Controlled release of dry reagents in porous media for tunable temporal and spatial distribution upon rehydration
Gina E.
Fridley
*,
Huy Q.
Le
,
Elain
Fu
and
Paul
Yager
Department of Bioengineering, University of Washington, Seattle, WA 98195, USA. E-mail: gfridley@uw.edu
First published on 24th August 2012
Abstract
Novel methods are demonstrated that enable controlled spatial and temporal rehydration of dried reagents in a porous matrix. These methods can be used in paper-based microfluidic assays to define reagent concentrations over time at zones downstream for improved performance, and can reduce costs by simplifying the manufacturing process with the use of a single porous substrate. First, the creation of uniform reagent pulses from patterned arrays of dried reagent is demonstrated. Second, reagents are stored dry in separate regions of the porous matrix so that they can be combined upon rehydration for immediate use in the device. Third, reagents are reconstituted sequentially from dry storage depots with tunable delivery times. Fourth, the total time for dissolution is varied to achieve a range of reagent delivery times to a downstream region. Finally, the utility of these control methods is demonstrated in the context of real-time reagent rehydration and mixing on a porous device.
Introduction
Lateral flow tests have been used for a variety of applications, ranging from home pregnancy tests to rapid diagnostic tests for infectious diseases in low-resource settings. These tests have been widely accepted because they are low-cost, rapid, easy to use, and require little to no instrumentation to interpret results. More recently, 2D and 3D paper-based devices have been demonstrated to extend capabilities to include multiplexed detection,1–4 multi-step processing capabilities,5–10 and the translation of classical microfluidics to paper-based systems.11,12A critical aspect of designing diagnostics for low-resource settings is to have as many of the required reagents as possible included on the device in dry form. The use of dry reagents reduces user steps, removes the need for a cold chain, provides a longer shelf life for the test, and facilitates device automation. Traditionally, reagents have been stored dry within separate pad material that is placed in line with the fluidic pathways for rehydration at the time of use. For example, antibodies conjugated to gold particle labels have been stored dry in polyester pads in both conventional microfluidic13 and lateral flow14,15 devices. However, the use of these separate pads has two significant disadvantages: it offers limited control over the spatial and temporal release of rehydrated reagent, and it requires additional materials and components that add to manufacturing costs.
Alternative techniques for storing dried reagents could expand the capabilities and lower the costs of paper-based devices. Here we develop methods to print reagents directly onto porous materials for later rehydration and use in on-paper assays. Piezoelectric inkjet printing has previously been used in the manufacture of a variety of biosensors,16,17 and Abe et al. demonstrated the rehydration of gold-labeled antibodies from hand-spotted storage regions.18Tunable control of the rehydration of reagents patterned on porous immunoassay devices has yet to be demonstrated, aside from the presentation of the initial results of this research at MicroTAS 2011.19 Similar techniques have been demonstrated in traditional channel-based microfluidic devices; Garcia et al. used cavities in channel walls to control the reconstitution of dried proteins20 and Hitzbleck et al. developed “reagent integrators” to store and subsequently release predetermined dilutions of reagents into microfluidic devices.21
In the research presented here, novel methods are demonstrated for printing patterns of reagents on porous membranes to achieve sophisticated control of the spatial and temporal concentration gradients that are created as the reagent dissolves during capillary flow. First, uniform reagent pulses were generated from patterned arrays of dried reagents. Second, reagents were stored dry in separate regions of the porous matrix and then combined upon rehydration for immediate use in the device, facilitating storage of reactive reagents. Third, a method was developed to delay the rehydration of individual reagent storage regions, allowing tunable staging of reagent delivery in an assay. Fourth, the rehydration time of individual reagent storage regions was extended, enabling a range in the duration of a given reagent pulse to the downstream assay. Finally, the utility of these control methods was demonstrated in the context of real-time reagent rehydration and mixing to achieve gold enhancement on a porous device.
