Insights in the (un)structural organization of Bacillus pasteurii UreG, an intrinsically disordered GTPase enzyme†‡
Barbara
Zambelli§
*a,
Nunilo
Cremades§
b,
Paolo
Neyroz
c,
Paola
Turano
de,
Vladimir N.
Uversky
fg and
Stefano
Ciurli
ad
aLaboratory of Bioinorganic Chemistry, University of Bologna, Via Fanin 40, Bologna, Italy. E-mail: barbara.zambelli@unibo.it; Fax: +39 051 209 6203; Tel: +39 051 209 6233
bDepartment of Chemistry, University of Cambridge, Lensfield Rd., CB21EW Cambridge, UK
cDepartment of Biochemistry “G. Moruzzi”, University of Bologna, Rimini, Italy
dMagnetic Resonance Center, University of Florence, Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy
eDepartment of Chemistry, University of Florence, Via della Lastruccia, 3, 50019 Sesto Fiorentino, Italy
fDepartment of Molecular Medicine, University of South Florida, 12901 Bruce B. Downs Blvd., MDC07, Tampa, USA
gInstitute for Biological Instrumentation, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia
First published on 16th September 2011
Abstract
In the past, enzymatic activity has always been expected to be dependent on overall protein rigidity, necessary for substrate recognition and optimal orientation. However, increasing evidence is now accumulating, revealing that some proteins characterized by intrinsic disorder are actually able to perform catalysis. Among them, the only known natural intrinsically disordered enzyme is UreG, a GTPase that, in plants and bacteria, is involved in the protein interaction network leading to Ni2+ ions delivery into the active site of urease. In this paper, we report a detailed analysis of the unfolding behaviour of UreG from Bacillus pasteurii (BpUreG), following its thermal and chemical denaturation with a combination of fluorescence spectroscopy, calorimetry, CD and NMR. The results demonstrate that BpUreG exists as an ensemble of inter-converting conformations, whose degrees of secondary structure depend on temperature and denaturant concentration. In particular, three major types of conformational ensembles with different degrees of residual structure were identified, with major structural characteristics resembling those of a molten globule (low temperature, absence of denaturant), pre-molten globule (high temperature, absence or presence of denaturant) and random coil (low temperature, presence of denaturant). Transitions among these ensembles of conformational states occur non-cooperatively although reversibly, with a gradual loss or acquisition of residual structure depending on the conditions. A possible role of disorder in the biological function of UreG is envisaged and discussed.
Introduction
The lack of a well-defined tertiary structure in many functional proteins is one of the most exciting discoveries of structural biology in the last decade.1–3 This concept led to rethink the sequence–structure–function paradigm, changing our perception of protein folding from a static view to a dynamic view. Generally, intrinsically disordered proteins (IDP) exert their function by interacting with specific partners, either small cofactors or other proteins, which often prompt a conformational transition of IDP to a more structured state.4,5 These interactions are usually promoted by a significant residual structure in the disordered proteins, whose degree can range from the molten globule-like state (collapsed disorder), with preserved secondary structural elements lacking a rigid orientation in the 3D space, to the random coil-like conformation (extended disorder).6While protein intrinsic disorder is well known to play a functional role in regulation of biological activities both in prokaryotes and eukaryotes, enzymatic activity has always been associated to overall protein rigidity. In particular, the first model for enzyme–substrate interaction involved a rigid “lock and key” mechanism, where the substrate conformation perfectly fits the catalytic site.7 This theory was subsequently extended to take into account the protein dynamics occurring during catalysis, with the “induced fit” model, in which structural rearrangement of the active site toward an optimal catalytic conformation is driven by substrate binding.8 Recently, the occurrence of a disorder-mediated induced fit mechanism has been proposed for a natural enzyme, the E. coliadenylate kinase, where the occurrence of an order-disorder-order transition, involving one domain of an otherwise folded protein, likely occurs upon substrate binding to the active site.9
Increasing evidence is now accumulating suggesting that disordered proteins are able to function as enzymatic catalysts. A folding intermediate of a mutant of ribonuclease T1, containing an extensive secondary structure but only partial packing of the hydrophobic core, showed ca. 40% of enzymatic activity with respect to the native folded protein,10 while two molten globular permutants of dihydrofolate reductase from E. coli retained 1–6% of enzymatic activity.11 Similarly, a double mutant of staphylococcal nuclease12 and the topologically redesigned Methanococcus jannaschiichorismate mutase,13 both existing in a molten globule-like fluxional behaviour in their apo-form, undergo structural reorganization and disorder-to-order transition upon substrate binding, in a mechanism that can be considered a further extension of the classical induced-fit model.12,14 The global catalytic efficiency of these proteins was not significantly affected by the disordered behaviour, remaining comparable to the wild-type levels. Disorder was found to accelerate the substrate binding to the molten globular chorismate mutase, in a step accompanied by a significant enthalpic gain compensating the unfavourable entropic term linked to the protein structural reorganization.15 A similar effect was observed for two mutants of the E. coliadenylate kinase, whose substrate binding domain has been structurally destabilized after mutations.9 Finally, deletion of one to seven residues in the C-terminal sequence of the capsid protein of the Semliki Forest virus produced an intrinsically disordered enzyme with a serine protease activity comparable to the structured wild-type protein. In this case, the folding upon substrate binding was not experimentally demonstrated.16
All these observations suggest that protein disorder plays a favourable role for substrate binding in artificially mutated enzymes that undergo global structural changes to accommodate the substrate in an optimal position for catalysis. On the other hand, the possibility that intrinsic disorder plays a role in catalysis is still questioned.17 The only known example of a possible role of disorder in a catalytic mechanism is represented by the transient molten globule-like folding intermediate of Sulfolobus solfataricusacylphosphatase, which features about 80% of the native-like catalytic activity despite the absence of a rigid tertiary structure. This activity is possibly driven by the close proximity of the catalytic residues, despite the structural heterogeneity of the active site.18 Binding of the substrate does not accelerate the conversion toward the native state, suggesting that the substrate induces a local rearrangement of the active site rather than a complete folding process.18 While these examples indicate that artificially created or induced protein disorder is compatible with catalytic activity, the only naturally occurring enzyme showing an intrinsically disordered conformation is UreG. This protein is a GTPase involved in the assembly of the Ni2+-dependent active site of urease,19,20 and proposed to catalyze, in the presence of CO2, the formation of carboxyphosphate, a carbamylation agent for the metal-binding lysine in the urease active site.21NMR data on Bacillus pasteurii UreG (BpUreG) revealed the presence of large conformational fluidity, while far-UV circular dichroism showed that this disordered state coexists with a significant secondary structure.22 This feature is conserved among UreG orthologs, such as the proteins from Klebsiella aerogenes (Ka),23Mycobacterium tuberculosis (Mt),24 and Helicobacter pylori (Hp).25 The folding behaviour of BpUreG at 25 °C was characterized using fluorescence spectroscopy experiments conducted in the absence and in the presence of chemical denaturant, supporting the hypothesis that BpUreG exists in solution in a stable flexible state, energetically similar to a molten globule.26
In their intrinsically disordered native state, BpUreG and MtUreG showed some enzymatic activity in vitro, which, even though small, resulted comparable to that reported for other homologues of GTPases.22,24BpUreG and HpUreG specifically bind Zn2+ ions, and this interaction was proposed to be functional for the protein interplay in the urease interaction network.22,25 A Zn2+-stabilized interaction of HpUreG with its cognate HpUreE, the chaperone generally considered responsible for Ni2+ ions delivery into the urease active site, was detected both in vitro and in vivo.27,28 A similar interaction was also reported for the KaUreG–KaUreE complex formation, enhanced by Ni2+ and Zn2+ ions.29
In vitro, under near-physiological conditions, addition of substrate analogues, metal ion cofactors, or UreE was not sufficient to induce the transition of UreG toward the completely structured form,26,27 indicating that some other interactions, possibly with additional urease chaperones, would be necessary to bring the protein to its fully active state in vivo. Theoretical structural models of the fully folded state of UreG revealed the possible catalytically active folded state of the protein.22,24,25
In this paper, we couple thermal and chemical denaturation to provide insights into the structural organization and stability of BpUreG. Using a combination of fluorescence spectroscopy, calorimetry, CD and NMR we provide evidence that the protein exists as an ensemble of inter-converting conformations containing different degrees of residual secondary and tertiary structures. The change in temperature and the presence of denaturant are able to shift the equilibrium toward differently structured ensembles through non-cooperative transitions.
Results and discussion
Intrinsic fluorescence spectroscopy monitors chemically-induced protein unfolding at different temperatures
Previously, intrinsic fluorescence spectroscopy was used to characterize the folding behaviour of BpUreG by taking advantage of the single tryptophan residue (W192) located in a predicted α-helix at the C-terminus.26 At 24 °C and in the presence of increasing concentrations of GuHCl denaturant, the wavelength of the maximum of the fluorescence spectrum (λmax) was progressively red-shifted, indicating a change of W192 from a buried environment to a more solvent exposed position upon protein unfolding.26 The resulting denaturation curve showed an apparent sigmoidal profile typical of a transition between two states. The fit of the data, performed under this assumption, provided a low value for the free energy of unfolding (ΔG0,un ≈ 1.5 kcal mol−1), suggesting the occurrence of a small molecular rearrangement, rather than a disruption of the structure of a well-folded protein, which normally presents much higher values for the free energy of unfolding.26 This atypical value was interpreted in terms of a non-completely folded structure of native BpUreG, a conclusion supported by the observation that small amounts of denaturant ([GuHCl] < 0.1 M) induce maximal exposure of the hydrophobic regions of the protein, as detected by protein binding to the fluorescent dye bis-8-anilino-1-naphthalene sulfonate (bis-ANS).26To confirm this behaviour and to further explore the structural stability of BpUreG, we performed the same GuHCl-induced unfolding analysis at different temperatures (Fig. 1). Under native conditions, the maximum of the fluorescence intensity gradually shifts toward the red part of the spectrum while the temperature increases (Fig. 1A). This is consistent with an increase in the solvent exposure of W192 at higher temperatures, suggestive of a thermal-induced protein unfolding. This transition does not reach a plateau at the highest temperature sampled (64 °C), indicating that the structural rearrangement is not complete and that the protein structure is resilient to thermal denaturation.
