
LMD and adaptor proteins as therapeutic targets
Intrinsic disorder and inhibition of protein–protein interactions, from a Gab2 perspective
IDP and PPI targets represent a largely untouched and potentially significant area for the development of novel therapeutic approaches.101,102 Gab2, for instance, is an attractive conceptual target for new drug intervention strategies in anticancer treatment. It performs its duty at the head of signalling networks, presumably computing inputs from cell-surface receptors to engender and orchestrate multiple biological actions. As such, uncoupling Gab2 from the receptor-driven signal flow may translate into the equivalent of having a cocktail of drugs that ‘hit’ several enzymes, as their docking and activation might be expected to be prevented. Secondly, these LMD proteins are heavily relied upon during embryonic and postnatal development, but their expression or functions in normal cells of mature individuals are apparently often suppressed. Therefore, there may be fewer side-effects associated with such drugs (see also the related discussion for FRS2103). Moreover, inhibitors of docking proteins could provide a useful complement in combination therapy with other drugs101 either in a ‘synthetic lethality’ scenario, or to elicit an additive effect onto the same pathway potentially providing a way of reducing or overcoming drug resistance. One suspected drawback lies in the limits of current feasibility.Is it possible to develop inhibitors that disrupt Gab protein–protein interactions in which one or more intrinsically disordered sequences participate? This is a general challenge confronted by every research endeavour into PPI inhibitors, confounded by the potentially variable nature of IDRs, yet emerging strategies aim to overcome this.102 It is known that PPI regions often have some preorganised structure and may transition to an ordered state upon binding104,105 although IDPs do exhibit variability in this regard.106 This is necessarily the case when the partner protein is a structured domain. In fact, 4 of 8 key PPI inhibitors reported to date target complexes comprising one disordered and a second ordered domain.107 The known docking proteins that couple to Gab proteins do so mostly by way of their globular domains (i.e. SH2 and SH3) and thus should present structured and quite conformationally invariant surfaces. The question may therefore be: can specificity be built into small-molecule inhibitors interfering with SH2 and SH3 interactions, as around 120 SH2 and 300 SH3 domains are encoded in the human genome and many of these contribute to regulating several signalling pathways? Interrupting Gab function does not need to be limited to the SH2 and SH3 domain interactions; however, without a precise view of the structural elements at play in other interactions, it may be challenging to design such inhibitors rationally.
The role of disorder in understanding target specificity
Characterising the location and types of disorder present in IDPs would permit more informed targeting of their interactions by small molecules. The disorder profile may therefore play a crucial part in understanding specificity issues. One illustrative example is presented by Arf (ADP-ribosylation factor) proteins, the small GTPases that are activated by their interaction with guanine-nucleotide exchange factors (GEFs). The naturally occurring fungal metabolite, brefeldin A (BFA), elicits its toxic properties by acting as an interfacial inhibitor of the ArfGDP/ArfGEF complex. The GTPase activity is uncompetitively inhibited upon BFA binding, locking the complex together in an abortive state.108,109A homologue of Arf1 is Arf6, sharing about 70% sequence identity. Crystal structures of both of these GTPases show high similarity.110,111 The binding sites for BFA are superimposable, with all residues contacting BFA identical in both structures, and yet BFA has no activity towards the Arf6/ArfGEF complex. It was therefore a puzzle for many years how BFA could elicit such a differential response to these Arf proteins. Although the picture is further complicated by differences in ArfGEF specificity between Arf1 and 6 it is now clear that the crystallographic state of the complex represents only one conformational possibility of the otherwise well-folded Arf protein. Further studies in solution using SAXS and 2D NMR on truncated Arf variants have revealed that the switch 1/interswitch region can transition between a folded and unfolded state, the latter never being captured in the crystallised form of the full-length protein, although observed in the truncated form.112 Thus, the local disorder in Arf6 most likely influences both its lack of BFA sensitivity and its ArfGEF specificity. Incidentally, the switch 1 region that can unfold is not predicted to be disordered (our finding, data not shown), underlining the importance of experimentation to determine such features, on an individual protein basis.
This leads to the possibility that, despite having such a large portfolio, even SH3 proteins may be divisible into subsets with differing solution states, which may contribute to specificity upon interaction with their protein partners. For instance, the solution dynamics of the SH3-N domain from the Drosophila adaptor protein Drk has been studied in detail by NMR, and it exists in roughly a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equilibrium between the folded and the unfolded states in vitro.113,114 Systematic solution studies would be necessary to determine the complete dynamical profile of SH3 domains, or other targets of interest. Alternatively, differences in the folding–unfolding characteristics of target proteins may be exploited in strategies to accommodate specificity.
:1 equilibrium between the folded and the unfolded states in vitro.113,114 Systematic solution studies would be necessary to determine the complete dynamical profile of SH3 domains, or other targets of interest. Alternatively, differences in the folding–unfolding characteristics of target proteins may be exploited in strategies to accommodate specificity.
