 Open Access Article
Open Access ArticleNon-emissive plastic colour filters for fluorescence detection
M.
Yamazaki
a,
S.
Krishnadasan
a,
A. J.
deMello
b and
J. C.
deMello
*a
aDept. Chemistry and Centre for Plastic Electronics, Imperial College London, Exhibition Road South Kensington, London SW7 2AY, UK
bDept. Chemistry and Applied Biosciences, ETH Zurich, HCI F 117, Wolfgang-Pauli-Strasse 10, CH-8093, Zurich, Switzerland. E-mail: j.demello@imperial.ac.uk
First published on 13th September 2012
Abstract
We report the fabrication of non-emissive short- and long-pass filters on plastic for high sensitivity fluorescence detection. The filters were prepared by overnight immersion of titania-coated polyethylene terephthalate (PET) in an appropriate dye solution – xylene cyanol for short-pass filtering and fluorescein disodium salt for long-pass filtering – followed by repeated washing to remove excess dye. The interface between the titania and the dye molecule induces efficient quenching of photo-generated excitons in the dye molecule, reducing auto-fluorescence to negligible values and so overcoming the principal weakness of conventional colour filters. Using the filters in conjunction with a 505 nm cyan light-emitting diode and a Si photodiode, dose-response measurements were made for T8661 Transfluosphere beads in the concentration range 1 × 109 to 1 × 105 beads μL−1, yielding a limit of detection of 3 × 104 beads μL−1. The LED/short-pass filter/T8661/long-pass filter/Si-photodiode combination reported here offers an attractive solution for sensitive, low cost fluorescence detection that is readily applicable to a wide range of bead-based immunodiagnostic assays.
Introduction
Microfluidic devices are attracting significant interest for point-of-care diagnostics due to their low unit cost, low reagent and sample usage, fast analysis times and small instrumental footprints.1 A typical microfluidic test comprises several assay steps (such as sampling, filtering, labeling, separation and isolation), followed by some method of analyte detection. Fluorescence is the most widely used detection method in microfluidics due to its high sensitivity, excellent dynamic range, ease of implementation and non-invasive nature.2 Fluorescence detection has accordingly been widely used for the interrogation of samples in microfluidic devices, with the vast majority of reports to date having used non-integrated laser excitation sources coupled with off-chip optics and photodetectors.For point-of-care applications, there is a growing need for self-contained systems that incorporate the microfluidic chip and all associated optical components into a compact, disposable, low-cost, monolithic structure.1,3–5 Highly integrated systems of this kind would eliminate the need for expensive dedicated bench-top instrumentation (requiring no more than some simple off-chip control electronics to drive the light-source, measure the response of the photodetector and process the signal into a usable format). As such they would find wide application in home, ambulance, hospital and GP surgery environments, where their ability to provide immediate and/or frequent testing would enable faster, more responsive and ultimately more successful treatment.1 In developing countries (such as those in sub-Saharan Africa) the consequences of such a technology would be far reaching, enabling governments to avoid the cost of establishing national networks of diagnostic laboratories (as exist in developed countries) and to instead move directly to a distributed healthcare model based entirely around point-of-care testing.6
To realise this vision, significant advances are needed in both microfluidic assay technology (allowing robust, quantitative tests to be performed on bodily fluids in a passively driven chip-based format) and in the development of integrated optics. The latter may be directly incorporated into the microfluidic chip itself or take the form of thin-film components laminated onto the planar faces of the microfluidic chip. In recent years a wide range of optical components have been successfully integrated into microfluidic devices, including both active components such as light-sources3,5,7,8 and photo-detectors3,5,7,9–11 and passive components such as prisms,12 lenses,13 filters,4 mirrors,14 gratings,15 waveguides,16,17 and polarisers.18,19
Optical filters play a particularly important role in microfluidic devices.4 In conventional fluorescence detection, the light source and detector are arranged at ninety degrees to one another to prevent direct illumination of the detector by the excitation light. This orthogonal geometry, however, is difficult and costly to implement in a microfluidic device since it requires optical-grade side-surfaces onto which the optical components must somehow be mounted. The light source and detector are most conveniently placed on the top and bottom faces of the microfluidic chip in a “face-on” geometry. Unfortunately this arrangement can lead to flooding of the detector with direct light from the excitation source, masking the much weaker analyte emission.4,20 Key to discriminating emission from excitation light in a face-on configuration is the use of a short-pass filter immediately after the light-source, together with a long-pass filter in front of the detector. The short-pass filter sharpens the emission spectrum of the light-source, removing any long-wavelength tail that would otherwise overlap the analyte signal (and so reduce sensitivity); while the long-pass filter blocks the sharpened excitation light, passing only the longer wavelength emission signal to the detector. For this approach to be successful the short- and long-pass filters must be chosen to display complementary transmission characteristics, i.e. the short-pass filter must block light when the long-pass filter transmits, and vice-versa.
