
Mutual effects of disorder and order in fusion proteins between intrinsically disordered domains and fluorescent proteins†
Abstract
Intrinsically disordered
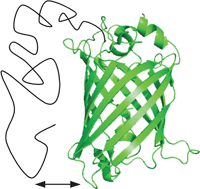
- This article is part of the themed collection: Intrinsically Disordered Proteins
 Please wait while we load your content...
Please wait while we load your content...