 | ||
| Fig. 1 Thermal and chemically-induced conformational transitions, followed by intrinsic fluorescence. (A) Thermal denaturation of BpUreG followed by the red-shift of the maximum of the intrinsic fluorescence spectra (λmax). (B) Representative emission spectrum of BpUreG at 25 °C and in the presence of different concentrations of GuHCl. The arrow indicates the red-shift of the fluorescence maximum upon protein unfolding. (C) Unfolding curves of BpUreG at different temperatures, monitored as changes in λmax as a function of denaturant concentration. The solid lines represent the fit of the data under the assumption of a two-state transition. (D) Stabilization curve of BpUreG. Each point represents ΔG0,un determined from an isothermal GuHCl-denaturation experiment. The solid line represents the fit of the experimental data to the equation described in the text. The melting temperature (Tm), corresponding to ΔG0,un = 0, and the temperature of maximal stability (Ts), corresponding to ΔS = 0, are indicated. | ||
A similar red shift of the λmax, coupled to the decrease of the fluorescence intensity, is observed when BpUreG is incubated with increasing concentrations of denaturant (Fig. 1B). All GuHCl-induced protein unfolding curves (Fig. 1C) were fitted to a two-state transition model, in analogy with the analysis previously reported,26 yielding relatively low ΔG0,un values in the 10–50 °C temperature range (Table S1, ESI‡). The stability of the protein was evaluated by plotting ΔG0,un as a function of temperature (Fig. 1D) and fitting the data to the protein stabilization curve (see Experimental).30 The fitting parameters obtained from this analysis are ΔHm = 22.8 kcal mol−1, Tm = 63.5 °C and ΔCp = 0.66 kcal mol−1 K−1.
Generally, ΔCp for protein unfolding is quite large and positive, and correlates to the unfolding-induced exposure of non-polar groups found in the hydrophobic core of the folded state.31 This parameter is approximately constant for all proteins and usually falls in the range 12–18 cal mol−1 K−1 per residue: therefore, the ΔCp calculated for BpUreG (211 residues), assuming a well-folded state, is 2.5–3.8 kcal mol−1 K−1.30 This value is much larger than determined from our unfolding studies, suggesting that a large portion of the hydrophobic core of the protein is already solvent-exposed in native BpUreG. This is consistent with the small ΔG0,un value (1.1 kcal mol−1) at the temperature of maximal stability (Ts = 30.7 °C).
Thermal denaturation of BpUreG is non-cooperative
The atypical thermodynamic parameters obtained for BpUreG unfolding from fluorescence experiments prompted us to investigate more deeply the folding behaviour of BpUreG using different techniques. Indeed, intrinsic fluorescence based on the single tryptophan in the protein yields only local information on the protein region neighbouring this residue. In order to explore the global properties of the protein, we carried out an unfolding study based on circular dichroism (CD) spectroscopy.The signal in the far-UV CD spectrum is an index of the secondary structure content in proteins. In particular, the CD spectrum of BpUreG at 10 °C shows negative deflections at 206 and 220 nm (Fig. 2A), in analogy with previously reported data for the protein at 25 °C.22 Upon increasing the temperature, the absolute value of the ellipticity decreases, as expected for an unfolding transition. However, even at the highest temperature recorded (90 °C), the protein retains a considerable amount of secondary structure, as the ellipticity at around 222 nm remains significantly negative, in contrast to a weak positive signal expected for a completely random coil conformation (Fig. 2A). Moreover, in these conditions, the CD signal is more pronounced than the one observed for the chemically-denatured BpUreG at 25 °C,26 therefore implying that the temperature-denatured protein does not reach the highest degree of disorder. The maintenance of the secondary structure in this state suggests that BpUreG is rather thermostable, confirming the fluorescence data obtained for BpUreG under native conditions and at different temperatures (Fig. 1A).
 | ||
| Fig. 2 Temperature-induced conformational transitions of BpUreG in the absence and presence of denaturant, followed by CD. (A) Far-UV CD spectra of BpUreG at 10 °C and 90 °C, in the absence and in the presence of 2.5 M GuHCl or 5 M urea. For the native protein, filled circles represent the apo-BpUreG, while crosses, empty squares and empty circles represent Zn-BpUreG, Ni-BpUreG and GTPγS-BpUreG respectively. (B) Changes in ellipticity at 222 nm in the 10 °C–90 °C temperature range for BpUreG, in the absence and in the presence of 2.5 M GuHCl and cofactors. | ||
The thermally-induced protein denaturation curve of BpUreG in native conditions was recorded by monitoring the ellipticity signal at 222 nm (Fig. 2B). The signal initially decreases to more negative values from 10 to 25 °C, indicating that the secondary structure content of the protein is maximum at 25 °C due to cold-denaturation at low temperatures. Subsequently, the ellipticity gradually changes to less negative values with increasing temperature, following a behaviour typical of non-cooperative thermal transition. The thermal denaturation of the protein is reversible (Fig. 2A) and independent of the presence or absence of tris(2-carboxyethyl)phosphine hydrochloride (TCEP), a reducing agent (Fig. S1, ESI‡). This indicates that the transition observed does not depend on the oxidation state of the single cysteine residue in BpUreG monomer, previously reported to be involved in an inter-monomeric disulfide bridge in the native BpUreG dimer.22 The thermal denaturation observed by CD is paralleled by the gradual increase of the fluorescence maximum in the 10–64 °C temperature range (Fig. 1A), which also suggests the lack of cooperativity for thermal-induced protein unfolding.