Optical filters have been successfully applied to fluorescence detection in a wide variety of formats, but considerable difficulties are encountered when (as required for monolithic integration) the filters are placed in close proximity to the detector.4 In such circumstances, any auto-fluorescence from the filter material will add to the analyte fluorescence, and so set a base-line beneath which it is difficult to measure a signal.20 Autofluorescence is a particular problem for low cost colour filters formed by dispersing pigments or dye molecules in a glass or polymer host.20 For low analyte concentrations, even very weak auto-fluorescence from the filters may obscure the analyte emission, so it is essential to select dyes that not only have acceptable transmission characteristics but also have extremely low photoluminescence quantum yields. The palette of such materials is needless-to-say extremely limited.
Auto-fluorescence can be largely avoided by switching to interference filters formed from an alternating stack of high and low refractive index materials.21 Interference filters typically have excellent characteristics in terms of strong blocking in the stop-band, high transmission in the pass-band, sharp roll-on and extremely low auto-fluorescence. However they are expensive to produce due to their complex multilayer structures and hence are ill-suited to low cost disposable applications. They also suffer from angular variability in their blocking/transmission characteristics, which degrades their performance in actual use.22
In a recent paper20 we reported a purely dye-based approach for fabricating high performance optical filters that avoids the expense and angular variability of interference filters, while overcoming the undesirable auto-fluorescence characteristics of conventional colour filters. Our approach exploited ultrafast electron-transfer between a photo-excited dye molecule and a transparent metal oxide onto which the molecule is absorbed23—the same process that forms the basis for the charge-generation step in dye-sensitised solar cells (DSSCs).24 In essence, a monolayer of the dye molecule is adsorbed onto the surface of a highly porous metal-oxide such as titania (TiO2), generating a diffuse interface between the two materials. The interface acts as an extremely efficient quenching site for the photo-excited exciton,25 reducing the fluorescence quantum yield of the dye to a negligible level without significantly perturbing its absorption characteristics. The oxide itself has a wide optical gap (ΔE > 3.2 eV for titania), so does not absorb over the visible wavelength range used for most diagnostic tests. Accordingly, if the crystallite size is significantly smaller than the wavelength of light so as to minimise optical scattering, the oxide serves as an invisible scaffold for the dye molecule.
The mechanism of the quenching process is well established,25 see Fig. 1. When an adsorbed dye molecule absorbs a photon, an electron is promoted to an unoccupied molecular orbital and from there undergoes ultrafast charge injection into the continuum of conduction band states in the titania. The electron relaxes by internal conversion to the conduction band-edge and then hops between titania sites until it reaches an interface with a dye-cation, whereupon it recombines non-radiatively to regenerate the dye. Importantly, the initial charge-transfer step from the dye to the oxide typically occurs on a very short time-scale (∼100 fs),25–28 meaning the photo-excited electron is removed from the dye molecule long before it has a chance to relax radiatively to the ground-state. In this way, it is possible to reduce the fluorescence quantum yield of the dye molecule to a negligible level and so quench the auto-fluorescence by many orders of magnitude.
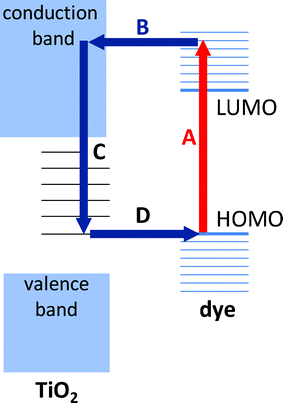 | ||
| Fig. 1 Electron transfer process in dye-sensitised titania. (A) Photo-excitation of dye. (B) Ultrafast electron transfer from the unoccupied molecular orbitals of the dye to the conduction band of titania (TiO2). (C) Thermalisation of the hot electron, followed by hopping between sub-gap titania sites. (D) Charge recombination with a proximate dye cation. | ||
Our original fabrication method used a commercial titania paste with a high organic mass content (WA-15DPT, Nanopac, South Korea). The precursor was deposited by blade-coating onto a glass substrate and thermally cured at 450 °C to remove the organic phase and so form a highly nanoporous film onto which the dye molecules could be readily attached by immersion in a dye solution. The requirement for high temperature curing, however, restricted the filters to niche applications on glass. Here we explore the possibility of extending the titania-based filters to plastic substrates, with a view to both reducing their cost and widening their applicability. We further investigate the feasibility of creating complementary short- and long-pass filters that, in combination with an appropriate bio-label, can enable sensitive fluorescence detection using low cost light-emitting diodes (LEDs) and silicon photodiodes as light sources and photodetectors. A sensitive diode-based fluorescence detection system is considered to be the only viable option for the low cost point-of-care applications identified above.