Materials and methods
Device patterning and construction
All porous devices consisted of untreated backed 8 μm pore diameter nitrocellulose membranes (Millipore Hi Flow Plus 135, Millipore, Billerica, MA) cut using a CO2 laser (Universal Laser Systems, Scottsdale, AZ). Reagents were patterned onto strips using a piezoelectric noncontact printer (SciFLEXARRAYER S3, Scienion AG, Berlin, Germany). After printing, strips were wrapped in foil to protect them from light, then dried in a desiccator overnight and stored until use.Spatial and temporal dissolution demonstrations
For the experiments demonstrating the spatial and temporal control of rehydration, the dimensions of strips were either 5 mm × 50 mm or 10 mm × 50 mm; AlexaFluor-488-labeled streptavidin (488SA) (Invitrogen, Carlsbad, CA) was used as a model protein. Unless otherwise noted, the 488SA was printed at 0.5 mg mL−1, in a solution also containing 1% BSA, 5% sucrose and 5% trehalose. Printed volumes were approximately 70 nL. Delayed pulses of reagent were created by patterning a 20% sucrose solution in barriers around dried spots, and the time duration of extended reagent pulses was controlled by patterning a 20% sucrose solution directly atop dried spots. In both of these cases, the patterned volume of sucrose solution was varied to achieve the total sucrose content described below. Extremely extended reagent pulses, which rehydrated over the course of several minutes, were achieved by patterning 200 nL of 20% sucrose directly atop each of 5 dried spots of 488SA. These 5 spots each contained 160 nL 488SA, and were patterned sequentially in the direction of flow.To set up experiments, nitrocellulose strips were affixed to a black poly(methylmethacrylate) (PMMA) substrate using double-sided tape (Scotch ®, 3 M, St. Paul, MN). An untreated cellulose pad (CFSP223000, Millipore, Billerica, MA), cut using the CO2 laser, was placed at the downstream end of each strip as a sink to drive continuous wicking over the timescale of the experiment. A polyester source pad (Grade 6613, Ahlstrom, Helsinki, Finland), also cut using the CO2 laser, was placed at the upstream end of each strip. The polyester was pre-treated with a solution composed of 0.25% Brij®, 0.5% BSA, and 100 mM Tris, adjusted to a pH of 8.2, then dried for 2 h at 37 °C and stored in a desiccator until use.
At the start of an experiment, 15 μL fetal bovine serum (FBS, Certified, One Shot™, US Origin, Gibco ®, 16000-077, Invitrogen, Carlsbad, CA) was applied to the polyester pad as a real-time blocker to prevent non-specific adsorption of reagents to nitrocellulose. Immediately following the addition of FBS, cellulose strips were used to connect the polyester pads to a well containing phosphate buffered saline (PBS), which provided continuous flow through the device. Small blocks of PMMA were placed over the cellulose strips to ensure consistent contact with the device.
Experiments were conducted in a light-tight box, illuminated by a pair of blue LEDs positioned to minimize reflections. Uncompressed videos for analysis were acquired with a web camera (Logitech, Fremont, CA) fitted with a 550 nm high-pass filter (FEL0550, Thor labs, Newton, NJ).
Gold enhancement using reagents dried on-strip
All porous devices were designed, cut, patterned, and stored as described above. Nitrocellulose strips were cut to a bottle-shaped silhouette—1 cm wide at the upstream end and 3 mm wide at the downstream end, with the taper halfway down the strip. A 0.375 μL line of gold-labelled streptavidin (OD 1.25, Arista Biologicals, Allentown, PA) was patterned at the downstream end of the strip, 1 cm from the end. GoldEnhance LM gold enhancement solution (Nanoprobes, Yaphank, NY) was used for enhancement; 2 μL total of each: “enhancer” solution, “activator” solution, and “initiator” solution were printed in three sequential 29-spot arrays on the upstream end of the device.As above, nitrocellulose strips were affixed to a PMMA substrate. However, no cellulose wicking pad was used. An untreated glass fiber pad (Ahlstrom, Helsinki, Finland), cut using the CO2 laser cutter was placed at the upstream end of each strip. To begin the experiment, 70 μL PBS was applied to the glass fiber pad, which wicked through the device in roughly 15 min. The device was allowed to incubate for another 15 min. Time-lapse .avi videos were acquired using a web camera (Logitech, Fremont, CA) for the full 30 min.