The shapes of the CD spectra (Fig. 2A) and of the protein denaturation curve (Fig. 2B) do not change significantly upon addition of excess of protein cofactors such as Ni2+, Zn2+, and the non-hydrolysable GTP analogue GTPγS (Fig. 2). This observation is consistent with previously reported data indicating that, at 25 °C, both the secondary structure of the protein, followed by CD, and the tertiary structure, followed by NMR, are insensitive to the presence of metal ions or substrate analogues.22,26
In order to further investigate the nature of this unfolding transition and to test its cooperativity, we performed differential scanning calorimetry (DSC) experiments. Usually, in the DSC experiments on melting of a well-ordered globular protein, a characteristic peak in the temperature profile of the heat capacity of the system is observed, corresponding to a first-order phase transition from the native to the fully unfolded state of the protein (for a two-state protein). However, in the case of BpUreG, the DSC thermogram shows the presence of a continuous and small increase in heat capacity, without any clear peak, in the 10–90 °C range (Fig. 3). The same result was observed in the presence of Zn2+ (data not shown), further confirming that binding of this cofactor to the protein does not influence its thermodynamic behaviour. This observation is consistent with the continuous loss of secondary structure upon increasing temperature observed by CD, indicating that the temperature-induced disordering of the residual structure of the protein is gradual and non-cooperative. Additionally, this suggests that the compact state of native BpUreG at low temperature does not represent a unique structure, but rather a broad conformational ensemble constituted by a large number of different conformational states. Throughout the gradual increase of heat capacity with temperature, the enthalpy of the system increases, as the distribution of denatured molecules progressively shifts toward the fully unfolded state. These data show that a simple two-state model is not able to describe the unfolding process of BpUreG.
 | ||
| Fig. 3 Temperature dependence of heat capacity of BpUreG (57 μM), followed by DSC in the 10–90 °C temperature range. | ||
The same spectroscopic and calorimetric behaviour has been reported for proteins such as α-lactalbumin32,33 and human lyzozyme.34 In both cases the protein undergoes a cooperative disruption of the native tertiary structure, followed by a non-cooperative transition in the denatured state that is characterized by different degrees of ordered residual structure depending on the temperature.
Chemically denatured BpUreG undergoes non-cooperative cold denaturation
A previous conformational analysis of BpUreG at 25 °C, followed by fluorescence and CD, showed that protein unfolding occurs upon incubation of the protein with concentrations of GuHCl larger than 2.5 M. This GuHCl-induced protein unfolding follows an apparent two-state mechanism.26 As the secondary structure of the protein is very resilient to thermal denaturation, we followed the effect of temperature over the unfolding transition induced by strong denaturants using CD. We found that, as expected, the absolute value of ellipticity in the far-UV region of the CD spectrum drops drastically upon incubation of the protein at low temperature (10 °C) and GuHCl concentrations of 2.5 M (Fig. 2A).On the other hand, the CD spectrum of the protein in the presence of 2.5 M GuHCl is significantly more negative at 90 °C as compared to the signal obtained at 10 °C. Furthermore, the signal in the presence of denaturant at high temperatures is similar to the spectrum of the native protein at the same temperature. These observations indicate that at high temperature and in the presence of denaturant the protein acquires a higher degree of structural order than at low temperatures, and that cold denaturation occurs for the GuHCl-induced disordered protein state. This effect is independent of the type of chemical denaturant used, as it is also observed with 5 M urea (Fig. 2A), therefore excluding the possibility that this event is due to an artefact of the increase of the ionic strength induced by GuHCl.
The ellipticity of the GuHCl-induced denatured state decreases linearly with temperature (Fig. 2B), indicating, also in this case, a non-cooperative transition. This value extrapolates to that of the temperature-induced denatured state in the absence of denaturant (116 °C, Fig. 2B), indicating that, at this temperature, the two denatured states are equivalent.
NMR spectroscopy provides structural evidence of uncooperative transitions
As previously reported, the NMR spectrum of BpUreG at 25 °C in the absence of denaturant is characterized by poorly resolved resonances with little dispersion in the 1H dimension of the backbone amides, most of which fall in the random coil region (7.6–8.5 ppm chemical shift range).22 This behaviour indicates the presence of large protein portions that lack a well-defined secondary and tertiary structure. In addition, observed resonances are extremely broad, suggesting that large portions of the protein backbone undergo exchange phenomena in the intermediate exchange regime. This situation does not change significantly in the temperature range from 10 to 50 °C (Fig. 4A).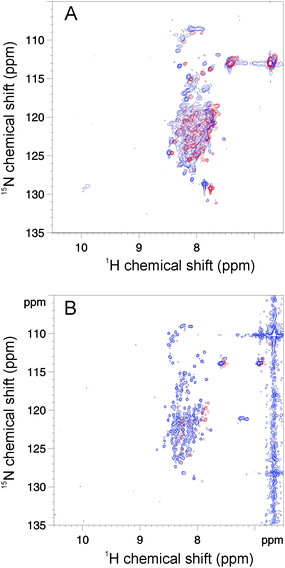 | ||
| Fig. 4 Temperature- and denaturant-induced changes of BpUreG. The 1H–15N HSQC NMR spectra of 15N-BpUreG recorded at 10 °C (blue) and 50 °C (red) are shown in the absence (A) and in the presence (B) of 2.5 M GuHCl. | ||
In the presence of 2.5 M GuHCl as denaturant, the 1H,15N-HSQC spectrum (Fig. 4B) shows about 140 peaks out of the 204 non-proline residues present in the protein primary structure, still in the random coil region, but with reduced signal line width as compared to the corresponding spectrum without denaturant. This observation indicates faster conformational exchange equilibria and/or faster exchange with bulk solvent. Both phenomena point to the absence of any defined structure for most of the protein residues. Increasing the temperature in the presence of 2.5 M GuHCl causes a progressive broadening and disappearing of the peaks, consistent with a transition to a state characterized by slower conformational equilibria. This transition is reversible, thus excluding irreversible protein aggregation with temperature. The volume of non-overlapping backbone signals decreases as a function of increasing temperature in a non-concerted fashion, indicating that the re-folding of the completely denatured state into a more structured state is largely non-cooperative.
Spectroscopic characterization of chemical- and thermal-induced denaturation of BpUreG shows the presence of three different structural states
This study reveals that, under native conditions, BpUreG structure is resilient to thermal unfolding. This apparent stability is not paralleled by an analogous resilience to chemical denaturation, as the protein reaches a fully unfolded conformation upon addition of moderate concentrations of chemical denaturant, such as urea or GuHCl. The chemically unfolded state undergoes a partial refolding upon temperature increase, with a partial formation of the secondary structure.BpUreG exhibits different thermal-dependent behaviours in the absence or presence of chemical denaturants, as the absolute value of the ellipticity decreases with temperature in native conditions, while it increases in the presence of denaturant. Therefore, we characterized in detail the combination of denaturant- and thermal-induced unfolding of the protein, by following GuHCl-induced changes in the CD spectrum at different temperatures (Fig. 5).
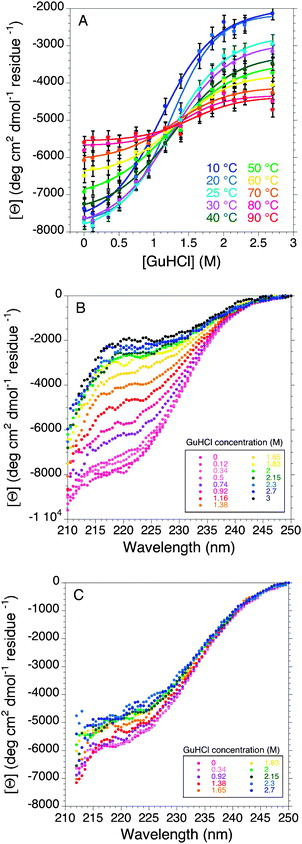 | ||
| Fig. 5 Combined effect of chemicals and temperature on BpUreG folding followed by far-UV CD (A) GuHCl-induced denaturation curves of BpUreG at different temperatures followed by far-UV CD at 222 nm. Bottom panels show the complete far-UV CD spectra of the GuHCl-induced denaturation of the protein at 10 °C (B) and at 90 °C (C). | ||
When the GuHCl concentration increases from 0 to 3 M at low temperatures, the CD spectra show an apparent two-state transition (Fig. 5A). While the intensities of the initial and final CD signal are different for each temperature sampled, the mid-point of these curves, representing the concentration of GuHCl at half-transition, does not vary significantly with temperature, suggesting that there are at least three main ensembles of structural conformations sampled by the protein, depending on the temperature and denaturant concentration. At low temperatures and low GuHCl concentrations, the protein ensemble shows the highest degree of residual structure, featuring some characteristics typical of a molten globule (MG-like), giving rise to undetectable or very broad NMR signals for all its residues, showing a blue-shifted maximum in the intrinsic fluorescence spectra, and retaining most of its secondary structure in the CD spectra. This structure is gradually lost upon increasing denaturant concentrations (Fig. 5B), reaching higher degree of disorder, close to the random coil (random coil-like) at 2.5–3 M GuHCl: in this state, the NMR spectra show narrower peaks still focused in the central part of the spectrum, the fluorescence maximum is red-shifted, and the secondary structure is severely decreased. On the other hand, at high temperatures and independently of the denaturant concentration, the BpUreG ensemble behaves differently (Fig. 5C), reaching, in average, an intermediate degree of residual structure similar to a pre-molten globule (pre-MG-like). In particular, as temperature increases in the absence of denaturant, BpUreG undergoes progressive and uncooperative loss of structure, even though it maintains a significant fraction of the secondary structure at high temperature, behaving like a pre-MG (Fig. S2, ESI‡). At intermediate denaturant concentrations (0.92 M GuHCl), the structural transition induced by temperature is minimal and the protein is in the intermediate folding state in the entire temperature range sampled (10–90 °C) (Fig. S2, ESI‡). When the experiment is conducted starting from a chemically-induced denatured state, the CD spectra show the presence of refolding upon temperature increase, moving from the random coil-like toward the intermediate pre-MG-like ensemble, consistently with a cold denaturation phenomenon (Fig. S2, ESI‡). Such a turn-out response to heat, where the temperature increase is accompanied by the formation of a more ordered secondary structure, has been reported and is expected for highly disordered IDPs. In fact, under physiological conditions, these proteins do not have an ordered structure because of the strong electrostatic repulsion (due to their high contents of charged and polar residues) and weak hydrophobic attraction (due to their low contents of hydrophobic residues). Therefore, the folding-promoting effects of elevated temperatures may be attributed to the increased strength of the hydrophobic interactions at higher temperatures, leading to a stronger hydrophobic attraction, known to be the major driving force for folding, and resulting in less compact conformations for cold-denatured states than for heat-denatured states.35
The DSC and CD experiments revealed that all these structural changes (heat-induced unfolding in native conditions or cold-induced unfolding in the presence of chemical denaturants) do not represent cooperative transitions, indicating that the protein never reaches a fully ordered state, rather existing as an ensemble of states, possessing different degrees of unfolding and separated by marginal energy barriers.
The transition between the three main ensembles of structural conformations identified is gradual and non-cooperative, as it can be visualized in the phase diagram shown in Fig. 6. According to this diagram, the temperature-induced structural transitions of the protein show different directions depending on the denaturant concentration, going from more structured to less structured conformations upon temperature increase at low GuHCl concentrations, and going from less structured to more structured conformations upon temperature increase at GuHCl concentrations higher than 1.25 M. The complete set of the far-UV CD-based experimental denaturation curves can be seen in Fig. S2 (ESI‡).
 | ||
| Fig. 6 Phase diagram of GuHCl- and temperature-induced structural disordering in BpUreG. The phase diagram was generated using the experimental information obtained for the GuHCl-induced denaturation of the protein at different temperatures followed by far-UV CD at 222 nm, assuming that the maximum and minimum absolute signal value at 222 nm corresponds to the maximum and minimum degree of structural order, represented as red and blue regions in the diagram. The black continuous lines represent the geometric sites of the points in the diagram at which the protein gives rise to a far-UV CD signal intermediate between its maximum, minimum and intermediate values. This corresponds to the half transition between the molten globule (MG)-like ensemble and the pre-molten globule (pre-MG)-like ensemble at low GuHCl concentrations; and to the half-transition between the pre-MG-like ensemble and the random coil-like ensemble at high GuHCl concentrations. | ||
Differently from CD and NMR, fluorescence experiments do not show the cold denaturation event in the presence of denaturant. Indeed, W192 fluorescence is significantly red-shifted when the protein moves from the native MG-like state to the pre-MG-like ensemble (increasing temperature in the absence of denaturant—Fig. 1A) or to the random-coil (increasing GuHCl concentration at low temperatures—Fig. 1B,C) ensembles, while this probe is insensitive to the transition from the random coil-like state to the pre-MG-like ensemble (increasing temperature at high GuHCl concentrations—Fig. 1C). These observations allowed us to conclude that W192 is found in a buried environment in the protein native state, while it is most likely fully exposed in the pre-MG-like and in the random coil conformations.
Experimental
Protein expression and purification
Protein expression and purification were carried out as described previously.22 The final protein prepared was diluted in 20 mM TrisHCl pH 8, containing 150 mM NaCl and 1 mM TCEP.Fluorescence spectroscopy
Steady-state fluorescence intensities and emission spectra were recorded using a PTI QuantaMaster C60/2000 (Photon Technology International, Inc., NJ) spectrofluorometer, operating in “photon counting” mode. Excitation and emission band-passes were set to 2.5 nm each. An excitation wavelength of 295 nm was used.To monitor the unfolding conformational transitions of BpUreG (15 μM) as a function of temperature and denaturant concentration, denaturation experiments were performed adding subsequent aliquots of GuHCl from a concentrated stock solution (6 M). The same starting and titrant solutions were used to measure the GuHCl equilibrium unfolding at six temperatures from 10 °C to 50 °C. The samples were equilibrated at each temperature before data collection. Fluorescence spectra were collected over time until no further change was observed. A spectrum of the buffer contribution to fluorescence emission was recorded at every temperature and subtracted to correct for the water Raman signal.
The free energy of unfolding at zero denaturant concentration (ΔG0,un) was determined from GuHCl-induced denaturation of BpUreG at different temperatures, assuming a two-state model and a linear relationship between ΔG0,un and GuHCl concentration, as previously described.26 An initial fit of all the denaturation curves showed that m (the denaturant concentration index) do not vary significantly with temperature (oscillating between 1.1 and 2.2 kcal mol−1 M−1), therefore the average value (1.57 kcal mol−1 M−1) was used as a fixed parameter in the fit.
The resulting free energies of unfolding were plotted as a function of temperature and these data were fitted according to the protein stabilization curve equation:30
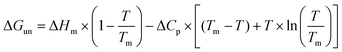
Differential scanning calorimetry
The heat capacity of BpUreG (at 57 μM and 23 μM) was measured as a function of temperature with a differential scanning VP-DSC microcalorimeter (Microcal LLC, Northampton, MA). Protein samples and reference solutions were degassed and carefully loaded into the cells to avoid bubble formation. The baseline of the instrument was routinely recorded before the experiments with both cells filled with buffer. Thermal denaturation scans of the protein were performed in the absence and in the presence of 0.12 M ZnSO4, at a scanning rate of 1 °C min−1 from 10 to 90 °C. Reversibility was checked by sample reheating after cooling inside the calorimetric cell.Circular dichroism
The far-UV CD spectra, as well as thermal unfolding scans, of BpUreG (15 μM) were measured at different temperatures using a Jasco-810 spectropolarimeter equipped with a Peltier holder, and cells of 0.1 cm path length. Experiments were conducted in the absence and in the presence of 45 μM ZnSO4, or 45 μM NiSO4, or 45 μM GTP-γS, 5 M urea, or different concentrations of GuHCl, as reported in the Results and discussion section. Spectra were normalized subtracting the ellipticity values recorded at 250 nm. Thermal runs were performed from 10 to 90 °C at a rate of 1–2 °C min−1, and protein unfolding was followed by far-UV circular spectroscopy at 222 nm. Data points were acquired every 0.1 °C with a response time of 2 s and a bandwidth of 1 mm. CD spectra of the buffer were also recorded as a control experiment (Fig. S3, ESI‡).NMR spectroscopy
NMR spectroscopy experiments were performed on a 15N-labeled BpUreG (500 μM) protein sample obtained growing transformed E. colicells in minimal medium, using (15NH4)SO4 as a sole nitrogen source, as previously described.22Phase-sensitive gradient-enhanced 1H,15N-HSQC spectra using water flip-back and echo–antiecho TPPI gradient selection36,37 were recorded on a Bruker Avance 800 spectrometer operating at 800.13 MHz equipped with a TXI cryoprobe. The spectra were obtained on samples containing 10% v/v D2O, in the absence and presence of 2.5 M GuHCl, in the temperature range 10–50 °C. The spectra consisted of 8 scans, spectral windows of 14 ppm in the proton dimension, and 36 ppm in the nitrogen dimension, with the carrier set at the water frequency and 118 ppm, respectively. Matrices of 1024 × 128 points were acquired and transformed into 2048 × 1024 points. NMR spectra were processed and visualized using TopSpin 2.0.
Conclusions
Ordered proteins contain a relatively stable three-dimensional structure, and undergo cooperative first-order folding/unfolding transitions between two distinct states. On the other hand, intrinsically disordered proteins exist as dynamic ensembles of conformers, and typically undergo non-cooperative changes in their relative populations.38 All evidences accumulated so far on BpUreG agree with the latter situation, with this protein featuring an intrinsic fluxional behaviour in the native state.This report confirms the intrinsically disordered state of this protein under native conditions and further clarifies the conformational landscape sampled by this protein as a function of temperature and denaturant concentration. Therefore it provides structural insights on the nature of this atypical enzyme, the first and, so far, unique natural enzyme with an intrinsically disordered tract. The reported results indicate that both the native and chemically-denatured states of BpUreG are essentially highly dynamic ensembles of conformations. The degree of structure and ordering found among these conformational ensembles depends on temperature and denaturant concentration. In other words, BpUreG unfolding transitions do not occur between two energetically discrete states separated by relatively high energy barriers, but rather unfolding of this protein can be described as a conformational shift among a continuum of states, relatively close in energy and separated by marginal barriers. For this reason, a simple two-state model, as the one applied to interpret the fluorescence data, fails to describe the fine complexity of the BpUreG unfolding process. The apparent sigmoidal protein denaturation curves, described in this work for intrinsic fluorescence experiments, derive from the fact that the parameter that follows the denaturation process (λmax) presents inherent fixed values, being independent from physical conditions such as temperature or solvent viscosity, and thus it remains relatively constant for the pure native and unfolded states. This produces linear baselines for the pre- and post-transition slopes. As a consequence, apparent sigmoidal curves can be retrieved more frequently and can be erroneously interpreted as indicative of two-state transitions, when not complemented with data derived by other techniques.
The data presented in this work are noteworthy, especially considering that the presence of native disorder in a natural enzyme implies that such overall structural flexibility may play a biological role and could have been selected by evolution. The available data do not prove the occurrence of an induced-fit mechanism with an overall structural transition upon substrate binding, and therefore they cannot clarify whether the observed residual activity in vitro arises from a small fraction of fully folded protein present in the sample, or it is compatible with the absence of a fully ordered enzyme and active site.
The fact that UreG proteins from different biological sources present various degrees of protein disorder suggests that disorder-to-order transition is a possible mechanism for UreG functioning in vivo and that the protein can be activated by the interaction with other protein partners, such as other urease chaperones that trigger its active form. The latter could require a global structural rearrangement toward the fully folded state, or a local rearrangement limited to the active site residues.
It is well known that GTP hydrolysis is regulated at different levels by cells in order to avoid unnecessary consumption of GTP. A protein interaction network, involving different effectors such as GTPase activating proteins (GAP) and/or guanine nucleotide exchange proteins (GEP), is very common among GTPases. A possible GAP for UreG has been identified in UreF.39 Furthermore, HpUreG was found to interact with other protein partners, such as HpUreE27 and HpHypB,40 the ortholog of UreG in the [Ni–Fe]-hydrogenase system. In addition to the substrate GTP, UreG interacts with metal ions, such as Ni2+ and Zn2+, with such an interaction playing a possible functional role.20
Intrinsically disordered proteins often act as hubs for protein–protein interaction networks, binding several different partners in regulative processes.41 UreG is able to bind different protein partners and cofactors, possibly forming a functional super-complex that also includes UreF and UreD.23 This suggests that UreG is part of an activation network in which this enzyme acts as a local hub or scaffold protein that coordinates multiple protein–protein or protein–cofactor interactions finally driving, through a switch represented by GTP hydrolysis, the entire process of Ni2+ ion trafficking toward urease activation. A similar role could also involve UreD.42 In this view, native disorder is a possible general mechanism that cells use to regulate enzymatic activity, allowing UreG, partially or totally inactive in the isolated state, to interact and to be regulated by different protein partners.
Acknowledgements
Work supported by a grant of Ministero Italiano dell'Università e della Ricerca (PRIN 2007—to B.Z., S.C. and P.T.), by a grant from the University of Bologna (RFO 2009—to B.Z. and S.C.), and by the Program of the Russian Academy of Sciences for the “Molecular and Cellular Biology” (to V.N.U). N.C. is a Human Frontiers Science Program (HFSP) Long-term Fellow (LT000795/2009). The authors thank Prof. Christopher M. Dobson for very helpful comments on earlier versions of this paper.Notes and references
- V. N. Uversky, Int. J. Biochem. Cell Biol., 2011, 43, 1090–1103 CrossRef CAS.
- P. Tompa, Curr. Opin. Struct. Biol., 2011, 21, 419–425 CrossRef CAS.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS.
- A. K. Dunker, C. J. Brown, J. D. Lawson, L. M. Iakoucheva and Z. Obradovic, Biochemistry, 2002, 41, 6573–6582 CrossRef CAS.
- P. Tompa, FEBS Lett., 2005, 579, 3346–3354 CrossRef CAS.
- V. N. Uversky, Protein Sci., 2002, 11, 739–756 CrossRef CAS.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1984, 27, 2985–2993.
- D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 98–104.
- U. Olsson and M. Wolf-Watz, Nat. Commun., 2010, 1, 111 Search PubMed.
- T. Kiefhaber, F. X. Schmid, K. Willaert, Y. Engelborghs and A. Chaffotte, Protein Sci., 1992, 1, 1162–1172 CrossRef CAS.
- V. N. Uversky, V. P. Kutyshenko, N. Protasova, V. V. Rogov, K. S. Vassilenko and A. T. Gudkov, Protein Sci., 1996, 5, 1844–1851 CrossRef CAS.
- Y. Li and G. Jing, J. Biochem., 2000, 128, 739–744 CAS.
- K. Vamvaca, B. Vogeli, P. Kast, K. Pervushin and D. Hilvert, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12860–12864 CrossRef CAS.
- K. Pervushin, K. Vamvaca, B. Vogeli and D. Hilvert, Nat. Struct. Mol. Biol., 2007, 14, 1202–1206 CrossRef CAS.
- K. Vamvaca, I. Jelesarov and D. Hilvert, J. Mol. Biol., 2008, 382, 971–977 CrossRef CAS.
- M. Morillas, H. Eberl, F. H. Allain, R. Glockshuber and E. Kuennemann, J. Mol. Biol., 2008, 376, 721–735 CrossRef CAS.
- M. Roca, B. Messer, D. Hilvert and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13877–13882 CrossRef CAS.
- F. Bemporad, J. Gsponer, H. I. Hopearuoho, G. Plakoutsi, G. Stati, M. Stefani, N. Taddei, M. Vendruscolo and F. Chiti, EMBO J., 2008, 27, 1525–1535 CAS.
- S. B. Mulrooney and R. P. Hausinger, FEMS Microbiol. Rev., 2003, 27, 239–261 CrossRef CAS.
- B. Zambelli, F. Musiani, S. Benini and S. Ciurli, Acc. Chem. Res., 2011, 44, 520–530 CrossRef CAS.
- A. Soriano and R. P. Hausinger, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11140–11144 CrossRef CAS.
- B. Zambelli, M. Stola, F. Musiani, K. De Vriendt, B. Samyn, B. Devreese, J. Van Beeumen, P. Turano, A. Dikiy, D. A. Bryant and S. Ciurli, J. Biol. Chem., 2005, 280, 4684–4695 CAS.
- M. B. Moncrief and R. P. Hausinger, J. Bacteriol., 1997, 179, 4081–4086 CAS.
- B. Zambelli, F. Musiani, M. Savini, P. Tucker and S. Ciurli, Biochemistry, 2007, 46, 3171–3182 CrossRef CAS.
- B. Zambelli, P. Turano, F. Musiani, P. Neyroz and S. Ciurli, Proteins: Struct., Funct., Bioinf., 2009, 74, 222–239 Search PubMed.
- P. Neyroz, B. Zambelli and S. Ciurli, Biochemistry, 2006, 45, 8918–8930 CrossRef CAS.
- M. Bellucci, B. Zambelli, F. Musiani, P. Turano and S. Ciurli, Biochem. J., 2009, 422, 91–100 CrossRef CAS.
- P. Voland, D. L. Weeks, E. A. Marcus, C. Prinz, G. Sachs and D. Scott, Am. J. Physiol. Gastrointest. Liver Physiol., 2003, 284, G96–G106 CAS.
- J. L. Boer, S. Quiroz-Valenzuela, K. L. Anderson and R. P. Hausinger, Biochemistry, 2010, 49, 5859–5869 CrossRef CAS.
- W. J. Becktel and J. A. Schellman, Biopolymers, 1987, 26, 1859–1877 CAS.
- K. P. Murphy, P. L. Privalov and S. J. Gill, Science, 1990, 247, 559–561 CAS.
- Y. V. Griko, E. Freire and P. L. Privalov, Biochemistry, 1994, 33, 1889–1899 CrossRef CAS.
- Y. V. Griko, J. Protein Chem., 1999, 18, 361–369 CrossRef CAS.
- A. Dhulesia, N. Cremades, J. R. Kumita, S. T. Hsu, M. F. Mossuto, M. Dumoulin, D. Nietlispach, M. Akke, X. Salvatella and C. M. Dobson, J. Am. Chem. Soc., 2010, 132, 15580–15588 CrossRef.
- V. N. Uversky, Protein J., 2009, 28, 305–325 CrossRef CAS.
- A. L. Davis, J. Keeler, E. D. Laue and D. Moskau, J. Magn. Reson., 1992, 98, 207–216 CAS.
- S. Grzesiek, J. Anglister, H. Ren and A. Bax, J. Am. Chem. Soc., 1993, 115, 4369–4370 CrossRef CAS.
- V. N. Uversky and A. K. Dunker, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1231–1264 CrossRef CAS.
- M. Salomone-Stagni, B. Zambelli, F. Musiani and S. Ciurli, Proteins: Struct., Funct., Bioinf., 2007, 68, 749–761 Search PubMed.
- K. Stingl, K. Schauer, C. Ecobichon, A. Labigne, P. Lenormand, J. C. Rousselle, A. Namane and H. de Reuse, Mol. Cell. Proteomics, 2008, 7, 2429–2441 CrossRef CAS.
- A. K. Dunker, M. S. Cortese, P. Romero, L. M. Iakoucheva and V. N. Uversky, FEBS J., 2005, 272, 5129–5148 CrossRef CAS.
- F. Musiani, M. Bellucci and S. Ciurli, J. Chem. Inf. Model., 2011, 51, 1513–1520 CrossRef CAS.
Footnotes |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M. Madan Babu. |
| ‡ Electronic supplementary information (ESI) available: Additional tables and plots of CD and fluorescence experiments. See DOI: 10.1039/c1mb05227f |
| § BZ and NC contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2012 |