PPI inhibition targeting SH3 domains
In general, SH3 domains have not proved to be easy targets for the development of high-affinity, specific compounds. However, the potential for specificity would seem more likely for the Grb2 SH3-C domain as it binds with a distinctive atypical charged interaction on Gab proteins. We note, though, that the proportion of SH3s receptive to the RxxK motif has not yet been determined. The two Grb2 binding sites on Gab are both relatively proline-rich regions, but the RxxK motif embedded in this context predominantly contributes towards their affinity: substitution by alanine of either the arginine or lysine abolishes binding, respectively, totally or nearly completely.6 An added problem is the issue of attaining potency sufficiently higher than the wild-type interaction and high enough to constitute a viable drug.115 Typically, SH3 domains interact with dissociation constants (Kd) of around 1–100 μM, although there are reports of sub-micromolar affinities e.g. for the amphiphysin SH3 binding to dynamin-1 determined by surface plasmon resonance (Kd = 10 nM),116 and the Grb2-related adaptor Mona/Gads SH3-C binding to SLP-76 determined by isothermal titration calorimetry, (Kd = 181 nM)117 or fluorescence polarisation (Kd = 240 nM).118 Mona/Gads also displayed a binding affinity of 170 nM to an interaction motif from the phosphatase-like protein, HD-PTP (PTPN23).6 These comparatively high affinities suggest that, unless they arise due to unique properties of these particular SH3 domains, there may be an opportunity with other SH3 domains to mimic or improve upon the native ligand's affinity by a chemical molecule. For Gab2, the interaction with Grb2 SH3-C has binding affinities that are low micromolar: ∼3 μM for the 310 helix site and ∼30 μM for the PP-II helix site.6 It is of course not predictable whether high potency could be achieved for this particular protein surface within the constraints of the chemical space corresponding to drug-like physico-chemical properties. Having said this, to our knowledge, early attempts to define small-molecule inhibitors were limited to SH3 domains binding to the originally identified PxxP motifs, and thus the RxxK-binding SH3 subset remains to be tested. Furthermore, the chemical libraries then available likely did not comprise representative molecules from the chemical subspace appropriate for these surfaces. It is possible that new approaches to construct chemical libraries more suited to PPI surfaces (as proposed in ref. 119) will yield greater success in the long run.One notable attempt to target more suitable SH3 ligands—a library of small aliphatic and aromatic hybrid peptide-peptoids—demonstrated that affinities as high as 40 nM are possible, in this case using Grb2 SH3-N.120 It would appear, therefore, that an exhaustive search space has not been covered and it is conceivable that a fragment-based screening approach could prove to be the most effective means to develop drug-like molecules against PPIs such as SH3s. Briefly, there has been some success in small-molecule inhibition of the SH3 domain from cortactin binding to AMAP1, an effector of Arf6-mediated invasion in breast cancer cells.121 However, the molecule UCS15A, a product of the bacterium Streptomyces, blocks the PPI probably not by adhering to the SH3 but by directly binding to the proline-rich sequence in the protein partner.122
Alternatively, there may be value in exploring peptido-mimetics for PPIs involving helical structures, as found in Gab1 and 2, since these can be readily made to match the geometries of polypeptide chains. Scaffolds based on terphenyl or teraryl groups are showing promise for other targets.123 It may also be possible to employ this strategy for non-helical or non-beta structures, indeed for less regular polypeptide conformations even, so long as detailed structural information is available, i.e. a co-complex of the two proteins involved in the PPI. Even in peptide–protein complexes, the conformation of the peptide is most likely to reflect the actual conformational state found in the folded protein.124 There is evidence to suggest that regions of intrinsically unfolded proteins that participate in protein–protein interactions begin with the residual structure adopted upon binding, or at least transiently form this structure dynamically when uncomplexed49,125 (an illustrative example is provided by the transient helix appearance prior to formation of the MYPT1–PP1 complex126). Thus, the structure of a peptide–protein co-complex should be usable as a reliable starting guide for mimetic design, expanding the chemical possibilities for targeting interactions involving a disordered protein binding to a structured surface. This has been demonstrated, for instance, by current Smac (second mitochondrial inhibitor of caspases) mimetics, which bind with nanomolar affinity to multiple anti-apoptotic XIAP (X-linked inhibitor of apopotosis) proteins. The apoptotic response elicited is equivalent to that induced by Smac itself both in vitro and in animal models, and hence the compounds are presently in phase I clinical trials.127
Conclusions
LMD proteins play crucial roles in the generation of signalling network hubs and presumably in computing several well-coordinated signalling outputs from multiple incoming signals, but they have remained largely unstudied on the level of protein structure. The dual literature and protein sequence analyses presented in this review, centered around LMD proteins of the Gab family and the Gab-mimicking virulence factor CagA from Helicobacter pylori, have revealed the varied predicted levels of disorder and hidden order within these structurally uncharacterised proteins. The combined approach of secondary structural analysis and disorder prediction, alongside correlation of published reports and data-mining from curated databases (SMART, Pfam), has allowed a fairly consistent view to emerge of ordered and disordered regions of interest in these proteins. These insights not only enhance the understanding of these proteins' functions, but also guide the way for the design of appropriate experiments to validate the predictions. The value of prediction tools needs no emphasis, yet predicting structural elements like PP-II and 310 helices—vital for mediating many protein–protein interactions (e.g. Grb2 SH3-C binding sites in Gab1–4)—still remains a computational challenge and hence they are notably lacking in secondary structural prediction programs.It is evident that while intrinsic disorder seems to be fully compatible with Gab functions in eukaryotes, the strong prediction of structure for the functional Gab-mimic CagA indicates that disorder is not a strict requirement for at least some Gab functions. There are significant differences in the functional repertoires of these human and bacterial proteins. For instance, intrinsic disorder may benefit Gab in mediating multiple interactions and signalling responses, whereas for CagA promiscuous binding is not necessarily the goal as it presumably performs only a subset of the roles accomplished by the endogenous Gab proteins. Its functional requirement is to elicit particular cellular responses (breakdown of intercellular junctions, cell scattering, apoptosis, etc.) that benefit the pathogen. Also, CagA must physically transition from a bacterial environment to the host cell, possibly adding further to constraints on its structure.
The NFN hypothesis for LMD protein compaction is an appealing model, which provides a convenient explanation of how Gab proteins and similarly structured LMD proteins might coordinate multiple downstream signalling events and segregate, or bring together, binding partners in a directional manner. This hypothesis by no means attempts to rebuild order and detract from the clearly established reality of protein intrinsic disorder, rather it serves to illustrate how versatile and varied protein surfaces and geometries can be, in order to carry out multiple cellular roles, e.g. ‘fly-casting’ to trap partners; moulding to different partners as in the case of hub proteins like p53; folding-back in autoregulatory processes, thereby protecting interaction sites. Thus, not only is there a wide spectrum of spatial forms within the proteome, individual proteins themselves can adopt a range of disordered and ordered states, which are context-dependent. This of course makes simple categorisation very tricky and potentially less meaningful. Whereas the traditional concept of protein structure encompassed globular proteins, molten and pre-molten globules, and those with inherent disorder, it is evident that disordered sequences themselves can be sub-classified into flexible disorder, constrained disorder and non-conserved disorder of no apparent function43 (though possibly fulfilling a basic role as linkers).
The fact that Gab-related proteins and many of their protein partners exist throughout the Metazoa kingdom, indicate that these mechanisms were primitively formed and probably represent one of diverse means to deal with the complexities of signalling systems in the context of multicellularity. Interestingly, individual Gab orthologues across species share more sequence identity than non-identity in the disordered tail, and are more closely related to one another than its paralogues are within a given species. This argues that the constraints on these supposedly intrinsically disordered sequences are nearly as great as for folded domains, although the PH domain is more highly conserved. Also, it suggests that constrained disorder is common in Gab proteins, a feature that may be related to their function as multi-site docking platforms and, consequently, the sheer number of conserved phosphorylation and protein binding sites within their sequences. Therefore, a simplistic classification of the Gab protein family is difficult as they constitute: a folded domain, followed by presumed constrained and some flexible disorder, punctuated with at least two defined secondary structural elements (PP-II and 310 helices) and potentially through compaction by NFN, a form of intrinsically disordered tertiary structure, with any number of uncharacterised putative disorder-to-order transitions. Outstanding issues surrounding the true geometric form of Gab proteins include understanding how their conformations might alter in receptor-stimulated or unstimulated cells. These questions feature among several currently under investigation in our laboratory.
Finally, while initial attempts to develop small molecules to inhibit PPIs were met with difficulty, modern fragment-based screening approaches are emerging as the strategy of choice. With this in mind, early high-throughput screening with larger molecules would most certainly not have covered a comprehensive chemical search space optimised for PPI surfaces. Furthermore, the future of IDPs as drug targets is more realistic as experimental data begin to become available revealing stable interaction surfaces, such as disordered sequences that adopt transient structure, capable of being stabilised or trapped by association to small molecules, or as a template for compounds based on peptido-mimetics. In view of this, the PPIs involving Gab-family proteins and their associated effectors and adaptors are certainly viable candidates. Validation of these propositions by focused experimentation is now essential.
Acknowledgements
We would like to thank Peter Tompa and Silja Wessler for useful comments on certain aspects of this work. SMF is indebted for his funding by the cancer charities Heads Up, Cancer Research UK and Breast Cancer Campaign and by the EU FP7 program. PCS acknowledges post-doctoral funding support from Cancer Research UK and the University of Oxford.References
- F. U. Wohrle, R. J. Daly and T. Brummer, Cell Commun. Signaling, 2009, 7, 22 CrossRef.
- K. Mardilovich, S. L. Pankratz and L. M. Shaw, Cell Commun. Signaling, 2009, 7, 14 CrossRef.
- N. Gotoh, Cancer Sci., 2008, 99, 1319–1325 CrossRef CAS.
- P. Di Stefano, M. P. C. Leal, G. Tornillo, B. Bisaro, D. Repetto, A. Pincini, E. Santopietro, N. Sharma, E. Turco, S. Cabodi and P. Defilippi, Am. J. Cancer Res., 2011, 1, 663–673 CAS.
- N. Tikhmyanova, J. L. Little and E. A. Golemis, Cell. Mol. Life Sci., 2010, 67, 1025–1048 CrossRef CAS.
- M. Harkiolaki, T. Tsirka, M. Lewitzky, P. C. Simister, D. Joshi, L. E. Bird, E. Y. Jones, N. O'Reilly and S. M. Feller, Structure, 2009, 17, 809–822 CrossRef CAS.
- N. Bisson, D. A. James, G. Ivosev, S. A. Tate, R. Bonner, L. Taylor and T. Pawson, Nat. Biotechnol., 2011, 29, 653–658 CrossRef CAS.
- U. Schaeper, N. H. Gehring, K. P. Fuchs, M. Sachs, B. Kempkes and W. Birchmeier, J. Cell Biol., 2000, 149, 1419–1432 CrossRef CAS.
- C. Bourgin, R. P. Bourette, S. Arnaud, Y. Liu, L. R. Rohrschneider and G. Mouchiroud, Mol. Cell. Biol., 2002, 22, 3744–3756 CrossRef CAS.
- S. H. Ong, Y. R. Hadari, N. Gotoh, G. R. Guy, J. Schlessinger and I. Lax, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6074–6079 CrossRef CAS.
- H. Gu and B. G. Neel, Trends Cell Biol., 2003, 13, 122–130 CrossRef CAS.
- Y. Liu and L. R. Rohrschneider, FEBS Lett., 2002, 515, 1–7 CrossRef CAS.
- T. Brummer, M. Larance, M. T. Herrera Abreu, R. J. Lyons, P. Timpson, C. H. Emmerich, E. D. Fleuren, G. M. Lehrbach, D. Schramek, M. Guilhaus, D. E. James and R. J. Daly, EMBO J., 2008, 27, 2305–2316 CrossRef CAS.
- M. Sachs, H. Brohmann, D. Zechner, T. Muller, J. Hulsken, I. Walther, U. Schaeper, C. Birchmeier and W. Birchmeier, J. Cell Biol., 2000, 150, 1375–1384 CrossRef CAS.
- M. Itoh, Y. Yoshida, K. Nishida, M. Narimatsu, M. Hibi and T. Hirano, Mol. Cell. Biol., 2000, 20, 3695–3704 CrossRef CAS.
- M. Seiffert, J. M. Custodio, I. Wolf, M. Harkey, Y. Liu, J. N. Blattman, P. D. Greenberg and L. R. Rohrschneider, Mol. Cell. Biol., 2003, 23, 2415–2424 CrossRef CAS.
- H. Gu, K. Saito, L. D. Klaman, J. Shen, T. Fleming, Y. Wang, J. C. Pratt, G. Lin, B. Lim, J. P. Kinet and B. G. Neel, Nature, 2001, 412, 186–190 CrossRef CAS.
- K. Nishida, L. Wang, E. Morii, S. J. Park, M. Narimatsu, S. Itoh, S. Yamasaki, M. Fujishima, K. Ishihara, M. Hibi, Y. Kitamura and T. Hirano, Blood, 2002, 99, 1866–1869 CrossRef.
- T. Wada, T. Nakashima, A. J. Oliveira-dos-Santos, J. Gasser, H. Hara, G. Schett and J. M. Penninger, Nat. Med., 2005, 11, 394–399 CrossRef CAS.
- Y. Zhang, E. Diaz-Flores, G. Li, Z. Wang, Z. Kang, E. Haviernikova, S. Rowe, C. K. Qu, W. Tse, K. M. Shannon and K. D. Bunting, Blood, 2007, 110, 116–124 CrossRef CAS.
- E. A. Bard-Chapeau, J. Yuan, N. Droin, S. Long, E. E. Zhang, T. V. Nguyen and G. S. Feng, Mol. Cell. Biol., 2006, 26, 4664–4674 CrossRef CAS.
- E. A. Bard-Chapeau, A. L. Hevener, S. Long, E. E. Zhang, J. M. Olefsky and G. S. Feng, Nat. Med., 2005, 11, 567–571 CrossRef CAS.
- Y. Lu, Y. Xiong, Y. Q. Huo, J. Y. Han, X. A. Yang, R. L. Zhang, D. S. Zhu, S. Klein-Hessling, J. Li, X. Y. Zhang, X. F. Han, Y. L. Li, B. Sheng, Y. L. He, M. Shibuya, G. S. Feng and J. C. Luo, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2957–2962 CrossRef CAS.
- W. Shioyama, Y. Nakaoka, K. Higuchi, T. Minami, Y. Taniyama, K. Nishida, H. Kidoya, T. Sonobe, H. Naito, Y. Arita, T. Hashimoto, T. Kuroda, Y. Fujio, M. Shirai, N. Takakura, R. Morishita, K. Yamauchi-Takihara, T. Kodama, T. Hirano, N. Mochizuki and I. Komuro, Circ. Res., 2011, 108, 664–675 CrossRef CAS.
- J. J. Zhao, W. Y. Wang, C. H. Ha, J. Y. Kim, C. Wong, E. M. Redmond, A. Hamik, M. K. Jain, G. S. Feng and Z. G. Jin, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 1016–U1147 CrossRef CAS.
- U. Schaeper, R. Vogel, J. Chmielowiec, J. Huelsken, M. Rosario and W. Birchmeier, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15376–15381 CrossRef CAS.
- R. D. Finn, J. Mistry, J. Tate, P. Coggill, A. Heger, J. E. Pollington, O. L. Gavin, P. Gunasekaran, G. Ceric, K. Forslund, L. Holm, E. L. Sonnhammer, S. R. Eddy and A. Bateman, Nucleic Acids Res., 2010, 38, D211–222 CrossRef CAS.
- I. Letunic, T. Doerks and P. Bork, Nucleic Acids Res., 2009, 37, D229–232 CrossRef CAS.
- P. C. Simister, F. Schaper, N. O'Reilly, S. McGowan and S. M. Feller, PLoS Biol., 2011, 9, e1000591 CAS.
- C. M. Gould, F. Diella, A. Via, P. Puntervoll, C. Gemund, S. Chabanis-Davidson, S. Michael, A. Sayadi, J. C. Bryne, C. Chica, M. Seiler, N. E. Davey, N. Haslam, R. J. Weatheritt, A. Budd, T. Hughes, J. Pas, L. Rychlewski, G. Trave, R. Aasland, M. Helmer-Citterich, R. Linding and T. J. Gibson, Nucleic Acids Res., 2009, 38, D167–180 CrossRef.
- S. Ren, V. N. Uversky, Z. Chen, A. K. Dunker and Z. Obradovic, BMC Genomics, 2008, 9(Suppl 2), S26 CrossRef.
- K. Bryson, L. J. McGuffin, R. L. Marsden, J. J. Ward, J. S. Sodhi and D. T. Jones, Nucleic Acids Res., 2005, 33, W36–38 CrossRef CAS.
- F. Orosz and J. Ovadi, Bioinformatics, 2011, 27, 1449–1454 CrossRef CAS.
- V. N. Uversky, J. Biomed. Biotechnol., 2010, 2010, 568068 CrossRef.
- P. Romero, Z. Obradovic, X. Li, E. C. Garner, C. J. Brown and A. K. Dunker, Proteins: Struct., Funct., Genet., 2001, 42, 38–48 CrossRef CAS.
- Z. R. Yang, R. Thomson, P. McNeil and R. M. Esnouf, Bioinformatics, 2005, 21, 3369–3376 CrossRef CAS.
- T. Ishida and K. Kinoshita, Bioinformatics, 2008, 24, 1344–1348 CrossRef CAS.
- B. Xue, R. L. Dunbrack, R. W. Williams, A. K. Dunker and V. N. Uversky, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 996–1010 CrossRef CAS.
- A. Schlessinger, M. Punta, G. Yachdav, L. Kajan and B. Rost, PLoS One, 2009, 4, e4433 Search PubMed.
- B. Titz, T. Low, E. Komisopoulou, S. S. Chen, L. Rubbi and T. G. Graeber, Oncogene, 2010, 29, 5895–5910 CrossRef CAS.
- Z. Dosztanyi, V. Csizmok, P. Tompa and I. Simon, Bioinformatics, 2005, 21, 3433–3434 CrossRef CAS.
- S. Vucetic, C. J. Brown, A. K. Dunker and Z. Obradovic, Proteins: Struct., Funct., Genet., 2003, 52, 573–584 CrossRef CAS.
- J. Bellay, S. Han, M. Michaut, T. Kim, M. Costanzo, B. J. Andrews, C. Boone, G. D. Bader, C. L. Myers and P. M. Kim, GenomeBiology, 2011, 12, R14 CrossRef CAS.
- R. Eulenfeld and F. Schaper, J. Cell Sci., 2009, 122, 55–64 CrossRef CAS.
- P. Tompa, M. Fuxreiter, C. J. Oldfield, I. Simon, A. K. Dunker and V. N. Uversky, BioEssays, 2009, 31, 328–335 CrossRef CAS.
- T. Sjoblom, S. Jones, L. D. Wood, D. W. Parsons, J. Lin, T. D. Barber, D. Mandelker, R. J. Leary, J. Ptak, N. Silliman, S. Szabo, P. Buckhaults, C. Farrell, P. Meeh, S. D. Markowitz, J. Willis, D. Dawson, J. K. Willson, A. F. Gazdar, J. Hartigan, L. Wu, C. Liu, G. Parmigiani, B. H. Park, K. E. Bachman, N. Papadopoulos, B. Vogelstein, K. W. Kinzler and V. E. Velculescu, Science, 2006, 314, 268–274 CrossRef.
- C. B. McDonald, K. L. Seldeen, B. J. Deegan, V. Bhat and A. Farooq, J. Mol. Recognit., 2011, 24, 585–596 CrossRef CAS.
- B. Meszaros, I. Simon and Z. Dosztanyi, PLoS Comput. Biol., 2009, 5, e1000376 Search PubMed.
- M. S. Cortese, V. N. Uversky and A. K. Dunker, Prog. Biophys. Mol. Biol., 2008, 98, 85–106 CrossRef CAS.
- A. Rath, A. R. Davidson and C. M. Deber, Biopolymers, 2005, 80, 179–185 CrossRef CAS.
- P. K. Vlasov, A. V. Vlasova, V. G. Tumanyan and N. G. Esipova, Proteins: Struct., Funct., Bioinf., 2005, 61, 763–768 CrossRef CAS.
- M. L. Wang, H. Yao and W. B. Xu, Comput. Biol. Chem., 2005, 29, 95–100 CrossRef CAS.
- M. J. Suskiewicz, J. L. Sussman, I. Silman and Y. Shaul, Protein Sci., 2011, 20, 1285–1297 CrossRef CAS.
- M. Harkiolaki, R. J. Gilbert, E. Y. Jones and S. M. Feller, Structure, 2006, 14, 1741–1753 CrossRef CAS.
- C. A. Galea, A. Nourse, Y. Wang, S. G. Sivakolundu, W. T. Heller and R. W. Kriwacki, J. Mol. Biol., 2008, 376, 827–838 CrossRef CAS.
- E. R. Lacy, I. Filippov, W. S. Lewis, S. Otieno, L. Xiao, S. Weiss, L. Hengst and R. W. Kriwacki, Nat. Struct. Mol. Biol., 2004, 11, 358–364 CAS.
- A. Akhmanova and M. O. Steinmetz, Nat. Rev. Mol. Cell Biol., 2008, 9, 309–322 CrossRef CAS.
- G. Lansbergen, Y. Komarova, M. Modesti, C. Wyman, C. C. Hoogenraad, H. V. Goodson, R. P. Lemaitre, D. N. Drechsel, E. van Munster, T. W. Gadella, Jr., F. Grosveld, N. Galjart, G. G. Borisy and A. Akhmanova, J. Cell Biol., 2004, 166, 1003–1014 CrossRef CAS.
- O. Hecht, H. Ridley, R. Boetzel, A. Lewin, N. Cull, D. A. Chalton, J. H. Lakey and G. R. Moore, FEBS Lett., 2008, 582, 2673–2677 CrossRef CAS.
- M. Hatakeyama, Microbes Infect., 2003, 5, 143–150 CrossRef CAS.
- C. M. Botham, A. M. Wandler and K. Guillemin, PLoS Pathog., 2008, 4, e1000064 Search PubMed.
- B. Hoy, M. Lower, C. Weydig, G. Carra, N. Tegtmeyer, T. Geppert, P. Schroder, N. Sewald, S. Backert, G. Schneider and S. Wessler, EMBO Rep., 2010, 11, 798–804 CrossRef CAS.
- T. Kwok, D. Zabler, S. Urman, M. Rohde, R. Hartig, S. Wessler, R. Misselwitz, J. Berger, N. Sewald, W. Konig and S. Backert, Nature, 2007, 449, 862–866 CrossRef CAS.
- L. F. Jimenez-Soto, S. Kutter, X. Sewald, C. Ertl, E. Weiss, U. Kapp, M. Rohde, T. Pirch, K. Jung, S. F. Retta, L. Terradot, W. Fischer and R. Haas, PLoS Pathog., 2009, 5, e1000684 Search PubMed.
- M. Selbach, S. Moese, C. R. Hauck, T. F. Meyer and S. Backert, J. Biol. Chem., 2002, 277, 6775–6778 CrossRef CAS.
- M. Poppe, S. M. Feller, G. Romer and S. Wessler, Oncogene, 2007, 26, 3462–3472 CrossRef CAS.
- H. Mimuro, T. Suzuki, J. Tanaka, M. Asahi, R. Haas and C. Sasakawa, Mol. Cell, 2002, 10, 745–755 CrossRef CAS.
- N. Tegtmeyer, S. Wessler and S. Backert, FEBS J., 2011, 278, 1190–1202 CrossRef CAS.
- T. Raabe, J. Riesgo-Escovar, X. Liu, B. S. Bausenwein, P. Deak, P. Maroy and E. Hafen, Cell, 1996, 85, 911–920 CrossRef CAS.
- S. M. Feller, H. Wecklein, M. Lewitzky, E. Kibler and T. Raabe, Mech. Dev., 2002, 116, 129–139 CrossRef CAS.
- L. Buti, E. Spooner, A. G. Van der Veen, R. Rappuoli, A. Covacci and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9238–9243 CrossRef CAS.
- F. Bagnoli, L. Buti, L. Tompkins, A. Covacci and M. R. Amieva, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16339–16344 CrossRef CAS.
- C. Pelz, S. Steininger, C. Weiss, F. Coscia and R. Vogelmann, J. Biol. Chem., 2011, 286, 8999–9008 CrossRef CAS.
- S. Steininger, C. Pelz and R. Vogelmann, Gut Microbes, 2011, 2, 167–172 CrossRef.
- S. Ren, H. Higashi, H. Lu, T. Azuma and M. Hatakeyama, J. Biol. Chem., 2006, 281, 32344–32352 CrossRef CAS.
- R. Herbst, X. Zhang, J. Qin and M. A. Simon, EMBO J., 1999, 18, 6950–6961 CrossRef CAS.
- D. Nesic, M. C. Miller, Z. T. Quinkert, M. Stein, B. T. Chait and C. E. Stebbins, Nat. Struct. Mol. Biol., 2010, 17, 130–132 CAS.
- M. Schwab, BioEssays, 1998, 20, 473–479 CrossRef CAS.
- R. J. Daly, H. Gu, J. Parmar, S. Malaney, R. J. Lyons, R. Kairouz, D. R. Head, S. M. Henshall, B. G. Neel and R. L. Sutherland, Oncogene, 2002, 21, 5175–5181 CrossRef CAS.
- M. Bentires-Alj, S. G. Gil, R. Chan, Z. C. Wang, Y. Wang, N. Imanaka, L. N. Harris, A. Richardson, B. G. Neel and H. Gu, Nat. Med., 2006, 12, 114–121 CrossRef CAS.
- S. H. Lee, E. G. Jeong, S. W. Nam, J. Y. Lee and N. J. Yoo, Pathology, 2007, 39, 326–329 CrossRef CAS.
- B. Horst, S. K. Gruvberger-Saal, B. D. Hopkins, L. Bordone, Y. Yang, K. A. Chernoff, I. Uzoma, V. Schwipper, J. Liebau, N. J. Nowak, G. Brunner, D. Owens, D. L. Rimm, R. Parsons and J. T. Celebi, Am. J. Pathol., 2009, 174, 1524–1533 CrossRef CAS.
- X. L. Xu, X. Wang, Z. L. Chen, M. Jin, W. Yang, G. F. Zhao and J. W. Li, Int. J. Biol. Sci., 2011, 7, 496–504 CAS.
- M. Sattler, M. G. Mohi, Y. B. Pride, L. R. Quinnan, N. A. Malouf, K. Podar, F. Gesbert, H. Iwasaki, S. Li, R. A. Van Etten, H. Gu, J. D. Griffin and B. G. Neel, Cancer Cell, 2002, 1, 479–492 CrossRef CAS.
- M. G. Mohi, I. R. Williams, C. R. Dearolf, G. Chan, J. L. Kutok, S. Cohen, K. Morgan, C. Boulton, H. Shigematsu, H. Keilhack, K. Akashi, D. G. Gilliland and B. G. Neel, Cancer Cell, 2005, 7, 179–191 CrossRef CAS.
- A. Zatkova, C. Schoch, F. Speleman, B. Poppe, C. Mannhalter, C. Fonatsch and K. Wimmer, Genes, Chromosomes Cancer, 2006, 45, 798–807 CrossRef CAS.
- K. Mood, C. Saucier, Y. S. Bong, H. S. Lee, M. Park and I. O. Daar, Mol. Biol. Cell, 2006, 17, 3717–3728 CrossRef CAS.
- J. P. Eder, G. F. Vande Woude, S. A. Boerner and P. M. LoRusso, Clin. Cancer Res., 2009, 15, 2207–2214 CrossRef CAS.
- A. E. Moran, D. H. Hunt, S. H. Javid, M. Redston, A. M. Carothers and M. M. Bertagnolli, J. Biol. Chem., 2004, 279, 43261–43272 CrossRef CAS.
- G. S. Kapoor, Y. Zhan, G. R. Johnson and D. M. O'Rourke, Mol. Cell. Biol., 2004, 24, 823–836 CrossRef CAS.
- L. Y. Bourguignon, P. A. Singleton, H. Zhu and F. Diedrich, J. Biol. Chem., 2003, 278, 29420–29434 CrossRef CAS.
- C. Haynes, C. J. Oldfield, F. Ji, N. Klitgord, M. E. Cusick, P. Radivojac, V. N. Uversky, M. Vidal and L. M. Iakoucheva, PLoS Comput. Biol., 2006, 2, e100 Search PubMed.
- R. R. Vallabhajosyula, D. Chakravarti, S. Lutfeali, A. Ray and A. Raval, PLoS One, 2009, 4, e5344 Search PubMed.
- M. Vidal, M. E. Cusick and A. L. Barabasi, Cell, 2011, 144, 986–998 CrossRef CAS.
- J. D. Han, N. Bertin, T. Hao, D. S. Goldberg, G. F. Berriz, L. V. Zhang, D. Dupuy, A. J. Walhout, M. E. Cusick, F. P. Roth and M. Vidal, Nature, 2004, 430, 88–93 CrossRef CAS.
- S. Agarwal, C. M. Deane, M. A. Porter and N. S. Jones, PLoS Comput. Biol., 2010, 6, e1000817 Search PubMed.
- D. Xu, S. Wang, W. M. Yu, G. Chan, T. Araki, K. D. Bunting, B. G. Neel and C. K. Qu, Blood, 2010, 116, 3611–3621 CrossRef CAS.
- B. T. Hennessy, A. M. Gonzalez-Angulo, K. Stemke-Hale, M. Z. Gilcrease, S. Krishnamurthy, J. S. Lee, J. Fridlyand, A. Sahin, R. Agarwal, C. Joy, W. Liu, D. Stivers, K. Baggerly, M. Carey, A. Lluch, C. Monteagudo, X. He, V. Weigman, C. Fan, J. Palazzo, G. N. Hortobagyi, L. K. Nolden, N. J. Wang, V. Valero, J. W. Gray, C. M. Perou and G. B. Mills, Cancer Res., 2009, 69, 4116–4124 CrossRef CAS.
- J. R. Adams, N. F. Schachter, J. C. Liu, E. Zacksenhaus and S. E. Egan, Oncotarget, 2011, 2, 435–447 Search PubMed.
- K. L. Everett, T. D. Bunney, Y. Yoon, F. Rodrigues-Lima, R. Harris, P. C. Driscoll, K. Abe, H. Fuchs, M. H. de Angelis, P. Yu, W. Cho and M. Katan, J. Biol. Chem., 2009, 284, 23083–23093 CrossRef CAS.
- M. R. Arkin and A. Whitty, Curr. Opin. Chem. Biol., 2009, 13, 284–290 CrossRef CAS.
- J. Gsponer and M. M. Babu, Prog. Biophys. Mol. Biol., 2009, 99, 94–103 CrossRef CAS.
- T. Sato and N. Gotoh, Expert Opin. Ther. Targets, 2009, 13, 689–700 CrossRef CAS.
- P. Tompa, FEBS Lett., 2005, 579, 3346–3354 CrossRef CAS.
- A. K. Dunker, I. Silman, V. N. Uversky and J. L. Sussman, Curr. Opin. Struct. Biol., 2008, 18, 756–764 CrossRef CAS.
- E. Hazy and P. Tompa, ChemPhysChem, 2009, 10, 1415–1419 CrossRef CAS.
- P. Tompa, Structure and function of intrinsically disordered proteins, CRC Press/Taylor and Francis, Boca Raton, 2009 Search PubMed.
- A. Peyroche, B. Antonny, S. Robineau, J. Acker, J. Cherfils and C. L. Jackson, Mol. Cell, 1999, 3, 275–285 CrossRef CAS.
- E. Mossessova, R. A. Corpina and J. Goldberg, Mol. Cell, 2003, 12, 1403–1411 CrossRef CAS.
- J. C. Amor, D. H. Harrison, R. A. Kahn and D. Ringe, Nature, 1994, 372, 704–708 CrossRef CAS.
- J. Menetrey, E. Macia, S. Pasqualato, M. Franco and J. Cherfils, Nat. Struct. Biol., 2000, 7, 466–469 CrossRef CAS.
- V. Biou, K. Aizel, P. Roblin, A. Thureau, E. Jacquet, S. Hansson, B. Guibert, E. Guittet, C. van Heijenoort, M. Zeghouf, J. Perez and J. Cherfils, J. Mol. Biol., 2010, 402, 696–707 CrossRef CAS.
- J. A. Marsh, C. Neale, F. E. Jack, W. Y. Choy, A. Y. Lee, K. A. Crowhurst and J. D. Forman-Kay, J. Mol. Biol., 2007, 367, 1494–1510 CrossRef CAS.
- J. A. Marsh and J. D. Forman-Kay, Biophys. J., 2010, 98, 2383–2390 CrossRef CAS.
- P. Chene, ChemMedChem, 2006, 1, 400–411 CrossRef CAS.
- E. Solomaha, F. L. Szeto, M. A. Yousef and H. C. Palfrey, J. Biol. Chem., 2005, 280, 23147–23156 CrossRef CAS.
- M. Harkiolaki, M. Lewitzky, R. J. Gilbert, E. Y. Jones, R. P. Bourette, G. Mouchiroud, H. Sondermann, I. Moarefi and S. M. Feller, EMBO J., 2003, 22, 2571–2582 CrossRef CAS.
- D. M. Berry, P. Nash, S. K. Liu, T. Pawson and C. J. McGlade, Curr. Biol., 2002, 12, 1336–1341 CrossRef CAS.
- C. Reynes, H. Host, A. C. Camproux, G. Laconde, F. Leroux, A. Mazars, B. Deprez, R. Fahraeus, B. O. Villoutreix and O. Sperandio, PLoS Comput. Biol., 2010, 6, e1000695 Search PubMed.
- J. T. Nguyen, C. W. Turck, F. E. Cohen, R. N. Zuckermann and W. A. Lim, Science, 1998, 282, 2088–2092 CrossRef CAS.
- S. Hashimoto, M. Hirose, A. Hashimoto, M. Morishige, A. Yamada, H. Hosaka, K. Akagi, E. Ogawa, C. Oneyama, T. Agatsuma, M. Okada, H. Kobayashi, H. Wada, H. Nakano, T. Ikegami, A. Nakagawa and H. Sabe, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7036–7041 CrossRef CAS.
- C. Oneyama, H. Nakano and S. V. Sharma, Oncogene, 2002, 21, 2037–2050 CrossRef CAS.
- N. T. Ross, W. P. Katt and A. D. Hamilton, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 989–1008 CrossRef CAS.
- P. Vanhee, F. Stricher, L. Baeten, E. Verschueren, T. Lenaerts, L. Serrano, F. Rousseau and J. Schymkowitz, Structure, 2009, 17, 1128–1136 CrossRef CAS.
- C. A. Galea, Y. Wang, S. G. Sivakolundu and R. W. Kriwacki, Biochemistry, 2008, 47, 7598–7609 CrossRef CAS.
- A. S. Pinheiro, J. A. Marsh, J. D. Forman-Kay and W. Peti, J. Am. Chem. Soc., 2011, 133, 73–80 CrossRef CAS.
- Q. Cai, H. Sun, Y. Peng, J. Lu, Z. Nikolovska-Coleska, D. McEachern, L. Liu, S. Qiu, C. Y. Yang, R. Miller, H. Yi, T. Zhang, D. Sun, S. Kang, M. Guo, L. Leopold, D. Yang and S. Wang, J. Med. Chem., 2011, 54, 2714–2726 CrossRef CAS.
- A. M. Waterhouse, J. B. Procter, D. M. Martin, M. Clamp and G. J. Barton, Bioinformatics, 2009, 25, 1189–1191 CrossRef CAS.
Footnote |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M. Madan Babu. |
| This journal is © The Royal Society of Chemistry 2012 |