Image analysis
The fluorescence of the 488SA was used as a visual marker of the protein's location after rehydration from its printed spot. Uncompressed .avi files were analysed with ImageJ and Matlab (Mathworks, Natick, MA). The green channel of each video was extracted and corrected for (i) background fluorescence by subtracting an image of the blank membrane, and (ii) non-uniformities caused by lighting or filter transmittance by normalizing with an image of fluorescein-flooded nitrocellulose.Delay times were quantified by subtracting the time at which the control spot reached maximum fluorescence intensity from the time at which the delayed spot reached maximum intensity, at a distance of 1 mm downstream from the initial spot location. The rehydration time of the extended-duration reagent storage regions was determined in the following way: the maximum value of the background intensity was quantified, and then doubled to yield a minimum signal threshold; then the time during which the fluorescence signal remained over this threshold was considered the duration of the elongated pulse. This was also quantified at 1 mm downstream from the initial spot location. The combination of individual rehydrated spots was measured by quantifying the fluorescence intensity over time of the rehydrated 488SA at 2.5 cm downstream from the initial spot locations. This distance was chosen to allow complete combination to occur.
Time-lapse videos of gold enhancement experiments were separated into image stacks using QuickTime 7 Pro (Apple Inc., Cupertino, CA, USA). The first image in the stack was used as the “pre-enhancement” image, and the image at the 30-minute time point was used as the “post-enhancement” image. Images were analysed using ImageJ. An enhancement ratio was determined by comparing background-subtracted average signal intensity between the pre- and post-enhancement images using a 13 pixel wide by 62 pixel high region of interest. The average signal intensity of this region was calculated for both images, and then background-subtracted using a background region 3 mm upstream of the signal line. The background-subtracted intensity was normalized by the intensity of the white background to account for any variation in lighting over the 30-minute video. The enhancement factor was calculated by dividing the post-enhancement signal by the pre-enhancement signal.
Results and discussion
Rehydration directly from nitrocellulose
Immunoassay developers have long utilized the powerful non-specific protein-binding capability of nitrocellulose to immobilize capture antibodies.14 In this work, however, the efficient non-specific binding of proteins was an obstacle that needed to be overcome to enable reagents to be patterned and stored directly on-strip for later rehydration and use in downstream applications.To mitigate non-specific binding of reagents to their initial patterned locations, we experimented with various additives, such as sugars and blocking proteins, mixed with our model protein prior to printing. By including sugars in the reagent solution that is dried on the paper, the viscosity of the rehydrated reagent solution is increased; this increase in viscosity has been used to provide control over the dissolution time of reagents dried in storage cavities in polydimethylsiloxane (PDMS) devices.20 In addition to imparting control over the dissolution of dried reagents, the sugars sucrose and trehalose have been used to stabilize dried proteins.13,20,22,23 The hydroxyl groups of the sugar molecules are believed to substitute for the waters of hydration of the protein that are lost upon drying, preserving the protein's native conformation.22 Additionally, trehalose has a high glass transition temperature relative to that of other sugars,20,22,24 which ensures that it remains an amorphous glass at elevated temperatures. The low molecular mobility of this glassy state contributes to protecting dehydrated proteins from damage such as chemical degradation or physical denaturation20,22,23 that may occur during storage or transport of point-of-care assays.20,22
The final mixture included 5% sucrose, 5% trehalose and 1% BSA added to the 488SA prior to patterning. Fig. 1 shows the different rehydration characteristics imparted by the additives. Without any additives, a significant amount of the printed 488SA remains non-specifically bound to the initial printed location, and the portion that does rehydrate into the fluid front leaves a long streak as it flows. The addition of 1% BSA reduces the amount of non-specifically bound 488SA, but causes a lag in the rehydration of the bulk. Adding only 5% sucrose and 5% trehalose appears to have the opposite effect—the 488SA is dissolved almost immediately into the fluid front, but more remains bound than with the addition of BSA. By adding all three components, a combination of the most desirable characteristics is observed; very little 488SA remains non-specifically bound, while the bulk of the rehydration occurs immediately at the wetting front.