Experimental
Most small molecule fluorophores have relatively small (< 40 nm) Stoke's shifts between their absorption and emission peaks, which requires the use of filters with very sharp roll-off – a characteristic that is hard to achieve using colour filters. This requirement can be avoided by deliberately selecting bio-labels with large Stoke's shifts, e.g. phosphorescent molecules, quantum dots, or energy transfer beads (ETBs).29–34 The latter comprise a series of two or more fluorophores dispersed in a solid polymer matrix with surface groups onto which bio-active molecules such as antibodies can be easily attached.35–37 The fluorophores are selected to have overlapping excitation and emission spectra such that excitation energy can be efficiently channelled in the direction of decreasing energy from the first dye in the series to the last. This enables Stoke's shifts of 100 nm or more to be obtained. ETBs are near-ideal candidates for analyses where a label size in excess of 100 nm is permissible since the polymer host can both protect the dyes from photo-degradation and provide (via appropriate surface functionalisation) the chemical selectivity required for the chosen assay. Various ETBs are available commercially and, for the current work, we selected T8861 TransFluoSphere beads (Invitrogen, US), which use a readily functionalised carboxylate-modified polystyrene polymer as the host. T8861 beads exhibit a 100 nm Stoke's shift between their excitation peak at 500 nm and their emission peak at 600 nm (Fig. 2) - wavelengths that are easily excitable and detectable using low cost, off-the-shelf LEDs and photodiodes.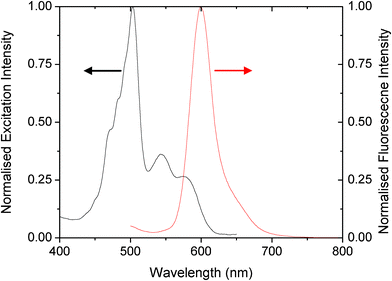 | ||
| Fig. 2 Excitation and emission spectra of the T-8861 Transfluosphere beads. | ||
As noted, conventional high temperature titania pastes contain organic surfactants that burn away at high temperatures, leaving behind a highly porous sintered network of titania with large internal surface area and good mechanical stability. By switching to surfactant-free slurries of nanocrystalline titania in alcohol/water mixtures the need for high temperature curing can be removed (since there is no organic phase to remove). The porosity of the resultant films however is unavoidably diminished due to the denser packing of the titania colloids, which disadvantageously reduces the surface area available for dye attachment.38
To assess whether the resultant films might nonetheless be suitable for filter applications, 4 μm films of titania were blade-coated onto polycarbonate (PC) substrates from a commercially available surfactant-free titania paste (Ti-Nanoxide-LT, Solaronix SA, Switzerland). For DSSCs, subsequent sintering at ∼100 °C is required to improve electrical connectivity between individual crystallites but, since conductivity was not a concern for the current application, both sintered and unsintered films were evaluated. Following our previous work,20 Fluorescein Disodium Salt (FDS) was selected as the component dye for the long-pass filters due firstly to its spectral compatibility with T8861 – it has a relatively sharp absorption cut-on midway between the emission and absorption peaks of the T8661 beads – and secondly for its ability to attach directly to titania via its carbonyl and carboxylate groups. Dye attachment was carried out by immersing the substrates overnight in a 0.01 M solution of FDS in ethanol and then washing repeatedly with ethanol to remove unattached dye molecules. The complete removal of ‘stray’ (loosely attached) dye molecules is critical to eliminating filter fluorescence since only those dye molecules in direct contact with the titania surface are guaranteed to undergo efficient fluorescence quenching.
Fig. 3 shows absorption spectra for two Ti-FDS filters on PC prepared from a surfactant-free slurry with and without thermal curing at 120 °C. (Note, PC has a relatively high glass transition temperature Tg of 150 °C). Also shown for comparison is the absorption spectrum of a Ti-FDS filter on glass, prepared using a high temperature surfactant-based precursor and stained and rinsed in an identical manner. The glass filter exhibited strong blocking characteristics with a peak absorbance of 1.8, consistent with the high internal surface area available for dye attachment. The absorption spectra of the two PC filters were similar in appearance to that of the glass filter but approximately half as strong at the peak. This is consistent with the denser packing of titania films obtained from surfactant free slurries, which leaves only the uppermost monolayers of the film available for dye attachment. Whilst the optical densities of the individual PC-supported films are certainly too low for use as high performance long-pass filters, improved blocking can be achieved by coating both sides of the PC substrates with titania and stacking several such double-sided filters until the requisite OD is achieved (although this is obviously achieved at the expense of transmission in the pass-band).
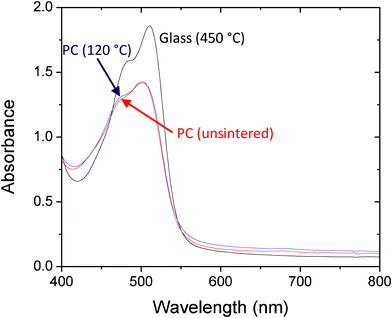 | ||
| Fig. 3 Absorption spectra for FDS-coated titania filters on glass and polycarbonate (PC). The glass filter (black line) was prepared using a titania paste with a high organic mass content, requiring prolonged curing at 450 °C. The polycarbonate filters were prepared from a surfactant-free slurry of titania with (blue) or without (red) thermal annealing at 120 °C. All titania films were subsequently immersed overnight in a 0.01 M solution of FDS in ethanol, and then rinsed thoroughly with clean ethanol to remove excess dye. | ||
The striking similarity between the absorption characteristics of the sintered and unsintered films suggests minimal changes in the film morphology during curing. Interestingly, the unsintered films were substantially more resistant to cracking than the sintered ones - a possible consequence of positively charged TiOH groups in the titania binding electrostatically to carbonyl groups on the PC substrate, increasing adhesion. (The hydroxide groups are lost after prolonged heating.39,40) We therefore decided to use all titania films in an unsintered form, allowing us in turn to switch substrate material from PC to cheaper and more easily handled polyethylene terephthalate (PET, Tg = 70 °C). The adhesion of the unsintered titania to PET was at least as good as to PC, and all data presented below were therefore obtained using unsintered titania films on PET substrates.
The selection of dye molecules for short-pass filtering is challenging. Whereas long-pass dyes must absorb continuously above the π–π* energy gap – common behaviour for organic chromophores – short-pass dyes require a window in the absorption spectrum at short wavelengths, greatly limiting the palette of available dyes. The choice of chromophore is further restricted by the requirement for functional groups that adhere strongly to titania. Indeed, so restrictive does the choice become that we were unable to identify any commonly available dyes with suitable anchor groups and transmission characteristics matched to our chosen T8661 beads. Relaxing the requirement for anchor groups, we identified two potential contenders for use as short-pass dyes: Remazol Brilliant Blue R (RBBR, Sigma Aldrich) and Xylene Cyanol FF (XC, Sigma Aldrich) - see Fig. 4 for solution spectra. Of the two dyes, XC had the preferred transmission characteristics and was therefore selected for further investigation.
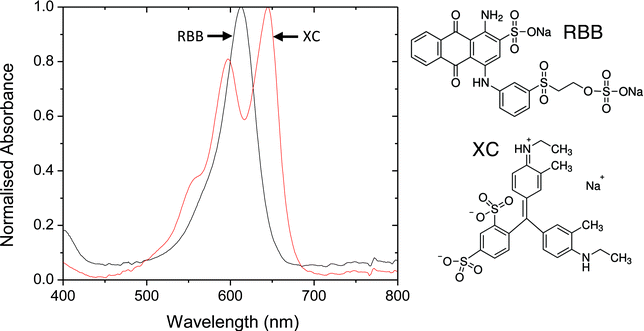 | ||
| Fig. 4 Solution-phase absorption spectra of candidate short-pass dyes in ethanol. XC = Xylene Cyanol (XC) and RBB = Remazol Brilliant Blue. | ||
Whilst in principle either of the aforementioned dyes could be derivatised with suitable anchor groups to allow them to attach to titania, such an approach is inconvenient and risks altering the absorption characteristics of the dyes. We therefore sought a more general approach for attaching the dyes to the titania surface. Immersion of titania films in an acidic/basic solution has previously been shown41 to yield a proton rich/deficient surface onto which anionic/cationic molecules may be electrostatically attached. Noting that both dyes are ionic in nature, this appeared to be a promising approach for achieving dye attachment.
To test the suitability of this approach for xylene cyanol, unsintered titania films on PET were immersed in 0.1 M HNO3 for 1 min. The films were removed from the acidic solution, dried in air, transferred to a 0.01 M solution of xylene cyanol in ethanol for 24 h, and rinsed thoroughly in ethanol before drying. Control films were prepared in the same way, omitting the acid treatment. Absorption spectra of the resultant filters are shown in Fig. 5. Visual inspection of the control films indicated substantial loss of XC during the washing step (consistent with the absence of anchor groups), with the weak absorption spectrum confirming the presence of only trace quantities of retained dye on the substrate. The acid-treated filters by contrast were permanently stained and had peak optical densities of similar magnitude to those obtained using FDS, suggesting a similar degree of surface coverage. (The spatial uniformity of the filters was also similar, with the absorbance of both the FDS and XC filters differing by < ± 5% over the area of the plastic substrate). Importantly, the mechanical stability of the XC films was comparable to the FDS films, indicating no significant reduction in titania adhesion as a consequence of the acid treatment. The filters could be further strengthened by applying a film of adhesive-coated acetate (“adhesive tape”, 3 M, US) to the upper dye, yielding a robust film that could be repeatedly flexed without significant risk of cracking. Fig. 6 shows absorption spectra for the encapsulated short- and long-pass filters under 0.04 mWcm−2, 500 nm monochromatic light over the course of three hours. The filters showed minimal changes in their spectral characteristics, indicating suitable stability for the envisaged “use-once” applications.
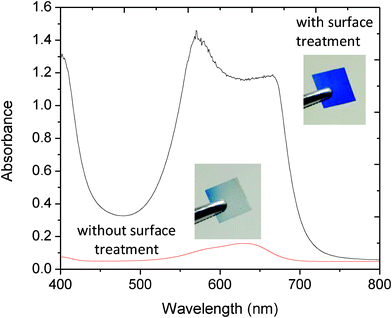 | ||
| Fig. 5 Absorption spectra of XC-sensitised titania films on polycarbonate with and without acid-treatment of the titania. (Spectra recorded after repeated rinsing in ethanol). Also shown inset are images of 1 cm × 1 cm coated films for the two cases. | ||
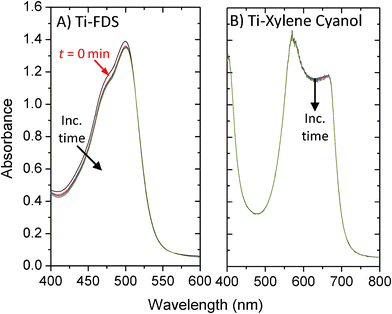 | ||
| Fig. 6 Normalised absorption spectra of filters based on Ti-FDS (A) and Ti-XC (B) over a period of 3 h under continuous illumination by 0.04 mWcm−2, 500 nm monochromatic light. In both cases films were encapsulated using acetate adhesive tape. | ||
Fig. 7A shows the emission characteristics of the Ti-FDS long-pass filter under 440 nm, 1 mW HeCd laser excitation – well within the stop band of the filter (see Experimental). Also shown for reference are the emission characteristics of a commercial long-pass Schott Glass filter (OG530). The Schott glass filter exhibited strong autofluorescence that would preclude its use in high sensitivity detection. The Ti-FDS filter by contrast exhibited extremely weak fluorescence that we were unable to detect even with a fifteen minute integration time on the CCD spectrometer. Significantly, within the detection limits of the experiment, the FDS filters on PET performed as well as our previous glass filters prepared using a high temperature paste. Fig. 7B shows the emission characteristics of the Ti-XC filter under 0.5 mW 633 nm HeNe laser excitation - again well within the stop band of the filter. The level of auto-fluorescence was again minimal and below the detection limit of the CCD. (We note that for completeness we also tested the Ti-FDS filter under 633 nm excitation and the Ti-XC filter under 488 nm excitation, i.e. using laser wavelengths in the pass band of the two filters. In neither case were we able to detect emission).
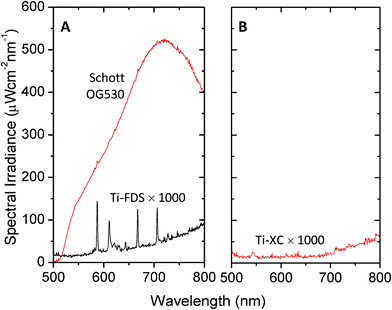 | ||
| Fig. 7 (A) Autofluorescence spectrum for an unsintered Ti-FDS long-pass filter on PET under 1 mW 442 nm HeCd excitation. Also shown for reference is the emission spectrum for a commercial Schott glass filter obtained under identical conditions. (B) Autofluorescence spectrum for an unsintered Ti-XC short-pass filter on PET under 0.5 mW 633 nm HeNe excitation. All other acquisition conditions were the same. Spectra for the titania-based filters have been multiplied by a factor of 1000 for clarity. | ||
FDS and XC serve as effective complementary filters with spectral characteristics that are well matched to the T8861 Transfluosphere beads: the 488 nm absorption peak of the beads is well within the pass band of the Ti-XC filters and the stop-band of the Ti-FDS filters; while the 605 nm emission peak is well within the pass band of the Ti-FDS filters and the stop band of the Ti-XC filters. To assess the performance of the filters in conjunction with the T8661 beads, a dose-response measurement was carried out, using a 505 nm LED operating at 20 mA (Luxeon REBEL LED Cyan, Philips Lumileds Lighting, N.V.) for excitation, a stack of four Ti-XC films for short-pass filtering, a holder for 1 mm path-length cuvettes, a stack of four Ti-FDS films for long-pass filtering and a silicon photodiode for optical detection (S1226-8BQ, Hamamatsu, Japan), see Fig. 8A. The LED was driven in a square-wave duty-cycle at 170 Hz, 20 mA using a home-made constant-current source. For optimal sensitivity the signal from the photodiode was measured using a digital lock-in amplifier (SR830, Stanford Instruments US), locking into the first harmonic of the LED drive signal. A time constant of 3 s was used for all measurements, with the signal being averaged over 100 measurements obtained at 1 s intervals. The use of lock-in amplification has a number of advantages: as well as providing excellent noise rejection that allows for the straightforward detection of pA-level signals, it also eliminates background signals due to static and slowly varying ambient light. In addition, although implemented here using a rather expensive digital lock-in amplifier, comparable levels of noise rejection can be readily achieved using inexpensive analogue lock-in detection, providing a clear route to low cost implementation.42–44
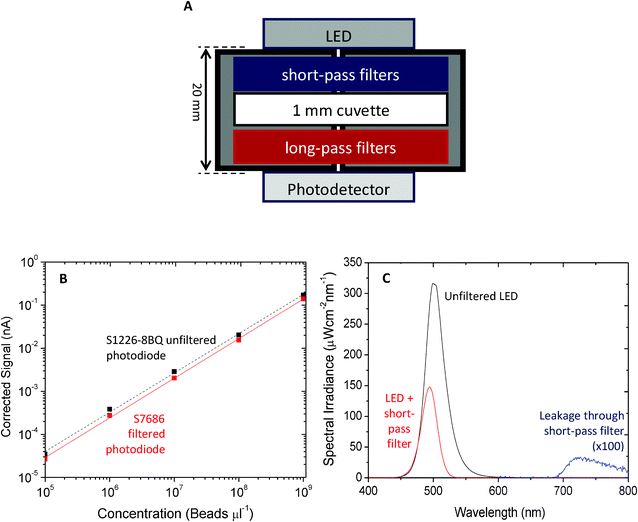 | ||
| Fig. 8 (A) Schematic of fluorescence detection configuration. The LED, filters, cuvette and photodetector were mounted in a custom-made holder designed to fix all components tightly in place. To achieve adequate blocking, four PET short-pass filters were placed between the LED and the cuvette and four PET long-pass filters were placed between the cuvette and the photodetector. A small hole was drilled through the holder along the optical axis to permit light to pass. The LED was driven by a pulsed constant-current driver at 170 Hz, 20 mA. The signal was detected using an (unfiltered) SR830 Stanford Lock-In amplifier, locked to the first harmonic frequency. (B) Signal versus transfluosphere bead concentration in distilled water using a S1226-8BQ filtered silicon photodiode and a filtered S7686 photopic Si photodiode; standard deviation of measured signals was < ± 1% relative to the mean. (C) Influence of a single Ti-XC filter on the emission spectrum of the 505 nm cyan LED. Weak but non-negligible leakage light is evident between 700 and 800 nm. | ||
To perform the dose-response measurements, filled 1 mm path-length cuvettes containing aqueous dispersions of the T8661 beads in the concentration range 1 × 109 to 1 × 105 beads μL−1 were prepared by sequential ten-fold dilutions from an initial as-received 1 × 1010 beads μL−1 stock solution. An additional reference cuvette was prepared containing water only. Placing this reference cuvette into the cuvette holder yielded a background signal of 440 pA compared to a signal of just 1.2 pA when the LED was switched off, indicating an appreciable degree of leakage due to incomplete blocking of the excitation light by the long-pass filter. Corrected signals for the various bead solutions were obtained by subtracting the 440 pA background. The corrected signal versus T8661 concentration is plotted in Fig. 8B (dotted line). Defining the limit of detection λ as the concentration of analyte that yields a signal equal to the background plus three times the standard deviation of the blank (3σ = 0.26 pA), we obtained λ = 4.9 × 105 beads μL−1.
The cause of the background signal may be understood from Fig. 8c which shows the influence of the short-pass filters on the emission spectrum of the LED: without filtering a long tail persists out to 600 nm and beyond, overlapping with the emission from the T8661 beads; with filtering the long wavelength tail is heavily suppressed and the signal intensity falls to < 0.045% of its peak value by 525 nm. Above 700 nm, however, the short-pass filter ceases to absorb and a weak “rump” of unfiltered LED light can be detected between 700 and 800 nm. Importantly this emission lies within the pass band of the long pass filter and, although minuscule in intensity compared to the main emission peak of the LED at 505 nm, it is nonetheless significant compared to the analyte emission at the lower bead concentrations. It therefore has a detrimental impact on the limit of detection.
To improve the sensitivity further it would in principle be possible to select a different short-pass dye with an absorption band that persists beyond 800 nm. As noted above however there is a relative dearth of usable short-pass dye molecules and an easier option is to insert an additional IR-blocking filter in front of the photodiode. (Note, the risk of autofluorescence from this filter is extremely low as virtually all the excitation light has already been removed by the intervening filters; hence it is not necessary to use a metal-oxide supported filter here). Instead of using a discrete IR-filter, here we opted to use a filtered silicon photodiode with an integrated band-pass filter, nominally intended to allow the photodiode to mimic the response of the eye (S7686, Hamamatsu Japan). Using the S7686 in place of the original unfiltered S1226-8BQ silicon photodiode, the background signal from the reference cuvette was reduced to just 22 pA (compared to a signal of 0.6 pA when the LED was switched off). The solid line in Fig. 8B shows the corrected signal versus T8661 concentration, using the S7686 photodiode. The 3σ (0.012 pA) limit of detection was reduced to λ = ∼3 × 104 beads μL−1. This value is highly promising for a purely colour filter-based detection method, and compares favorably to detection limits of ∼6 × 104 beads μL−1 that were previously reported using a combination of colour filters and polarisers.19 The latter approach, although an effective one, is dependent on precise and reproducible alignment of crossed-polarisers, significantly adding to manufacturing cost and complexity. No such alignment criteria apply to the current method. Moreover IR filtering by the S7686 photodiode was only partial (incident photon conversion efficiency of ∼0.83% between 700 and 800 nm) and further reductions in the background signal should be achievable through the addition of discrete IR filters in front of the photodetector, perhaps offering a further ten-fold improvement in the limit of detection using the current components. (Still better detection limits could, of course, be achieved using a laser diode and avalanche-photodiode for the light-source and detector but this would not satisfy our objective of delivering an ultra-low cost detection platform).
In conclusion, we have developed a complementary set of short- and long-pass filters on plastic that are well matched to the spectral characteristics of T8661 Transfluosphere beads. The filters are prepared by overnight immersion of a titania-coated PET substrate in an appropriate dye solution – xylene cyanol for short-pass filtering and fluorescein disodium salt for long-pass filtering – followed by repeated washing to remove excess dye. The interface between the titania and the dye molecule induces efficient quenching of the photo-generated excitons in the dye molecule, reducing the photoluminescence to a negligible value. Using the filters in conjunction with a 505 nm cyan LED and a photopic Si photodiode, dose-response plots were measured for T8661 in the concentration range 1 × 109 to × 105 beads μL−1, yielding a limit of detection of 3 × 104 beads μL−1. This compares favourably with a previous value of 6 × 104 beads μL−1 obtained using a combination of colour filters and polarisers.19 The LED/Ti-XC/T8661/Ti-FDS/Si-photodiode combination reported here offers a highly attractive solution for sensitive, low cost, integrated fluorescence detection in a microfluidic chip that is readily applicable to a wide range of bead-based immunodiagnostic assays.
Experimental details
The transmission characteristics of the filters were measured with a dual-beam UV/Vis spectrometer (Philips Unicam, UK), keeping the reference path empty so as to determine the overall transmission properties of the filters (dye plus substrate as opposed to the dye alone). Autofluorescence spectra for the Ti-FDS long-pass filters were obtained by directing a 1.1 mW, 1.5 mm diameter beam from a 442 nm HeCd laser (IK5552R-F, Kimmon, Japan) onto the front side of each filter and placing a fibre-optic coupled to a CCD spectrometer (QE65000, Ocean Optics US) against the rear-side of the filter immediately behind the laser spot. This configuration broadly mimics the typical situation in a microfluidic device where the light-source and photodetector are directly opposite one another, and a long-pass filter is placed immediately in front of the photodetector to block the excitation light. Integration times of up to 15 min were used to collect the spectra due to the weak emission from the dye-sensitised titania films. Autofluorescence spectra for the Ti-XC short-pass filters were obtained in the same way using a 0.5 mW, 0.48 mm diameter beam from a 633 nm HeNe laser (Standard JDS Uniphase HeNe Laser, JDS Uniphase Corporation, UK).Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement No. 248052 (PHOTOFET).References
- G. M. Whitesides, The origins and the future of microfluidics, Nature, 2006, 442(7101), 368 CrossRef CAS.
- G. S. Fiorini, Disposable microfluidic devices: fabrication, function, and application, BioTechniques, 2005, 38(3), 429 CrossRef CAS.
- O. Hofmann, et al., Thin-film organic photodiodes as integrated detectors for microscale chemiluminescence assays, Sens. Actuators B, 2005, B106(2), 878 CAS.
- O. Hofmann, et al., Monolithically integrated dye-doped PDMS long-pass filters for disposable on-chip fluorescence detection, Lab Chip, 2006, 6(8), 981 RSC.
- O. Hofmann, et al., Towards microalbuminuria determination on a disposable diagnostic microchip with integrated fluorescence detection based on thin-film organic light emitting diodes, Lab Chip, 2005, 5(8), 863 RSC.
- P. Yager, et al., Microfluidic diagnostic technologies for global public health, Nature, 2006, 442(7101), 412–418 CrossRef CAS.
- X. Wang, et al., Integrated thin-film polymer/fullerene photodetectors for on-chip microfluidic chemiluminescence detection, Lab Chip, 2007, 7(1), 58 RSC.
- J. B. Edel, et al., Thin-film polymer light emitting diodes as integrated excitation sources for microscale capillary electrophoresis, Lab Chip, 2004, 4(2), 136 RSC.
- D. Brennan, et al., Emerging optofluidic technologies for point-of-care genetic analysis systems: a review, Anal. Bioanal. Chem., 2009, 395(3), 621 CrossRef CAS.
- S. Moon, et al., Integrating microfluidics and lensless imaging for point-of-care testing, Biosens. Bioelectron., 2009, 24(11), 3208–3214 CrossRef CAS.
- E. Thrush, et al., Monolithically integrated semiconductor fluorescence sensor for microfluidic applications, Sens. Actuators, B, 2005, 105(2), 393 CrossRef.
- Q. Kou, On-chip optical components and microfluidic systems, Microelectron. Eng., 2004, 73–74, 876 CrossRef CAS.
- N. Chronis, Tunable liquid-filled microlens array integrated with microfluidic network, Opt. Express, 2003, 11(19), 2370 CrossRef.
- S. Kostner, Microsystems for Optical Cell Detection: Near versus Far Field, Part. Part. Syst. Charact., 2008, 25(1), 92 CrossRef.
- K. Hosokawa, K. Hanada and R. Maeda, A polydimethylsiloxane (PDMS) deformable diffraction grating for monitoring of local pressure in microfluidic devices, J. Micromech. Microeng., 2002, 12(1), 1 CrossRef CAS.
- S. Balslev, et al., Lab-on-a-chip with integrated optical transducers, Lab Chip, 2006, 6(2), 213 RSC.
- R. Irawan, et al., Integration of optical fiber light guide, fluorescence detection system, and multichannel disposable microfludic chip, Biomed. Microdevices, 2007, 9(3), 413 CrossRef CAS.
- A. Banerjee, A polarization isolation method for high-sensitivity, low-cost on-chip fluorescence detection for microfluidic lab-on-a-chip, IEEE Sens. J., 2008, 8(5), 621 CrossRef CAS.
- G. Ryu, et al., Highly sensitive fluorescence detection system for microfluidic lab-on-a-chip, Lab Chip, 2011, 11(9), 1664–1670 RSC.
- M. Yamazaki, et al., Non-emissive colour filters for fluorescence detection, Lab Chip, 2011, 11(7), 1228–1233 RSC.
- B. S. Wherrett, Optically bistable interference filters: optimization considerations, J. Opt. Soc. Am. B, 1986, 3(2), 351 CrossRef CAS.
- C. Richard, et al., An integrated hybrid interference and absorption filter for fluorescence detection in lab-on-a-chip devices, Lab Chip, 2009, 9(10), 1371 RSC.
- M. Gratzel, Conversion of sunlight to electric power by nanocrystalline dye-sensitized solar cells, J. Photochem. Photobiol., A, 2004, 164(1–3), 3 CrossRef CAS.
- S. Y. Huang, et al., Charge Recombination in Dye-Sensitized Nanocrystalline TiO2 Solar Cells, J. Phys. Chem. B, 1997, 101(14), 2576–2582 CrossRef CAS.
- R. Huber, The role of surface states in the ultrafast photoinduced electron transfer from sensitizing dye molecules to semiconductor colloids, J. Phys. Chem. B, 2000, 104(38), 8995 CrossRef CAS.
- P. V. Kamat, Interfacial charge transfer processes in colloidal semiconductor systems, Progr. React. Kinet., 1994, 19(3), 277 CAS.
- T. Hannappel, B. Burfeindt and W. Storck, Measurement of ultrafast photoinduced electron transfer from chemically anchored Ru-dye molecules into empty electronic states in a colloidal anatase TiO2 film, J. Phys. Chem. B, 1997, 101(35), 6799 CrossRef CAS.
- Y. Tachibana, et al., Subpicosecond interfacial charge separation in dye-sensitized nanocrystalline titanium dioxide films, J. Phys. Chem., 1996, 100(51), 20056 CrossRef CAS.
- A. Joshi, Temperature dependence of the band gap of colloidal CdSe/ZnS core/shell nanocrystals embedded into an ultraviolet curable resin, Appl. Phys. Lett., 2006, 89, 131907 CrossRef.
- V. V. Nikesh, Highly photoluminescent ZnSe/ZnS quantum dots, Semicond. Sci. Technol., 2001, 16, 687 CrossRef CAS.
- Y. Fu, Solid substrate room temperature phosphorescence immunoassay based on an antibody labeled with tetramethylrhodamine B isothiocyanate, Anal. Chim. Acta, 2002, 470(2), 121 CrossRef CAS.
- P. Huhtinen, Synthesis, characterization, and application of Eu (III), Tb (III), Sm (III), and Dy (III) lanthanide chelate nanoparticle labels, Anal. Chem., 2005, 77(8), 2643 CrossRef CAS.
- M. Seydack, Nanoparticle labels in immunosensing using optical detection methods, Biosens. Bioelectron., 2005, 20(12), 2454 CrossRef CAS.
- G. Liu, Nanomaterial labels in electrochemical immunosensors and immunoassays, Talanta, 2007, 74(3), 308 CrossRef CAS.
- Y. M. Zhang, Key residues responsible for acyl carrier protein and β-ketoacyl-acyl carrier protein reductase (FabG) interaction, J. Biol. Chem., 2003, 278(52), 52935 CrossRef CAS.
- M. B. Meza, Bead-based HTS applications in drug discovery, Drug Discovery Today, 2000, 5, 38–41 CrossRef.
- D. Moll, Biomolecular interaction analysis in functional proteomics, J. Neural Transm., 2006, 113(8), 1015–1032 CrossRef CAS.
- T. Miyasaka, M. Ikegami and Y. Kijitori, Photovoltaic Performance of Plastic Dye-Sensitized Electrodes Prepared by Low-Temperature Binder-Free Coating of Mesoscopic Titania, J. Electrochem. Soc., 2007, 154(5), A455–A461 CrossRef CAS.
- F. M. Fowkes, et al., Acid–base properties of glass surfaces, J. Non-Cryst. Solids, 1990, 120(1–3), 47–60 CrossRef CAS.
- F. M. Fowkes, Acid–Base Interactions in Polymer Adhesion, in Tribology Series, J.M. Georges, Editor. 1981, Elsevier. p. 119-137 Search PubMed.
- H. Lachheb, Photocatalytic degradation of various types of dyes (Alizarin S, Crocein Orange G, Methyl Red, Congo Red, Methylene Blue) in water by UV-irradiated titania, Appl. Catal., B, 2002, 39(1), 75 CrossRef CAS.
- S. Roy, A. Chakravarti and S. Sil, A simple phase-sensitive amplifier with automatic offset nulling, Indian Journal of Physics, 2012, 86(2), 117–124 CrossRef CAS.
- P. A. Temple, An introduction to phase-sensitive amplifiers: An inexpensive student instrument, Am. J. Phys., 1975, 43(9), 801–807 CrossRef.
- G. Ferri, et al., A low-voltage integrated CMOS analog lock-in amplifier prototype for LAPS applications, Sens. Actuators, A, 2001, 92(1–3), 263–272 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |