Mutual effects of disorder and order in fusion proteins between intrinsically disordered domains and fluorescent proteins†
Marina
Lotti
*a and
Sonia
Longhi
*b
aDipartimento di Biotecnologie e Bioscienze, Università Statale Milano-Bicocca, Piazza della Scienza 2, 20126 Milan, Italy. E-mail: marina.lotti@unimib.it; Fax: +39 02 64 48 35 69; Tel: +39 02 64 48 35 27
bArchitecture et Fonction des Macromolécules Biologiques, UMR 6098 CNRS et Universités d'Aix-Marseille I et II, Avenue de Luminy, 163, Case 932, 13288 Marseille Cedex 09, France. E-mail: Sonia.Longhi@afmb.univ-mrs.fr; Fax: +33 4 91 26 67 20; Tel: +33 4 91 82 55 80
First published on 18th August 2011
Abstract
Intrinsically disordered proteins are being paid an increasing amount of interest due to the understanding of the crucial role that flexible regions play in molecular recognition and in signaling. Accordingly, reports focusing on the structural and functional characterization of intrinsically disordered proteins or regions are growing exponentially. Relatively few studies have however been reported on the mutual effects of ordered and disordered moieties in artificial fusion proteins. In this review, we focus on the few available experimental data based on the use of chimeras in which fluorescent proteins were fused to disordered domains of different lengths, compactness and propensity to form secondary structures. The impact of the artificial fusion on the conformational and functional properties of the resulting proteins is discussed.
 Marina Lotti | Marina Lotti is a professor of Biochemistry and a Head of the Department of Biotechnology and Biosciences at the University of Milano-Bicocca. She obtained her PhD in Science at the Max-Planck-Institute for Molecular Genetics of Berlin and was then a researcher of the Italian National Research Council and of the State University of Milano. She became a professor at the University of Milano-Bicocca in 2000, where she leads the group of Protein Engineering and Industrial Enzymology. Her current research interests are cold-adapted enzymes, aggregation of recombinant proteins in bacterial cells, functional and conformational aspects of intrinsically disordered proteins. |
 Sonia Longhi | Sonia Longhi is a Director of Research at the Center for the National Scientific Research (CNRS), heading the “Structural Disorder and Molecular Recognition” group within the AFMB laboratory. She obtained a PhD in molecular biology from the Universitá degli Studi of Milano in 1993. She got a HDR in structural virology from the University of Aix-Marseille I in 2003. Her scientific focus is on intrinsically disordered proteins (IDPs) and the mechanistic and functional aspects of the interactions they establish with partners. She has authored more than seventy scientific publications, edited a book on measles virus nucleoprotein and co-edited (with Prof. Vladimir Uversky) a book on the experimental approaches to characterize IDPs and the folding coupled to binding events they undergo in the presence of partners. |
Introduction
Since the importance of structural disorder for the function of several biologically relevant proteins has been acknowledged, information available about intrinsically disordered proteins (IDPs) keeps steadily growing. Several excellent reviews have appeared over the last few years concerning the characterization, the function, the interaction mechanisms and the regulation of IDPs, and an updated overview can be found in the most recent review articles.1–10 IDPs inherently escape atomistic description by X-ray crystallography and their structural characterization requires the combination of several approaches.11,12 The plasticity associated with the conformational freedom characteristic of IDPs provides the ground for their varied biological roles and for their ability to interact with multiple partners, all features that engage IDPs in signaling and regulation within the cells and, as a consequence, also in various pathologies, such as cancer, as well as cardiovascular and neurodegenerative diseases.13–16 Due to their functional and interaction promiscuity however, over-expression of IDPs could lead to dosage sensitivity17 and hence tight regulation of IDPs is critical for the cell.18Consistent with their central biological function, IDPs are predicted to be abundant in all living beings, with their percentage over the total cell proteins growing with the complexity of proteomes. Half of the proteins in eukaryotic cells are predicted to contain long regions of disorder and about 25–30% to be mostly disordered.19 This percentage is even higher (70%) when only signaling proteins are considered.20 Disorder is also largely represented in viral proteins, probably because it is of advantage in terms of genetic compaction and pleiotropy, since disordered regions allow the establishment of multiple interactions thereby leading to multiple biological effects. Besides, it has also been proposed that disorder may play a role in buffering the deleterious effects of mutations21–23 and in alleviating evolutionary constraints within proteins encoded by overlapping reading frames (see ref. 24 and 25 and references therein cited).
IDPs show an extremely wide diversity in their structural properties: indeed they can attain extended conformations (random coil-like) or remain globally collapsed with regions of fluctuating secondary structure (molten globule-like). Conformational and spectroscopic analyses showed that random coil-like proteins can be subdivided in their turn into two major groups. While the first group consists of proteins with extended maximum dimensions typical of random coils with no (or little) secondary structure, the second group comprises the so-called pre-molten globules, which are more compact (but still less compact than globular or molten globule proteins) and retain some residual secondary structure.1,26–30
Some polypeptide chains, for example the yeast Sic 1 protein31–34 and the N-terminal region of the vesicular stomatitis virus phosphoprotein,35 are mainly disordered, even though they possess segments endowed with a certain propensity to form transient structural elements. In other proteins, disordered regions and folded domains coexist within the same polypeptide. In such proteins, the disordered moiety is often involved in interaction with partners and exerts a regulatory role. Such a modular organization, with alternating ordered and disordered domains, has been reported for a few viral proteins, as for instance the nucleoprotein (N) and the phosphoprotein (P) from measles36–41 and Sendai viruses,42–47 as well as the P proteins from Rhabdoviridae members.48–52 Alternating disordered and ordered regions have also been described in two Saccharomyces cerevisiaeproteins, namely Knr4 and Ure2. Knr4 participates in cell wall formation and cell cycle regulation and is constituted by a central globular domain flanked by two disordered regions,53,54 whereas the prion protein, Ure2, is built up by a largely disordered N-terminus bound to a globular GST-like C-terminal domain.55
Since the disordered part of such modular proteins is the one engaged in molecular interactions, numerous studies have focused on the characterization of isolated disordered domains both in isolation and upon interaction with partner proteins. Nevertheless, a detailed description of the conformational effect that a disordered domain and a domain endowed with a stable fold produce on each other when they are covalently bound is still lacking and only a few groups have directly or indirectly tackled this issue. This information, however, is of importance since the properties (binding ability, regulation, participation in protein networks) of the disordered moiety might well be affected by the flanking region(s), and, inversely, the features of the globular part of the polypeptide could at least partly depend on the attached less structured regions. This is particularly relevant in the case of disordered domains that may occur in different contexts, i.e. that can be flanked by different regions as a result of different alternative splicing and/or of mRNA editing events.
Sparse data are available about the functional consequences of swapping or removing disordered regions from such mixed proteins. In one such a study, the authors swapped the position of the N- and C-terminal regions of Ure2, with or without an intervening peptide linker, to create the CLN-Ure2 and CN-Ure2 variants respectively.55 Although the secondary structure content and the stability of the variants were the same as those of wtUre2, their ability to form amyloid-like fibrils was found to be either delayed (CLN-Ure2) or substantially reduced (CN-Ure2). In another similar study, the interaction abilities of various Knr4 deletion variants were evaluated both in vivo and in vitro.54 While the disordered N-terminal domain of Knr4 was shown to be indispensable for the interactions, the disordered C-terminal domain was found to have a negative effect on the interaction strength, thus clearly showing that large disordered regions do not always promote protein–protein interactions, but in some cases, can rather inhibit them.
Other pieces of information are provided by electron microscopy and atomic force microscopy studies of fusion proteins where the IDP is linked to small globular domains at either the N- or the C-terminus. This approach was applied to enable measurement of the end-to-end distances and persistence lengths of the unstructured segments (for examples see ref. 56 and 57), as well as to substantiate that the predicted tumbling times of globular protein domains are affected by linked disordered regions.58 While these studies support the hypothesis of the existence of reciprocal functional effects, they inherently cannot provide a comparison of the physico-chemical properties of the IDP alone and in the context of the fusion.
A more direct contribution to this issue can be obtained through the analysis of chimeric proteins in which a disordered part is artificially linked to a folded domain and by the evaluation of the properties of the whole polypeptide and of its components in isolation. This approach offers unique advantages when the folded moiety is a protein very well characterized in terms of both structure and function and when it has a biological activity that can be tuned by subtle conformational changes. This review article focuses on the results reported in a few works published in recent years and based on the use of chimeras in which fluorescent proteins were fused to disordered domains of different lengths, compactness and propensity to form secondary structures. As fluorescent proteins are broadly used as tags, a non-negligible outcome of these studies is the possibility to evaluate whether information obtained by this experimental strategy is reliable in case the fusion partner is a disordered protein. In other words, these studies provide hints on the extent to which fluorescent proteins may artefactually affect the conformational and binding properties of the disordered moiety.
Fusions with disordered viral proteins
Two studies focused on the conformational and functional effects arising from the covalent linkage of the GFP to the nucleoprotein (N) and the phosphoprotein (P) of measles virus (MeV) have been reported. N is responsible for the encapsidation of the viral genome, for recruiting the viral polymerase and is also involved in virus assembly (see ref. 40 and 59 and references therein cited). Structurally, it is organized in a globular N-terminal core (residues 1–400) and a flexible C-terminal domain (NTAIL, residues 401–525).38,60,61 The P protein, beyond being an essential co-factor of the viral polymerase, acts as a chaperone for the N protein by preventing its illegitimate self-assembly in the absence of ongoing viral RNA synthesis. Its N-terminal domain (PNT, aa 1-230) possesses sequence and biochemical features typical of intrinsically disordered regions.36In a study aimed at characterizing the mutual conformational effects of disordered and ordered regions within fusion proteins, the GFP was linked at the C-terminus of NTAIL and PNT domains.62 In this case, the availability of detailed information about both the fluorescent protein and the two disordered components in isolation allowed to highlight changes arising from the covalent linkage. The far-UV spectra of the fusion proteins were clearly not the averages of the spectra of the ordered and disordered components in isolation, and were also different from the spectra of equimolar IDP + GFP mixtures (see Fig. 1). These findings indicate that the covalent association elicited conformational effects in the fusion partners. Surprisingly enough however, the properties of the chimeras were not those expected from the properties of their disordered parts. In particular, the secondary structure content of the PNT–GFP protein was higher than predicted and close to that of GFP alone, although PNT in isolation is less structured than NTAIL. Based on the inherent propensity of PNT to undergo a disorder-to-order transition,36 the authors hypothesized that the increase in order in PNT–GFP likely depends on a gain of structure within PNT. Conversely, the less ordered nature of the NTAIL–GFP fusion protein with respect to the average of the secondary structure contents of the two components was ascribed either to partial unfolding of GFP or to loss of residual structure by NTAIL, with the transiently populated α-helical regions of the latter61,63–68 adopting preferentially an extended (e.g. disordered) conformation when linked to GFP. Relevant differences in the fusions containing NTAIL and PNT were also evident in experiments aimed at evaluating protein compactness, i.e.size exclusion chromatography, and near-UV and visible CD spectra, all of which consistently showed that PNT–GFP was more compact than the fusion with NTAIL. All these results converged toward the conclusion that the conformation of the fusion protein was IDP-specific, however in a way hardly predictable from the properties of the disordered moiety in isolation. Based on these observations, it was tentatively suggested that the covalent association with GFP would endow the IDP with folding propensities different from those reported for the isolated disordered domain. However, no final conclusions could be drawn about which moiety dominates in determining the structural properties of the fusion protein. In fact, neither stability to denaturation nor resistance to proteolysis of the globular GFP was significantly reduced by the fusion with the disordered domain and, on the other hand, the two IDPs were only marginally protected from proteolysis by fusion with the compact GFP. Moreover, it was observed that PNT–GFP was not more resistant to proteases than NTAIL–GFP, in spite of its higher compactness. The authors speculated that possible differences in the structural rearrangements elicited by the fusion in the two chimeras might be delocalized because of the high flexibility of IDPs and therefore might have escaped detection by the techniques applied.62
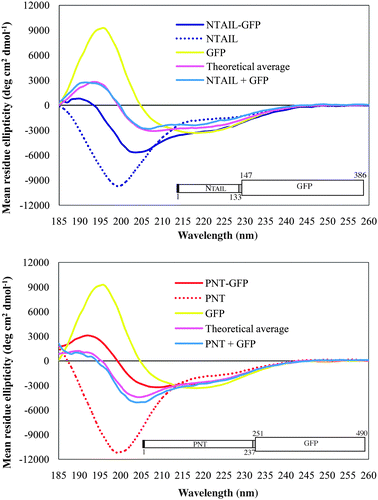 | ||
| Fig. 1 Far-UV CD spectra of NTAIL–GFP and PNT–GFP. The CD spectrum of each of the fusion proteins is compared with that of individual proteins, with the calculated average spectrum and with the spectrum of equimolar protein mixtures. Spectra were recorded in 10 mM sodium phosphate (pH 7.5) at 20 °C. The schematic representation of the constructs is also shown. The N-terminal hexahistidine tag and the linker region containing the TEV cleavage site are shown by a black and grey box, respectively. The disordered moiety is depicted as a narrow box, while the GFP is shown as a large box. Modified from ref. 62. | ||
In another study, the GFP was added either N-terminally or C-terminally to the whole N and P proteins69 and the functional impact of the fusion was examined in the context of mutated viruses. The authors built recombinant MeV genome constructs encoding hybrid proteins in addition to the standard N or P proteins (see Fig. 2). The four viruses (MeV-addN/GFP, MeV-addGFP/N, MeV-addP/GFP, and MeV-addGFP/P) were all rescued, and were found to efficiently propagate and to express GFP indicating that interference effects from the hybrid proteins, if any, were small. However, although all the fusion proteins were expressed, only the N/GFP and the GFP/P proteins were incorporated in virus particles. MeV-addGFP/P particles incorporated similar levels of P and GFP/P, suggesting structural and functional equivalence between the unmodified and the hybrid protein. To address the question as to whether N/GFP and GFP/P could functionally replace the standard proteins, the authors replaced the standard N and P genes in the MeV genome with the genes coding for the hybrid proteins, producing the recombinant genomes MeV-N/GFP and MeV-GFP/P (see Fig. 2). Only the MeV-GFP/P could be rescued, although it propagated less efficiently than the native virus. The inability to rescue MeV-N/GFP showed that N/GFP does not functionally replace N. Altogether, these results indicate that while addition of GFP to the amino terminus of P did not significantly compromise function, when GFP was linked to either the N protein termini or to the P protein carboxyl terminus, it strongly impaired the protein stability and/or function. These observations are well in line with the structural organization of the N and P proteins and with their interaction profile. Indeed, since the extreme carboxyl-terminal X domain (XD) is the region of P responsible for the interaction and the induced folding of the N protein carboxyl-terminal tail,37,38,61,70 it is not surprising that GFP addition to this terminus interferes with its function. On the other hand, the P protein amino-terminal domain is natively unfolded36 and is therefore more tolerant to modification. Likewise, when GFP was added to the N protein carboxyl terminus, which is intrinsically disordered37 and exposed on the outer surface of the nucleocapsid,60 it did not cause protein instability although it was less efficiently incorporated than N. Interestingly however, N/GFP did not functionally replace N, likely because the presence of GFP at the C-terminus of N prevents binding to P.
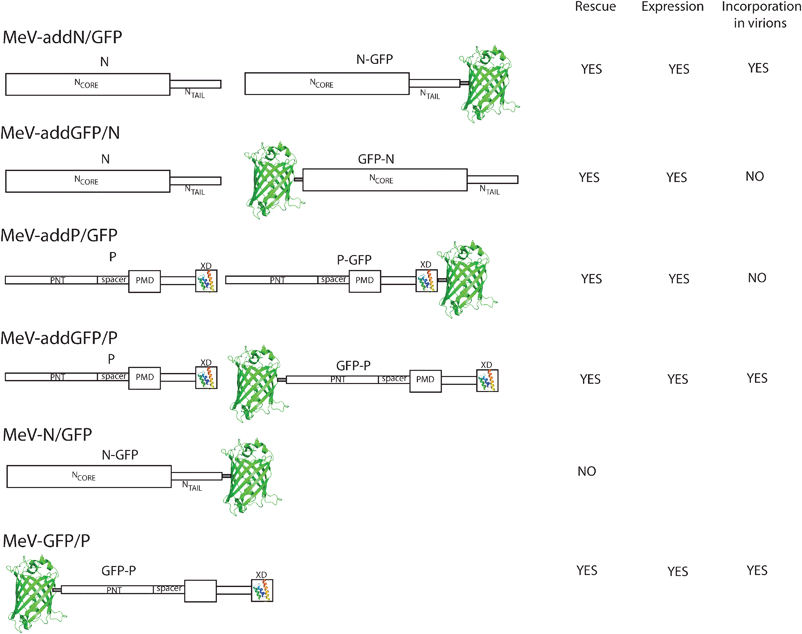 | ||
| Fig. 2 Schematic illustration of the functional impact of GFP fusions to either the N- or the C-terminus of measles virus (MeV) nucleoprotein (N) and phosphoprotein (P). Large and narrow boxes correspond to structured and disordered regions, respectively. NCORE: N-terminal region of N responsible for N–N interactions. NTAIL: intrinsically disordered domain. PNT: intrinsically disordered domain of P. PMD: P multimerization domain. XD: X domain of P. The structures of XD (PDB code 1OKS)37 and of GFP (pdb code 3GJ2)83 are shown (not drawn to scale). | ||
Fusions with disordered metal-responsive proteins
Following previous studies that made use of GFP-based FRET assays to explore the end-to-end separation of both structured and unstructured proteins,71 the group of Scott Banta has recently investigated the calcium-induced folding of the intrinsically disordered RTX domain of the adenylate cyclase toxin (CyaA) from Bordetella pertussis in the context of a fusion protein.72,73RTX (repeats-in-toxins) motifs are common to proteins secreted through the bacterial type 1 secretion system.72,74–77 In these proteins, the C-terminal secretion signal is preceded by multiple RTX motifs that fold to form β-roll domains upon low-affinity interaction with calcium. Since calcium is lower in the cell than in the extracellular environment, calcium-induced folding can be seen as a strategy to avoid the toxin folding into the active conformation within the producing cells.In a first study, the fifth RTX domain of CyaA78 was inserted between two fluorescent proteins, namely the cyan fluorescent protein (CFP) and the yellow fluorescent protein (YFP) (see Fig. 3A). The resulting fusion protein was used in FRET studies, where energy transfer efficiency critically depends on the distance between the chromophores and therefore reports on conformational changes of the RTX domain. Previous studies carried out on isolated RTX in the absence of calcium highlighted the presence of some residual structure, as the addition of a denaturant was required for complete unfolding.76 Strikingly, in the absence of calcium, the FRET-derived average inter-chromophore distance of the RTX FRET construct was found to be smaller than if the protein behaved as an ideal Gaussian chain-like polymer, arguing for the presence of some residual structure also in the context of the fusion with the fluorescent proteins.72 Addition of the fluorescent proteins at the two ends of RTX did not prevent calcium-induced folding. Indeed, addition of Ca2+ ions caused a fast conformational change, as shown by the decrease in the average distance between the fluorescent proteins observed using FRET, as well as by an increase in the intrinsic Trp fluorescence.72 CD studies showed that the sequences flanking the RTX domain are necessary for the Ca2+-induced conformational change, since an RTX construct lacking those sequences was not responsive to Ca2+.72 Surprisingly, when the RTX domain devoid of flanking sequences but linked to the fluorescent proteins was studied with FRET, a significant calcium-induced conformational change was observed, which resulted in approximately the same FRET efficiency as in the RTX construct containing the natural flanks. The affinity and cooperativity for Ca2+ binding were, however, reduced. These results indicated that the presence of the fluorescent proteins at both RTX termini was sufficient to restore the Ca2+-dependent ability of RTX to fold into a β-roll structure, though at higher Ca2+ concentrations. This means that not only the flanking CFP and YFP did not prevent Ca2+-induced folding of RTX, but they even acted as end caps functionally replacing the natural flanking sequences. On this basis it was concluded that in the context of this fusion the RTX domain adopts a conformation similar to that of other calcium-bound β-roll containing proteins, arguing for the lack of major structural effects related to the flanking fluorescent proteins. Similarly, the occurrence of an efficient energy transfer in the presence of Ca2+ demonstrated that the chromophores of CFP and YFP were located in a proper tertiary environment, which is possible only in case the presence of the disordered RTX domain does not affect the overall fold of the fluorescent proteins.
 | ||
| Fig. 3 Calcium-induced folding capabilities of RTX constructs. (A) Schematic image of the RTX-FRET construct showing the Ca2+-dependent β-roll structure formation that alters the distance between the FRET pair of CFP and YFP (not drawn to scale). The structures of GFP (pdb code 3GJ2)83 and of the Ca2+-bound state of an RTX domain from the Serratia marcescens serralysin enzyme (PDB code 1SAT)75 are shown. In the β roll, Ca2+ ions are shown as red spheres, and β strands are shown as yellow arrows. (B) Schematic illustration of the various RTX constructs and their ability to undergo a Ca2+-induced conformational change. The structure of MBP (PDB code 3LC8)84 is shown. The region encompassing residues 1529–1612 of the fifth RTX domain is shown in black, whereas the flanking regions are shown in grey. Modified from ref. 72. | ||
A subsequent study was aimed at further exploring the role of the natural flanking sequences and of non-native fusion partners in the formation of the calcium-responsive β-roll structure. The authors examined various RTX forms bearing truncations on either the N- or the C-terminal flanking sequences, as well as fusions at both RTX ends with non-native partners, such as the maltose binding protein (MBP) and fluorescent proteins (see Fig. 3B).73 As a first step, CD and fluorescence spectroscopy studies identified the minimal calcium-responsive element within the C-terminal flank. Both fluorescent proteins, as well as the MBP, were found to be able to replace the native flanking sequence and to confer calcium-induced folding when fused at both RTX ends. Very interestingly, this effect was retained also when fusion partners were linked to the C-terminus only,73 whereas fusions at the N-terminus failed to restore calcium sensitivity. Interestingly, calcium-sensitivity of the RTX construct devoid of the flanking sequences was also restored in the presence of the crowding agent PEG. The structural diversity between the natural flank, MBP and YFP was interpreted as the lack of precise structural requirements for enabling calcium responsiveness of RTX. This observation is in good agreement with other studies reporting the successful use of serralysin RTX-repeats as a calcium-switch to control the spatial separation of a bi-terminally fused β-galactosidase.79 Furthermore, changes in bis-ANS binding were similar for native and non-native flanks, suggesting that they all undergo similar conformational changes.73 An explanation for the need of natural or non-natural sequences at the carboxy terminus of the repeat was suggested by the observation that the folding and stability of proteins containing repeats are dominated by short-range interactions between adjacent repeats. These short-range interactions are not strong enough to overcome the high entropic penalty associated with folding and, as such, require capping groups at both ends to lower the energy barrier and nucleate folding. Without a flanking group, the RTX-repeats are unable to fold into the calcium-bound β-roll structure. Since PEG, the native flanking sequence, and two unrelated proteins (YFP and MBP) all enabled calcium-induced RTX folding, the authors proposed that β roll formation likely does not require any enthalpic interaction (salt bridges or hydrogen bonds) with specific residues or elements of the secondary structure in the flanks and rather relies on an entropically-driven mechanism. In calcium-poor environments the RTX-repeats are disordered77 and are thus flexible and have greater entropy than the calcium bound β roll. The finding that RTX without C-terminal flanking groups cannot undergo calcium-responsive folding suggested that the enthalpic contribution of calcium binding to RTX combined with the entropic contribution of the liberation of water from hydrophobic hydration is not large enough to overcome the entropic cost of the reduction in RTX flexibility associated with β roll formation. In this scenario, entropy loss has to be reduced in order to allow calcium-induced RTX folding. The authors refer to this entropy reduction as “entropic stabilization”. The role of the flank (with which the RTX does not establish any specific contact) would be to introduce a steric constraint favorable to the establishment of short-range interactions between adjacent repeats, thereby favoring folding. The term “entropic stabilization” reflects the fact that this latter effect does not rely on a precise 3D scaffold or pattern of amino acids in the flank. Entropy-driven stabilization is consistent with the ability of RTX-repeats with no capping groups to undergo calcium-induced conformational change in the presence of 25% PEG 8000,79,80 where PEG is known to enhance molecular crowding therefore reducing conformational freedom and protein entropy.
These studies converge to show that the disordered RTX-repeats are not able per se to undergo calcium-induced folding but they require the contribution of flanking regions irrespective of whether they are disordered (case of natural regions) or folded (case of fusions with fluorescent proteins and MBP). The presence of a folded protein at either the N- or the C-terminus of the RTX domain not only does not prevent the RTX domain to adopt its native β-roll conformation in the presence of calcium, but also provides entropic stabilization. Although the effects exerted on the globular part by the unstructured moiety were not investigated in detail, when fluorescent proteins were used as the fusion partners, no specific impact on the fluorescence emission or behavior was observed.
Fusions with unstructured photoreceptors
In another similar study, the functional impact of GFP N-terminally or C-terminally added to Arabidopsis thaliana cryptochrome 2 (CRY2) was investigated (see Fig. 4).81 Cryptochromes are blue-light photoreceptors that regulate a variety of responses such as growth and circadian rhythms in organisms ranging from bacteria to humans. A. thalianaCRY2 mediates photoperiodic promotion of floral initiation and blue light inhibition of hypocotyl elongation. Cryptochromes have two domains, an N-terminal PHR (photolyase homologous region) domain that is responsible for photon absorption, and a C-terminal effector domain. The closely related A. thalianaCRY1 was shown to consist of a structured N-terminal region and of a C-terminal domain that is intrinsically disordered.82 The C-terminal domain of CRY1 was shown to undergo a light-dependent conformational change resulting in its disengagement from the N-terminal PHR domain thereby leading to activation.82 While the biochemical and physiological activities of GFP–CRY2 were light-dependent similarly to those of the endogenous CRY2, CRY2–GFP was constitutively activated. In particular, CRY2–GFP was found to be constitutively phosphorylated, to promote detiolation both in the dark and in the light, and to activate floral initiation in both long-day and short-day photoperiods. The authors proposed that the attachment of GFP to the C-terminal domain of the cryptochrome may cause its partial disengagement from PHR, resulting in phosphorylation and activation of the photoreceptor in the absence of blue light. The observation that attachment of GFP to the N terminus of cryptochrome did not elicit loss of light responsiveness suggested that the constitutive activity of CRY2–GFP is most likely due to a conformational change rather than to the GFP sequence per se.81 In addition to differences in their regulation, GFP–CRY2 and CRY2–GFP also showed distinct activity in developing nuclear bodies in response to blue light. In fact, CRY2–GFP, like CRY2, but contrary to GFP–CRY2, formed nuclear bodies in response to blue light. The authors hypothesized that the different propensity of the different CRY2 proteins to form nuclear bodies could be due to their different stabilities in blue light. In fact, CRY2–GFP degradation in response to blue light was significantly delayed as compared with that of GFP–CRY2 or of the endogenous CRY2, suggesting that the deposition of nuclear bodies may result from accumulation of photoexcited CRY2–GFP. Consistent with this interpretation, both GFP–CRY2 and endogenous CRY2 were shown to build nuclear bodies in the presence of inhibitors of the 26S proteasome that block blue light-dependent CRY2 degradation.81 | ||
| Fig. 4 Models depicting light-dependent conformational change of CRY2, GFP–CRY2 and CRY2–GFP. The structure of the PHR domain of the related A. thalianaCRY1 is shown (pdb code 1U3C).85 The disordered C-terminal domain of CRY2 has been drawn arbitrarily. The structure of GFP (pdb code 3GJ2)83 is shown in green. The red squares depict phosphorylated residues of the CRY2 C-terminal domain. Modified from ref. 81. | ||
Taken together these results indicated that while addition of GFP to the N-terminus of the structured domain does not alter the function of CRY2, its fusion to the C-terminal disordered domain abolishes the ability of CRY2 to undergo light-dependent conformational changes and forces the protein to adopt a constitutively activated conformation. In addition, the delay detected in the degradation of CRY2–GFP points out a protective role of bound GFP towards proteolysis. This effect might be accounted for by a partial folding of the disordered domain induced by contacts established with GFP.
Conclusions
The few studies available on the conformational and functional impact of fusions between globular fluorescent proteins and disordered domains undergoing folding upon binding to a partner/ligand or following light-induced activation have pointed out a variety of effects.In the case of fusions with viral proteins, the conformational properties observed in vitro were found to at least partly correlate with the behavior observed in vivo for the constructs bearing GFP. In in vitro studies, the GFP was C-terminally fused to the disordered domains of the N and P viral proteins. The fusion proteins were shown to differ in their overall conformation and compactness depending on the fused IDP and to exhibit unique features not accounted for by the properties of the unstructured moieties in isolation. Conformational effects appeared to be subtle and to spread over the whole polypeptide chain, since neither the biological activity and stability of GFP were compromised by fusion with the disordered moiety, nor the latter was significantly more resistant to proteolysis in the context of the chimera. Since in this study only the overall conformational properties of the fusion proteins were addressed, a possible impact of the fusion on the function of the IDP, such as its binding and (induced) folding abilities, could not be ruled out. Some hints on this issue could be obtained from studies focused on viruses containing N and P proteins covalently linked at either terminus by GFP. In those studies, only the GFP-P protein was found to be functionally equivalent to the unmodified protein and GFP seemingly did not interfere with function. Lack of stability and/or function for the other constructs was accounted for by hindrance of specific protein–protein interactions crucial for transcription and replication of the viral genome. In these chimeras the conformation of the folded domain appeared relatively unaffected by the fusion, as judged from fluorescent emission and biochemical studies. As a consequence, the peculiar conformational properties of the fusion proteins were mainly attributed to the disordered parts that, on the other hand, were hypothesized to undergo subtle and delocalized (i.e. inter-dispersed) rearrangements.
In the toxin repeats, fusion of fluorescent proteins at both extremities did not prevent calcium-induced folding, and the presence of the globular proteins functionally replaced the natural RTX flanking sequences that are strictly necessary for calcium responsiveness. The lack of any structural requirement for the domain fused downstream the RTX was interpreted by the authors as linked to a reduction of the entropy of folding. Also in these studies, the folded domain remained unaffected upon fusion, though this aspect was not analyzed in detail. Major effects were observed on the disordered part but they were ascribed to thermodynamic aspects of calcium-induced folding.
Likewise, in the case of the photoreceptor protein, different effects were observed depending on whether the fluorescent protein was bound to the N-terminal region (the structured part of the protein) or to the C-terminus, the unstructured part that undergoes a light-dependent conformational change. While the first construct behaved very similarly to the wild type protein, the latter was constitutively activated and formed nuclear bodies as an effect of higher stability.
Examples available in the literature are still too scarce to try to generalize the mutual effects of ordered and disordered parts within fusion proteins. However, data available so far suggest that the nature (either structured or unstructured) of the two moieties is globally maintained in the fusion, although some subtle, local structural rearrangements can take place.
On the other hand, if perturbations in the emission properties of the fluorescent proteins were not reported, the disordered moiety appeared to be more affected by the presence of a linked folded domain, though this effect might be rather non-specific, i.e. it may result from the presence of a globular domain regardless of its specific scaffold.
The so far available studies point out that in spite of the relative tolerance of IDPs, the addition of GFP may have a strong impact on the binding abilities of the IDP and/or may lead to constitutive activation. As such, fusions with fluorescent proteins can exert a non-negligible impact in vivo. On the other hand, as only little structural perturbations are observed in the disordered moiety, fusions with GFP remain a valuable approach in in vitro studies aimed at assessing the conformational properties of IDPs. This is for instance the case of (single-molecule) electron microscopy and atomic force microscopy studies that make wide use of GFP fusions and that are expected to provide invaluable insights into the conformational properties of IDPs.
Acknowledgements
We would like to thank Ilaria Sambi and Pietro Gatti-Lafranconi for their contribution to the studies focused on the NTAIL–GFP and PNT–GFP constructs. The studies focused on PNT and NTAIL fusions with GFP herein discussed have been supported by a grant from the University Milano-Bicocca (Fondo di Ateneo per la Ricerca) to M.L. and by grants of the Agence Nationale de la Recherche, specific programs “Microbiologie et Immunologie” (ANR-05-MIIM-035-02) and “Physico-chimie du vivant” (ANR-08-PCVI-0020-01) to S.L.References
- P. Radivojac, L. M. Iakoucheva, C. J. Oldfield, Z. Obradovic, V. N. Uversky and A. K. Dunker, Biophys. J., 2007, 92, 1439–1456 CrossRef CAS.
- A. K. Dunker, C. J. Oldfield, J. Meng, P. Romero, J. Y. Yang, J. W. Chen, V. Vacic, Z. Obradovic and V. N. Uversky, BMC Genomics, 2008, 9((Suppl 2)), S1 CrossRef.
- A. K. Dunker, I. Silman, V. N. Uversky and J. L. Sussman, Curr. Opin. Struct. Biol., 2008, 18, 756–764 CrossRef CAS.
- P. E. Wright and H. J. Dyson, Curr. Opin. Struct. Biol., 2009, 19, 31–38 CrossRef CAS.
- V. N. Uversky, J. Biomed. Biotechnol., 2010, 2010, 568068 CrossRef.
- V. N. Uversky and A. K. Dunker, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1231–1264 CrossRef CAS.
- K. K. Turoverov, I. M. Kuznetsova and V. N. Uversky, Prog. Biophys. Mol. Biol., 2010, 102, 73–84 CrossRef CAS.
- V. N. Uversky, Int. J. Biochem. Cell Biol., 2011, 43, 1090–1103 CrossRef CAS.
- P. Tompa, Structure and Function of Intrinsically Disordered Proteins, CRC Press, Taylor & Francis Group, Boca Raton, Florida, 2010 Search PubMed.
- P. Tompa, Curr. Opin. Struct. Biol., 2011, 21, 419–425 CrossRef CAS.
- V. Receveur-Bréchot, J. M. Bourhis, V. N. Uversky, B. Canard and S. Longhi, Proteins: Struct., Funct., Bioinf., 2006, 62, 24–45 CrossRef.
- V. N. Uversky and S. Longhi, Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation, John Wiley and Sons, Hoboken, New Jersey, 2010 Search PubMed.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, Annu. Rev. Biophys., 2008, 37, 215–246 CrossRef CAS.
- V. N. Uversky, C. J. Oldfield, U. Midic, H. Xie, B. Xue, S. Vucetic, L. M. Iakoucheva, Z. Obradovic and A. K. Dunker, BMC Genomics, 2009, 10(Suppl 1), S7 CrossRef.
- U. Midic, C. J. Oldfield, A. K. Dunker, Z. Obradovic and V. N. Uversky, BMC Genomics, 2009, 10(Suppl 1), S12 CrossRef.
- V. N. Uversky, Expert Rev. Proteomics, 2010, 7, 543–564 CrossRef CAS.
- T. Vavouri, J. I. Semple, R. Garcia-Verdugo and B. Lehner, Cell, 2009, 138, 198–208 CrossRef CAS.
- J. Gsponer, M. E. Futschik, S. A. Teichmann and M. M. Babu, Science, 2008, 322, 1365–1368 CrossRef CAS.
- C. J. Oldfield, Y. Cheng, M. S. Cortese, C. J. Brown, V. N. Uversky and A. K. Dunker, Biochemistry, 2005, 44, 1989–2000 CrossRef CAS.
- L. M. Iakoucheva, C. J. Brown, J. D. Lawson, Z. Obradovic and A. K. Dunker, J. Mol. Biol., 2002, 323, 573–584 CrossRef CAS.
- N. Tokuriki, C. J. Oldfield, V. N. Uversky, I. N. Berezovsky and D. S. Tawfik, Trends Biochem. Sci., 2009, 34, 53–59 CrossRef CAS.
- B. Xue, R. W. Williams, C. J. Oldfield, G. K. Goh, A. K. Dunker and V. N. Uversky, Protein Pept. Lett., 2010, 17, 932–951 CrossRef CAS.
- V. N. Uversky and S. Longhi, Flexible Viruses: Structural Disorder in viral Proteins, John Wiley and Sons, Hoboken, New Jersey 2011 Search PubMed.
- J. Habchi, L. Mamelli, H. Darbon and S. Longhi, PLoS One, 2010, 5, e11684 Search PubMed.
- J. Habchi, L. Mamelli and S. Longhi, Structural disorder within the nucleoprotein and phosphoprotein from measles, Nipah and Hendra viruses, in Flexible Viruses Structural Disorder in viral Proteins, ed. V. N. Uversky and S. Longhi, John Wiley and Sons, Hoboken, New Jersey, 2011, , in press Search PubMed.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS.
- A. K. Dunker and Z. Obradovic, Nat. Biotechnol., 2001, 19, 805–806 CrossRef CAS.
- V. N. Uversky, Protein Sci., 2002, 11, 739–756 CrossRef CAS.
- V. N. Uversky, Cell. Mol. Life Sci., 2003, 60, 1852–1871 CrossRef CAS.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS.
- T. Mittag, S. Orlicky, W. Y. Choy, X. Tang, H. Lin, F. Sicheri, L. E. Kay, M. Tyers and J. D. Forman-Kay, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17772–17777 CrossRef CAS.
- S. Brocca, M. Samalikova, V. N. Uversky, M. Lotti, M. Vanoni, L. Alberghina and R. Grandori, Proteins: Struct., Funct., Bioinf., 2009, 76, 731–746 CrossRef CAS.
- T. Mittag, J. Marsh, A. Grishaev, S. Orlicky, H. Lin, F. Sicheri, M. Tyers and J. D. Forman-Kay, Structure (London), 2010, 18, 494–506 CAS.
- S. Brocca, L. Testa, F. Sobott, M. Samalikova, A. Natalello, E. Papaleo, M. Lotti, L. De Gioia, S. M. Doglia, L. Alberghina and R. Grandori, Biophys. J., 2011, 100, 2243–2252 CrossRef CAS.
- C. Leyrat, M. R. Jensen, E. A. Ribeiro, Jr., F. C. Gerard, R. W. Ruigrok, M. Blackledge and M. Jamin, Protein Sci., 2011, 20, 542–556 CrossRef CAS.
- D. Karlin, S. Longhi, V. Receveur and B. Canard, Virology, 2002, 296, 251–262 CrossRef CAS.
- K. Johansson, J. M. Bourhis, V. Campanacci, C. Cambillau, B. Canard and S. Longhi, J. Biol. Chem., 2003, 278, 44567–44573 CrossRef CAS.
- S. Longhi, V. Receveur-Brechot, D. Karlin, K. Johansson, H. Darbon, D. Bhella, R. Yeo, S. Finet and B. Canard, J. Biol. Chem., 2003, 278, 18638–18648 CrossRef CAS.
- D. Karlin, F. Ferron, B. Canard and S. Longhi, J. Gen. Virol., 2003, 84, 3239–3252 CrossRef CAS.
- S. Longhi, Curr. Top. Microbiol. Immunol., 2009, 329, 103–128 CrossRef CAS.
- S. Longhi and M. Oglesbee, Protein Pept. Lett., 2010, 17, 961–978 CrossRef CAS.
- N. Tarbouriech, J. Curran, C. Ebel, R. W. Ruigrok and W. P. Burmeister, Virology, 2000, 266, 99–109 CrossRef CAS.
- N. Tarbouriech, J. Curran, R. W. Ruigrok and W. P. Burmeister, Nat. Struct. Biol., 2000, 7, 777–781 CrossRef CAS.
- D. Marion, N. Tarbouriech, R. W. Ruigrok, W. P. Burmeister and L. Blanchard, J. Biomol. NMR, 2001, 21, 75–76 CrossRef CAS.
- L. Blanchard, N. Tarbouriech, M. Blackledge, P. Timmins, W. P. Burmeister, R. W. Ruigrok and D. Marion, Virology, 2004, 319, 201–211 CrossRef CAS.
- K. Houben, D. Marion, N. Tarbouriech, R. W. Ruigrok and L. Blanchard, J. Virol., 2007, 81, 6807–6816 CrossRef CAS.
- M. R. Jensen, P. Bernado, K. Houben, L. Blanchard, D. Marion, R. W. Ruigrok and M. Blackledge, Protein Pept. Lett., 2010, 17, 952–960 CrossRef CAS.
- R. Assenberg, O. Delmas, B. Morin, S. C. Graham, X. De Lamballerie, C. Laubert, B. Coutard, J. M. Grimes, J. Neyts, R. J. Owens, B. W. Brandt, A. Gorbalenya, P. Tucker, D. I. Stuart, B. Canard and H. Bourhy, Antiviral Res., 2010, 87, 149–161 CrossRef CAS.
- F. C. Gerard, A. Ribeiro Ede, Jr., C. Leyrat, I. Ivanov, D. Blondel, S. Longhi, R. W. Ruigrok and M. Jamin, J. Mol. Biol., 2009, 388, 978–996 CrossRef CAS.
- I. Ivanov, T. Crepin, M. Jamin and R. W. Ruigrok, J. Virol., 2010, 84, 3707–3710 CrossRef CAS.
- E. A. Ribeiro, Jr., A. Favier, F. C. Gerard, C. Leyrat, B. Brutscher, D. Blondel, R. W. Ruigrok, M. Blackledge and M. Jamin, J. Mol. Biol., 2008, 382, 525–538 CrossRef.
- E. D. Ribeiro, Jr., C. Leyrat, F. C. Gerard, A. A. Albertini, C. Falk, R. W. Ruigrok and M. Jamin, J. Mol. Biol., 2009, 394, 558–575 CrossRef.
- F. Durand, A. Dagkessamanskaia, H. Martin-Yken, M. Graille, H. Van Tilbeurgh, V. N. Uversky and J. M. Francois, Yeast, 2008, 25, 563–576 CrossRef CAS.
- A. Dagkessamanskaia, F. Durand, V. N. Uversky, M. Binda, F. Lopez, K. El Azzouzi, J. M. Francois and H. Martin-Yken, Protein Sci., 2010, 19, 1376–1385 CrossRef CAS.
- Y. Yu, H. Y. Wang, M. Bai and S. Perrett, Biochem. J., 2011, 434, 143–151 CrossRef CAS.
- H. Li, A. F. Oberhauser, S. D. Redick, M. Carrion-Vazquez, H. P. Erickson and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10682–10686 CrossRef CAS.
- T. Ohashi, C. A. Hale, P. A. de Boer and H. P. Erickson, J. Bacteriol., 2002, 184, 4313–4315 CrossRef CAS.
- S. H. Bae, H. J. Dyson and P. E. Wright, J. Am. Chem. Soc., 2009, 131, 6814–6821 CrossRef CAS.
- S. Longhi, Measles Virus Nucleoprotein, Nova Publishers Inc., Hauppage, NY, 2007 Search PubMed.
- D. Karlin, S. Longhi and B. Canard, Virology, 2002, 302, 420–432 CrossRef CAS.
- J. Bourhis, K. Johansson, V. Receveur-Bréchot, C. J. Oldfield, A. K. Dunker, B. Canard and S. Longhi, Virus Res., 2004, 99, 157–167 CrossRef CAS.
- I. Sambi, P. Gatti-Lafranconi, S. Longhi and M. Lotti, FEBS J., 2010, 277, 4438–4451 CrossRef CAS.
- J. M. Bourhis, V. Receveur-Bréchot, M. Oglesbee, X. Zhang, M. Buccellato, H. Darbon, B. Canard, S. Finet and S. Longhi, Protein Sci., 2005, 14, 1975–1992 CrossRef CAS.
- B. Morin, J. M. Bourhis, V. Belle, M. Woudstra, F. Carrière, B. Guigliarelli, A. Fournel and S. Longhi, J. Phys. Chem. B, 2006, 110, 20596–20608 CrossRef CAS.
- V. Belle, S. Rouger, S. Costanzo, E. Liquiere, J. Strancar, B. Guigliarelli, A. Fournel and S. Longhi, Proteins: Struct., Funct., Bioinf., 2008, 73, 973–988 CrossRef CAS.
- S. Gely, D. F. Lowry, C. Bernard, M. Ringkjobing-Jensen, M. Blackledge, S. Costanzo, H. Darbon, G. W. Daughdrill and S. Longhi, J. Mol. Recognit., 2010, 23, 435–447 CrossRef CAS.
- A. Kavalenka, I. Urbancic, V. Belle, S. Rouger, S. Costanzo, S. Kure, A. Fournel, S. Longhi, B. Guigliarelli and J. Strancar, Biophys. J., 2010, 98, 1055–1064 CrossRef CAS.
- M. Ringkjøbing Jensen, G. Communie, E. D. Ribeiro, Jr., N. Martinez, A. Desfosses, L. Salmon, L. Mollica, F. Gabel, M. Jamin, S. Longhi, R. W. Ruigrok and M. Blackledge, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9839–9844 CrossRef.
- P. Devaux and R. Cattaneo, J. Virol., 2004, 78, 11632–11640 CrossRef CAS.
- R. L. Kingston, D. J. Hamel, L. S. Gay, F. W. Dahlquist and B. W. Matthews, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8301–8306 CrossRef CAS.
- T. Ohashi, S. D. Galiacy, G. Briscoe and H. P. Erickson, Protein Sci., 2007, 16, 1429–1438 CrossRef CAS.
- G. R. Szilvay, M. A. Blenner, O. Shur, D. M. Cropek and S. Banta, Biochemistry, 2009, 48, 11273–11282 CrossRef CAS.
- M. A. Blenner, O. Shur, G. R. Szilvay, D. M. Cropek and S. Banta, J. Mol. Biol., 2010, 400, 244–256 CrossRef CAS.
- U. Baumann, S. Wu, K. M. Flaherty and D. B. McKay, EMBO J., 1993, 12, 3357–3364 CAS.
- U. Baumann, J. Mol. Biol., 1994, 242, 244–251 CrossRef CAS.
- C. Bauche, A. Chenal, O. Knapp, C. Bodenreider, R. Benz, A. Chaffotte and D. Ladant, J. Biol. Chem., 2006, 281, 16914–16926 CrossRef CAS.
- C. Angkawidjaja, D. J. You, H. Matsumura, K. Kuwahara, Y. Koga, K. Takano and S. Kanaya, FEBS Lett., 2007, 581, 5060–5064 CrossRef CAS.
- A. Chenal, J. I. Guijarro, B. Raynal, M. Delepierre and D. Ladant, J. Biol. Chem., 2009, 284, 1781–1789 CrossRef CAS.
- P. Ringler and G. E. Schulz, Science, 2003, 302, 106–109 CrossRef CAS.
- H. Lilie, W. Haehnel, R. Rudolph and U. Baumann, FEBS Lett., 2000, 470, 173–177 CrossRef CAS.
- X. Yu, R. Sayegh, M. Maymon, K. Warpeha, J. Klejnot, H. Yang, J. Huang, J. Lee, L. Kaufman and C. Lin, Plant Cell, 2009, 21, 118–130 CrossRef CAS.
- C. L. Partch, M. W. Clarkson, S. Ozgur, A. L. Lee and A. Sancar, Biochemistry, 2005, 44, 3795–3805 CrossRef CAS.
- J. N. Henderson, R. Gepshtein, J. R. Heenan, K. Kallio, D. Huppert and S. J. Remington, J. Am. Chem. Soc., 2009, 131, 4176–4177 CrossRef CAS.
- Y. Zhang, X. Gao and R. Michael Garavito, Biochem. Biophys. Res. Commun., 2011, 407, 674–679 CrossRef CAS.
- C. A. Brautigam, B. S. Smith, Z. Ma, M. Palnitkar, D. R. Tomchick, M. Machius and J. Deisenhofer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12142–12147 CrossRef CAS.
Footnote |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M Madan Babu. |
| This journal is © The Royal Society of Chemistry 2012 |