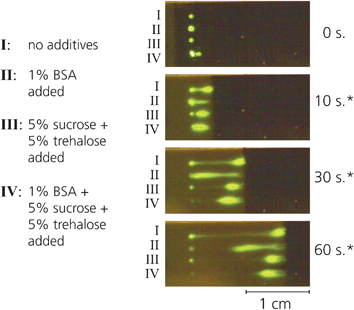 | ||
| Fig. 1 Mixing protein with different additives prior to printing yields dramatically different rehydration profiles. The combination of 5% sucrose, 5% trehalose and 1% BSA minimizes nonspecific binding at the site of printing, while also facilitating rapid rehydration into the wetting front. | ||
Spatial and temporal manipulation of dissolution
After demonstrating an effective method to enable drying reagent solutions directly onto nitrocellulose for later rehydration, we turned to developing techniques to control the spatial and temporal position of rehydrated reagents. These techniques enable the flexibility afforded by printing reagents on-strip rather than using separate conjugate pads for storage.First, spatially uniform pulses of reagent were created that spanned the width of a porous strip. Pulses such as these must be used in devices to ensure consistency of reagent delivery to downstream locations such as a capture zone. Uniform pulses can be achieved by patterning protein across the nitrocellulose strip, i.e. 90° to the direction of flow. As the wicking fluid front reaches these protein spots, the protein is dissolved with the liquid to yield a consistent, near-uniform pulse to a downstream region (Fig. 2). By varying the amount of reagent printed in each spot and the number of spots arrayed across the strip, the amount of reagent delivered by the pulse can be tuned to suit the needs of a particular assay. It is important to note that the initial pattern of spots does not affect the resulting pulse: all spots rehydrate into the wetting front, and diffuse together as long as the y-separation of spots is small, here 0.5 mm. This distance between spots, as well as the amount of reagent printed within each storage spot, will affect the uniformity of the resultant pulse. For instance, a y-separation of 1 mm or greater between 70 nL spots will yield individual streaks of reagent, whereas y-separations less than 0.5 mm will increase the uniformity of the pulse across the width of the strip.
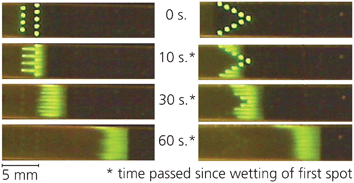 | ||
| Fig. 2 Protein patterned perpendicular to the direction of flow rehydrates to form a spatially uniform pulse. The initial pattern does not influence the shape of the resulting pulse, however transverse spot spacing, number of spots and total protein contained in each spot controls the uniformity of the pulse. | ||
The second method we developed was the controlled combination of reagents from separate regions of a device. Printing reagents separately and later combining them facilitates the storage of reagents that must not interact until they are mixed immediately before use in an assay. We accomplished this by patterning protein spots sequentially along a nitrocellulose strip. Here, a single strip contained an experiment in triplicate, alternating “combined spots”—two sequential spots each containing 17.5 ng 488SA that mix to form a uniform spot—with “single spots” that contained 35 ng 488SA. As mentioned above, a y-separation of 1 mm was sufficient to prevent transverse inter-diffusion (within these distances at these flowrates), and results in several distinct streaks downstream. We observed that sequentially patterned spots mix by diffusion with previously dissolved spots to form a single pulse of mixed reagents (Fig. 3a), and the combination of spots yields a pulse that is indistinguishable from a pulse that results from a single dissolved spot (Fig. 3b).
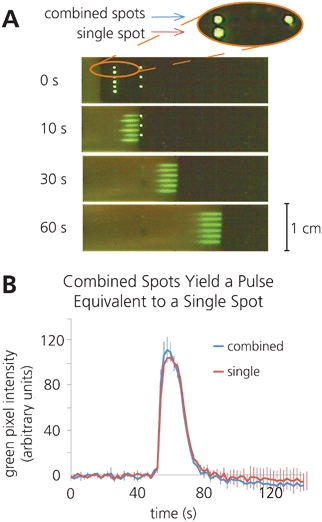 | ||
| Fig. 3 Reagents can be printed separately and combined upon rehydration by patterning sequential spots along the direction of flow. A) Time lapse images illustrating the combination of sequential rehydrated 17.5 ng 488SA spots, alongside rehydration of a single 35 ng 488SA spot. B) Plot of average fluorescence intensity vs. time at 2.5 cm downstream from the initial spot location, illustrating that separately printed spots combine to form a pulse equivalent to that created by a single rehydrated spot. (n = 3). | ||
Third, the timing of rehydration was manipulated to enable sequential release of dried reagents patterned on porous membranes. This delay in rehydration was achieved using barriers of sucrose surrounding individual spots, similar to methods previously developed in our group.6 These barriers were created by printing varying volumes of a 20% sucrose solution directly around the dried protein spot, which provided a time lag as the sucrose dissolved before the reagent could be released (Fig. 4). These different volumes were achieved by printing different numbers of drops of the sucrose solution using the piezoelectric printer, as described above. Dramatically different delays, from 19 s to 12 min, were achieved by printing sucrose barriers containing different amounts of total sucrose. Mid-delay sucrose barriers had the highest variability in delay times (Fig. 4b), likely because these barriers are the most sensitive to slight variations in the printing of sucrose barriers that lead to anomalous weak points in the barrier.
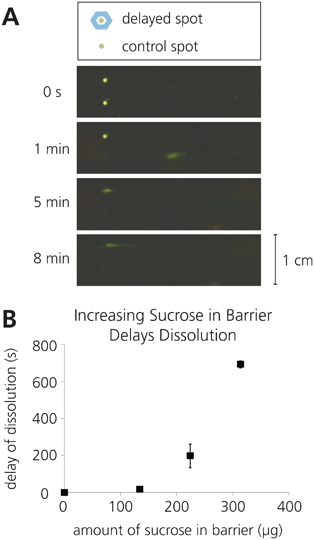 | ||
| Fig. 4 Rehydration of a printed reagent can be delayed by patterning a sugar barrier encircling the reagent spot. A) Schematic of sugar barrier patterning and time-lapse images of delayed rehydration enabled by a sugar barrier. B) Plot showing the variable delay times achieved by varying the sugar content within the sugar barrier (n = 3). | ||
Fourth, the time span over which a reagent dissolves from a dried reagent depot can be extended to yield longer time pulses of protein delivered to a downstream region. Extended pulses allow for increased interaction times between a reagent pulse and a capture or reaction zone, and could improve assay performance. We found that by printing sucrose solutions directly on top of dried protein spots, the duration of the dissolution time can be lengthened from less than 2 s in the control case, to over 50 s with 47 μg of extra sucrose added (Fig. 5a–c). Including different amounts of sucrose directly on top of dried reagent spots can tune the duration of the reagent pulse; however, high amounts of sucrose leads to larger variability in pulse duration.
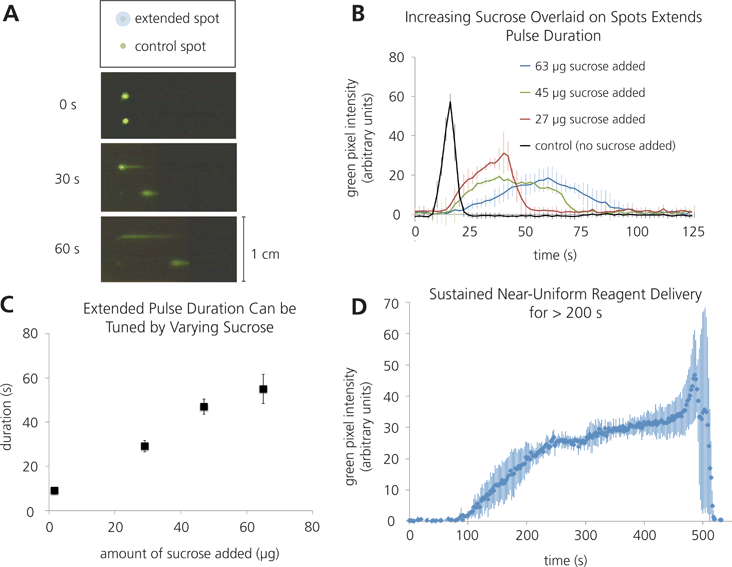 | ||
| Fig. 5 Extended rehydration times can be achieved by patterning sucrose directly on top of a reagent spot. A) Schematic and time-lapse images illustrating extended rehydration times compared to a control. B) Plot of fluorescence intensity vs. time of varying extended reagent pulses (n = 3). C) Plot illustrating the tunability of pulse duration using varying amounts of sucrose (n = 3). D) Plot of fluorescence intensity vs. time for an extremely extended pulse, using larger volumes of 488SA and sucrose (n = 3). | ||
Furthermore, exploration into these methods for increasing the duration of rehydration yielded a technique for extremely extended pulses, over which a dried reagent depot could deliver a nearly consistent stream for close to 4 min. As can be seen in Fig. 5d, there is a roughly 30 s “ramp up” time, as the concentration reaches its plateau, and then after the 4 min of consistent concentration, a 30 s “end burst” occurs, in which the last portion of the depot is rehydrated very rapidly, yielding a very short pulse of very high concentration. Both the “ramp up” and “end burst” behaviors are very reproducible in this system, but the “end burst” is currently not well understood. Ongoing work is aimed at elucidating possible causes for this behaviour.
Overall, these techniques to control the spatial and temporal dissolution of reagents have expanded the toolkit available to researchers and developers of paper-based microfluidic analytical devices. These methods have broad applicability in a variety of paper-based devices, but to illustrate one possible implementation, we have demonstrated immunoassay signal amplification by gold enhancement using reagents printed directly on-strip.
Gold enhancement by rehydrating reagents printed on nitrocellulose
One application of these methods is the patterning, storage, and on-strip combination of a multicomponent gold enhancement system that is commonly used to improve sensitivity in immunoassays. These experiments show the utility of storing reagents separately and then combining them immediately prior to use, and gold enhancement is just one example of a system in which these methods could be used.The multi-component gold enhancement system that we used requires the components to be stored separately and only mixed together immediately before use. Specifically, these components lose functionality if they are dried for storage after they have been mixed (data not shown). Thus, they must be stored dry in separate regions and recombined on-device for delivery to downstream regions of the assay. Previous work has shown effective rehydration and combination of these gold enhancement components from storage on glass fiber pads in the context of a malaria assay.15 Here, we show that gold enhancement reagents printed separately can be combined upon rehydration to yield a bolus of complete gold enhancement solution, which is then capable of enhancing previously patterned gold nanoparticles.
We achieved a 4.1 (± 0.2) fold gold signal enhancement (n = 3) after 30 min, using just 6 μl total printed gold enhancement reagents (Fig. 6). Though the enhancement ratio here is modest, it is a clear demonstration of the viability of patterning and drying reagents onto a paper device as a method for dry reagent storage in paper-based assays. Additionally, it is apparent in Fig. 6 that incomplete rehydration of the gold ion component of the enhancement solution is still an issue in this system. It is likely that the gold ions stick non-specifically to the nitrocellulose; further exploration into specific additives to the enhancement solutions could improve rehydration.
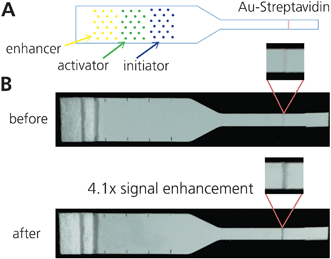 | ||
| Fig. 6 Combining gold enhancement reagents patterned on strip. A) Schematic of patterning. Yellow = enhancer, green = activator, blue = initiator. Spots were arrayed for minimal disruption to flow. B) Images of pre- and post-enhancement of a 1.25 OD gold-streptavidin line. After running for 30 min, the enhanced signal was 4.1x the initial signal, using grayscale intensity. | ||
One interesting question raised by this demonstration is the reason for a 30 min run time in this reaction, when our previously mentioned results showed relatively rapid rehydration of dry reagents. A possible explanation for the long run time is the need for an incubation step when such low volumes of gold enhancement reagents are used, particularly because the concentrations may be below optimal levels in this case, due to the rehydration volume being much larger than the printed volume. We are currently exploring the precise distributions of the gold enhancement reagents in further detail.
This model system is a simple and effective demonstration of the addition of multiple reagents patterned sequentially on a porous device, and is just one example of the potential applications enabled by printing reagents directly on porous devices for controlled rehydration and use in downstream assays. Other uses for these methods include, but are not limited to, enzymatic signal generation (e.g. HRP-TMB or HRP-DAB).
Conclusions
Here we have demonstrated a suite of methods that enable manipulation of the spatial and temporal distribution of reagents rehydrated from storage depots printed and dried directly on a porous matrix. To illustrate the utility of these methods, we have demonstrated signal enhancement using patterned and dried gold enhancement reagents. Gold enhancement is just one of several applications that can take advantage of these methods however, enzymatic signal generation using HRP and DAB can also be implemented with these techniques. Controlled reagent rehydration has direct applicability to extending the capabilities of paper-based diagnostic tests for use at the point of care by improving consistency, facilitating automation, and removing the need for cold storage. Additionally, by patterning reagents directly on the assay membrane, a complete assay could be fabricated using only one material, which could dramatically reduce manufacturing costs. Future work will demonstrate these techniques implemented in a full assay, and examine the long-term storage viability of reagents patterned directly on porous membranes.Acknowledgements
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship for G.E. Fridley under Grant No. DGE—0718124, as well as NIH (EB000252). Additional funding was provided by the University of Washington Bioengineering Department. The authors would also like to thank Peter Kauffman for his support and assistance with fluorescence imaging, Luke Allpress for his contributions to reagent dissolution studies, and Carly Holstein, Jen Osborn, and Shefali Oza for valuable discussions.References
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angewandte Chemie-International Edition, 2007, 46, 1318–1320 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Analytical Chemistry, 2010, 82, 3–10 CrossRef CAS.
- E. M. Fenton, M. R. Mascarenas, G. P. Lopez and S. S. Sibbett, ACS Applied Materials & Interfaces, 2009, 1, 124–129 CAS.
- S. Mendez, E. M. Fenton, G. R. Gallegos, D. N. Petsev, S. S. Sibbett, H. A. Stone, Y. Zhang and G. P. Lopez, Langmuir, 2010, 26, 1380–1385 CrossRef CAS.
- E. Fu, P. Kauffman, B. Lutz and P. Yager, Sensors and Actuators B-Chemical, 2010, 149, 325–328 CrossRef.
- E. Fu, B. Lutz, P. Kauffman and P. Yager, Lab on a Chip, 2010, 10, 918–920 RSC.
- E. Fu, T. Liang, J. Houghtaling, S. Ramachandran, S. A. Ramsey, B. Lutz and P. Yager, Analytical Chemistry, 2011, 83, 7941–7946 CrossRef CAS.
- E. L. Fu, S. Ramsey, P. Kauffman, B. Lutz and P. Yager, Microfluidics and Nanofluidics, 2011, 10, 29–35 CrossRef CAS.
- B. R. Lutz, P. Trinh, C. Ball, E. Fu and P. Yager, Lab on a Chip, 2011, 11, 4274–4278 RSC.
- I.-H. Cho, S.-M. Seo, E.-H. Paek and S.-H. Paek, Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences, 2010, 878, 271–277 CrossRef CAS.
- P. Kauffman, E. Fu, B. Lutz and P. Yager, Lab on a Chip, 2010, 10, 2614–2617 RSC.
- J. L. Osborn, B. Lutz, E. Fu, P. Kauffman, D. Y. Stevens and P. Yager, Lab on a Chip, 2010, 10, 2659–2665 RSC.
- D. Y. Stevens, C. R. Petri, J. L. Osborn, P. Spicar-Mihalic, K. G. McKenzie and P. Yager, Lab on a Chip, 2008, 8, 2038–2045 RSC.
- R. Wong and H. Tse, Lateral Flow Immunoassay, Humana Press, New York, 2009 Search PubMed.
- E. Fu, T. Liang, P. Spicar-Mihalic, J. Houghtaling, S. Ramachandran and P. Yager, Anal Chem, 2012 Search PubMed.
- M. S. Hasenbank, T. Edwards, E. Fu, R. Garzon, T. F. Kosar, M. Look, A. Mashadi-Hossein and P. Yager, Analytica Chimica Acta, 2008, 611, 80–88 CrossRef CAS.
- L. Gonzalez-Macia, A. Morrin, M. R. Smyth and A. J. Killard, Analyst, 2010, 135, 845–867 RSC.
- K. Abe, K. Kotera, K. Suzuki and D. Citterio, Analytical and Bioanalytical Chemistry, 2010, 398, 885–893 CrossRef CAS.
- G. E. Fridley, H. Le, E. Fu and P. Yager, in MicroTAS: 15th International Conference on Miniaturized Systems for Chemistry and Life Sciences, ed. J. P. Landers, Seattle, Washington, USA, 2011, pp. 981–983 Search PubMed.
- E. Garcia, J. R. Kirkham, A. V. Hatch, K. R. Hawkins and P. Yager, Lab on a Chip, 2004, 4, 78–82 RSC.
- M. Hitzbleck, L. Gervais and E. Delamarche, Lab on a Chip, 2011, 11, 2680–2685 RSC.
- J. H. Crowe, J. F. Carpenter and L. M. Crowe, Annual Review of Physiology, 1998, 60, 73–103 CrossRef CAS.
- F. Franks, R. H. M. Hatley and S. F. Mathias, Biopharm-the Technology & Business of Biopharmaceuticals, 1991, 4, 38 CAS.
- M. E. Elias and A. M. Elias, Journal of Molecular Liquids, 1999, 83, 303–310 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |