Rapid, sensitive, and multiplexed on-chip optical sensors for micro-gas chromatography†
Karthik
Reddy
ab,
Yunbo
Guo
a,
Jing
Liu
ac,
Wonsuk
Lee
ab,
Maung Kyaw
Khaing Oo
a and
Xudong
Fan
*ac
aDepartment of Biomedical Engineering, University of Michigan, 1101 Beal Avenue, Ann Arbor, MI 48109, USA. E-mail: xsfan@umich.edu; Fax: +1 734-647-4834; Tel: +1 734-763-1273
bDepartment of Electrical Engineering and Computer Science, University of Michigan, 1301 Beal Avenue, Ann Arbor, MI 48109, USA
cCenter for Wireless Integrated Microsensing and Systems, University of Michigan, 1301 Beal Avenue, Ann Arbor, MI 48109, USA
First published on 16th January 2012
Abstract
We developed and characterized a rapid, sensitive and integrated optical vapor sensor array for micro-gas chromatography (μGC) applications. The sensor is based on the Fabry–Pérot (FP) interferometer formed by a micrometre-thin vapor-sensitive polymer layer coated on a silicon wafer. The thickness and the refractive index of the polymer vary in response to the vapor analyte, resulting in a change in the reflected intensity of the laser impinged on the sensor. In our study, four different polymers were coated on four wells pre-etched on a silicon wafer to form a spatially separated sensor array. A CMOS imager was employed to simultaneously monitor the polymers' response, thus enabling multiplexed detection of a vapor analyte passing through the GC column. A sub-second detection time was demonstrated. In addition, a sub-picogram detection limit was achieved, representing orders of magnitude improvement over the on-chip vapor sensors previously reported.
1. Introduction
Rapid and in situ analysis of volatile organic compounds (VOCs) is of importance in a variety of fields including environmental monitoring, homeland security, and healthcare.1,2 Micro-gas chromatography (μGC) systems are ideally suited for on-site monitoring due to their low power consumption and small size. Typical μGC systems consist of a pre-concentrator, micro-fabricated separation columns, a pump and a detector to detect separated analytes. While tremendous progress has been made in μGC development, there are still a few problems in the current μGC systems that need major improvement. First, long sampling and pre-concentration times are required for detection of low concentrations or masses of VOCs. Second, due to the short columns, μGC suffers from the low chromatographic resolution. Several analytes may co-elute within one separation peak, making the analysis and identification of VOCs much more difficult than with a conventional GC system having excellent separation capability. To overcome these drawbacks, it is urgent to develop μGC sensors that are highly sensitive to reduce the sampling time, able to qualitatively analyze VOCs embedded in a co-eluted peak, and compatible with other μGC components for easy device integration and miniaturization.In recent years there has been an increasing focus on the use of arrays of partially sorptive sensors that may have the potential to achieve the above sensor design goals. Chemi-resistor arrays have been shown to effectively discriminate between individual analytes in a mixture.3–6 However, they are inherently susceptible to electromagnetic interference and have a detection limit of only a few nanograms.7 Surface acoustic wave (SAW) sensors coated with polymers have also been demonstrated for vapor discrimination,8–12 but they suffer from the interference between neighboring sensors.9,10 Furthermore, while a detection limit of 10 pg was reported with an uncoated SAW sensor,13 the detection limit for the polymer coated sensor increases to the nanogram range.14 Therefore, the SAW device is still not ideal for vapor sensor array development.
As compared to the chemi-resistor and SAW sensor, the optical vapor sensor is immune to electromagnetic interference and can operate without crosstalk, thus making it a promising candidate for use in μGC sensor arrays. Recent developments in optical vapor sensor technology have seen the implementation of Bragg15–17 and long period grating sensors,18–20surface plasmon resonance (SPR) sensors,21,22 localized surface plasmon resonance (LSPR) sensors,23,24 ring resonator sensors25–28 and photonic crystal fiber (PCF) sensors29 for the detection of VOCs. However, those vapor sensors are either incompatible with μGC components, difficult to fabricate, or complicated in optical design.
The Fabry–Pérot (FP) cavity based optical vapor sensor avoids the pitfalls of the above mentioned optical sensors due to its simple optical configuration, ease of fabrication and high sensitivity.30–36 As illustrated in Fig. 1(A), an FP sensor is formed by a thin layer of vapor sensitive polymer coated on a substrate. The light reflected by the air/polymer and polymer/substrate interface results in an interference spectrum (see Fig. 1(B)). The interaction of the VOC and polymer causes a change in the polymer thickness and/or refractive index, which in turn leads to a spectral shift in the characteristic interference spectrum corresponding to the extent of vapor sorption. Therefore, the FP sensor is able to provide quantitative and kinetic information about the vapor flowing inside a microfluidic channel. Previous work has shown that these sensors are capable of rapid sub-second VOC detection with a detection limit in the range of a few tens to a few hundreds of picograms.34,36
 | ||
| Fig. 1 (A) Schematic of the Fabry–Pérot (FP) sensor. The absorption of analytes results in a change in the thickness and RI of the polymer film, which in turn leads to a change in the interference pattern. R1 is the light reflected from the polymer–substrate interface. R2 and R2′ are the light reflected from the air–polymer interface before and after the polymer change, respectively. (B) Example of the interference pattern generated by an FP sensor and the effect of analyte absorption. At a fixed wavelength the resonance spectral shift can be recorded as an increase or decrease in the reflected intensity. | ||
Here we developed an FP sensor array on chip with significantly improved sensing capability for μGC applications. As shown in Fig. 2, using microfabrication technology, we were able to assemble four FP sensors inside a μGC fluidic channel for on-column detection. A CMOS imager was used to simultaneously monitor the FP sensor array in real-time. The FP sensor array described here offers several distinct advantages compared to those previously demonstrated. First, using a sensor array, both qualitative and quantitative analysis of VOC mixtures is possible to enhance the μGC's capability in analyte identification. Second, the sensor is capable of performing on-column multiple polymer interrogation, with no crosstalk between signals, using a single imager. Third, one to two orders of magnitude improvement in the detection limit can be achieved, tremendously reducing the amount of time needed for pre-concentration of vapors. Finally, the sensor is robust, cost effective and highly reproducible. In this paper we used four different polymers (OV-1, OV-73, OV-215 and OV-1701) to fabricate and characterize the FP sensor array on chip. Then four analytes (acetone, methanol, heptane and toluene) were employed as model systems to test the FP sensor array and establish a method to analyze VOC mixtures.
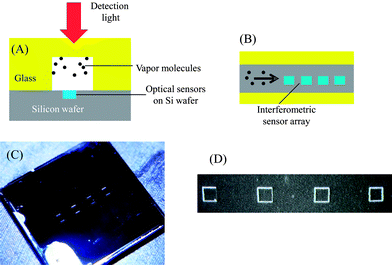 | ||
| Fig. 2 (A) Cross-sectional view of the FP sensor array fabricated on etched silicon wafer inside a 1 mm deep and 450 μm wide microfluidic channel. (B) Top-view of the FP sensor array. Four different polymer solutions were dropped into the etched wells using a micro-dropper. Dimensions are not to scale. (C) Image of an etched silicon chip containing the sensor array. The overall device was 8 mm × 6 mm and has an inlet and an outlet, to which a capillary column could be inserted for fluidic connection. (D) Image of 4 wells on chip. Each well was 200 μm × 200 μm and 1.3 μm deep. The wells were separated by 800 μm. | ||
2. Material and methods
2.1 Materials
All the analytes and solvents used in the experiments were purchased from Sigma (St Louis, MO) and had purity greater than 97%. GC guard column (part no. 22335, inner diameter 250 μm), RTX-1 column (part no. 40101, inner diameter 180 μm) and RTX-Wax column (part no. 12423, inner diameter 250 μm) were purchased from Restek (Bellefonte, PA). Universal quick seal column connectors were purchased from Varian (Palo Alto, CA). Silicon wafers were purchased from University Wafer (South Boston, MA). OV-1 (poly(dimethylsiloxane) or PDMS) was purchased from Fluka (St Louis, MO). OV-73 (diphenyldimethylsilicone), OV-215 (trifluoropropylmethylsilicone) and OV-1701 (dimethylphenyl cyano substituted) were purchased from Ohio Valley Specialty (Marietta, OH). Glass slides were purchased from VWR (Radnor, PA). UV-curable optical glue was purchased from Dymax (Torrington, CT). All materials were used as received.2.2 Sensor preparation
For studies of individual FP sensors, each sensor was prepared using the spin-coating method. First, a silicon wafer was diced to an 8 mm × 10 mm piece using an ADT 7100 dicing saw, which was subsequently immersed overnight in sulfuric acid–dichromate solution to oxidize any contaminants, followed by a rinse with deionized water. Finally, it was placed under UV light for an hour to ensure removal of any residues.Then OV-1, OV-73, OV-215 or OV-1701 was used as the vapor sensing layer. These polymers are commonly used in many GC applications like stationary phase of a column. The polymer solution was prepared by dissolving the polymer gum in their corresponding solvent. OV-1 was diluted with toluene (PDMS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene = 1:5), OV-73 and OV-1701 were diluted with pentane (OV-73:pentane = 1:6 and OV-1701:pentane = 1:5), and OV-215 was diluted with ethyl acetate (OV-215:ethyl acetate = 1:5). The polymer was then coated using a spin coater, with spin speed calibrated such that the polymer thickness for all four sensors was in the range of 1–1.2 μm. The polymer was first spun at 1500–2000 rpm for 10 seconds and then at 6600–7600 rpm for 30 seconds. The initial spin spreads the polymer across the entire silicon chip and the second step removes excess polymer and solvent. The spin-coated chip was then heated for 30 seconds at 80 °C to completely remove the solvent. Finally, an open-bottom microfluidic channel assembled from glass slides and UV-curable optical glue was sealed on top of the coated chip. The resultant channel was approximately 1 mm deep and 450 μm wide.
:toluene = 1:5), OV-73 and OV-1701 were diluted with pentane (OV-73:pentane = 1:6 and OV-1701:pentane = 1:5), and OV-215 was diluted with ethyl acetate (OV-215:ethyl acetate = 1:5). The polymer was then coated using a spin coater, with spin speed calibrated such that the polymer thickness for all four sensors was in the range of 1–1.2 μm. The polymer was first spun at 1500–2000 rpm for 10 seconds and then at 6600–7600 rpm for 30 seconds. The initial spin spreads the polymer across the entire silicon chip and the second step removes excess polymer and solvent. The spin-coated chip was then heated for 30 seconds at 80 °C to completely remove the solvent. Finally, an open-bottom microfluidic channel assembled from glass slides and UV-curable optical glue was sealed on top of the coated chip. The resultant channel was approximately 1 mm deep and 450 μm wide.
For sensor array preparation, we used the drop-coating method. The overall sensor array layout is illustrated in Fig. 2(A) and (B). First, four 1.3 μm deep wells were etched into a prime grade silicon wafer (8 mm × 6 mm) using an MA-6 and STS Pegasus-4 tools for lithography and etching, respectively (Fig. 2(C) and (D)). Each well was 200 μm × 200 μm and was separated by 800 μm so that the entire length can be imaged with a CMOS imager. The previously created polymer solutions were diluted, with the corresponding solvent, to one-tenth of their initial concentrations, and then drop coated into each well using a pulled capillary micro-dropper made in-house. The well acted as a containment barrier for the polymer, ensuring no cross-contamination between sensors, and that the sensing surface is nearly flush with the silicon, thereby minimizing disturbance to the gas flow. Finally, an open-bottom microfluidic channel assembled from glass slides and UV-curable optical glue was sealed on top of the coated chip. The resultant channel was approximately 1 mm deep and 450 μm wide.
2.3 Experimental setup
The experimental setup is illustrated in Fig. 3. To test each individual sensor, individual vapor analyte was injected at the GC injection port and delivered to the sensor module using a 4 m long GC guard column. A Toptica 785 nm laser was used to generate the optical detection beam. The light was aligned using an FC/APC terminated optical fiber and a beam collimator. The reflected beam was collected by a Thorlabs CMOS imager (product no. DCC1545M), with an acquisition rate of 16 frames per second. To acquire information regarding the interference spectrum of the FP sensors and to tune the beam incident angle to increase sensitivity of the sensors, a white light source was placed co-linearly with the laser beam and a spectrometer (Ocean Optics HR-2000) was used at the reflection side to monitor the interference spectrum (see the dashed lines in Fig. 3(B)).36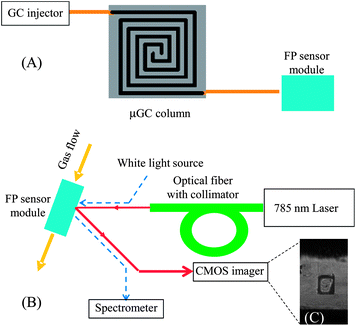 | ||
| Fig. 3 (A) Schematic of the μGC setup. The FP sensor module shown in Fig. 1(A) was connected to a GC injection port via a standard capillary GC column or a microfabricated GC column. (B) Schematic of the optical detection setup. A 785 nm laser was used to interrogate the change in reflected intensity caused by the presence of vapor analyte inside the channel. The incident angle could be adjusted to maximize the sensitivity. The CMOS imager provides quantitative and kinetic information about polymers' response to the vapor analytes. Dashed lines show the path of the white light, which was used to optimize the 785 nm laser alignment. (C) Image of an FP sensor with polymer in an etched well. | ||
To test the sensor array and to examine its collective response to VOCs, a mixture containing different mass combinations of the four vapor analytes was injected at the GC injection port. It was then delivered to a 25 cm long microfabricated GC column (400 μm × 100 μm) coated with OV-1, a 2.5 m long Carbowax column, and a 1 m long OV-1 column to separate a mixture of acetone, methanol, heptane and toluene before entering the sensor module (the fabrication and the subsequent coating of the microfabricated GC column can be found in ref. 37 and 38). The overall optical detection setup remained the same as previously described, except that a lens (VZM450 from Edmund Optics) was added between the sensor array and the imager, whose field of view is sufficiently large to capture all four sensors so that the response of all sensors to the vapor analyte flowing inside the channel could be obtained instantaneously and simultaneously. The data for each sensor were saved separately and could be processed to form chromatograms for each individual sensor. The reference signal was acquired from the light reflected from bare silicon and used to remove any long term amplitude drifts or false peaks caused by laser instability.
All experiments were carried out at room temperature with no heating of the columns or sensors. Mass of injected analytes was calibrated using the splitter and mass spectroscopy system. Helium was used as the carrier gas with a flow rate of 8 mL min−1.
3. Results and discussion
Sensor characterization
Each individual FP sensor was tested with four different analytes, acetone, methanol, heptane and toluene, injected individually. The insets in Fig. 4 show examples of the chromatogram for each analyte. The signal rises rapidly with the increasing presence of analyte and rapidly falls back to the baseline as it is purged from the polymer. Since the CMOS imager was operated at 16 frames per second, a system time resolution of 60 ms can be achieved. During the experiment analyte vapor was sampled using a gas syringe, allowing for control over the volume, and hence mass, of injected analytes. The peak response of each sensor to each injected mass is shown in Fig. 4 (and S1†). At low analyte concentrations, the sensor peak response is linear to the injected mass. When the injected mass increases, the saturation effect occurs and the response curve levels off. | ||
| Fig. 4 Response of four different polymers OV-1 (squares), OV-73 (triangles), OV-215 (circles) and OV-1701 (inverted triangles) to various injected masses of (A) acetone, (B) methanol, (C) heptane and (D) toluene. Insets show the chromatogram corresponding to the circled data point in each figure. Analytes were delivered to the sensor using a 4 m long guard column. | ||
The sensitivity of each sensor depends on the interaction between the analyte and the polymer, which in turn depends on a variety of factors, including polarity of the analyte and the polymer, volatility of the analyte, functional groups, molecular weight, etc. For example, as shown in Fig. 4(A), there is a clear difference in the responses for each polymer to the same analyte (acetone), with OV-215 showing the strongest interaction and OV-1 showing the weakest interaction. The same phenomenon occurs for the other three analytes. To estimate the detection limit, we use the lowest data point (usually in the picogram range) in combination with the sensor noise level of approximately 0.1 counts. Note that different sensors may have slightly different noise levels because of surface roughness of the polymer and scattering of the optical beam. However, these differences are very small and are not a major factor in the different detection limits of each sensor. The detection limit for each analyte is listed in Table S1†. Generally, our FP sensor shows a detection limit a few orders of magnitude better than previously reported.35,36 In particular, with OV-73 a sub-picogram detection limit of 0.64 pg and 0.79 pg was achieved for heptane and toluene, respectively. Based on the retention time (4 s) and the peak width (0.125 s) for heptane obtained from the inset in Fig. 4(C), as well as the inner diameter (250 μm) and length (4 m) of the GC column, the above detection limit corresponds to about 25 ppb in concentration at atmospheric pressure. Similarly, a concentration detection limit of 28 ppb for toluene can also be derived. Both are a few times better than the best results reported for chemi-resistors6 and SAW sensors,11 and at least one order of magnitude better than previously reported FP sensors.34–36
Demonstration of functional sensor array and pattern analysis
From Fig. 4 it is clear that the sensitivities of different polymers to analytes vary greatly, which allows the use of response patterns of those polymers to better resolve vapor analytes. The sensor array was constructed by drop-coating the etched wells on a silicon chip, which confines the polymers and prevents cross-contamination. The wells were closely arranged so that they can be imaged with a CMOS imager. Fig. 5 shows the chromatograms obtained by each sensor for two different mass combinations of the four analytes. Due to the high linear speed of the analyte traveling inside the microfluidic channel, all FP sensors in the array were able to detect the same analyte virtually simultaneously.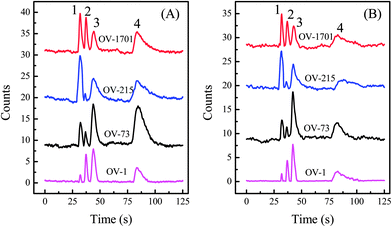 | ||
| Fig. 5 Chromatographic response of four FP sensors to a mixture of acetone (#1), methanol (#2), heptane (#3), and toluene (#4). The injected mass ratio for acetone, methanol, heptane, and toluene was (A) 1.4:14:1:2.6 and (B) 0.7:8.6:1:0.7, respectively. Chromatograms are vertically shifted for clarity. Analytes were delivered to the sensors using a series of OV-1 and Carbowax columns. | ||
The peak heights are used to extract the response patterns for each analyte. The response pattern shown in Fig. 6 corresponds to the injected mixture for Fig. 5(A). The response patterns clearly differ for each injected analyte, and match the initial testing results shown in Fig. 3. The error bars show a variation of less than 15 percent between runs, which will not impede the use of response patterns as a method of analysis. The response pattern for chromatogram in Fig. 5(B) that used a different mass combination is shown in Fig. S2†. Although the absolute peak height is different between Fig. 5(A) and (B), the corresponding response patterns agree very well with each other.
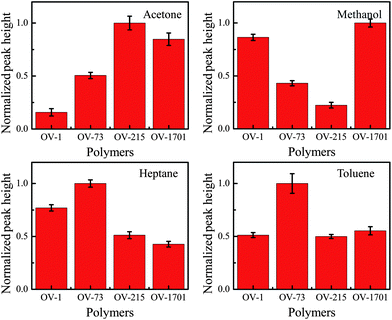 | ||
| Fig. 6 Normalized response patterns of each analyte with respect to the four polymers on chip derived from chromatograms shown in Fig. 5(A). Error bars show the standard deviation measured over 5 runs. Clear differences can be seen between the response patterns of each analyte, forming a basis of analyte identification. | ||
4. Conclusion and future work
We have fabricated and characterized a highly sensitive FP sensor array with orders of magnitude improvement in the VOC detection limit. The array is robust, reproducible, fast in response and compatible with μGC components. In particular, it has the ability to simultaneously gather information from multiple sensors to conduct pattern analysis for qualitative and quantitative analysis of VOC mixtures. Future work will focus on the development of a complete FP sensor based μGC system comprising micro-preconcentrators, micro-pumps, micro-valves, and micro-fabricated GC columns to address the actual needs in environmental monitoring, homeland security, and healthcare.Acknowledgements
This work was supported by the National Science Foundation (IOS 0946735) and the Lurie Nanofabrication Facility at the University of Michigan, a member of the National Nanotechnology Infrastructure Network (NNIN) funded by the NSF.References
- E. J. Staples and S. Viswanathan, IEEE Sens. J., 2005, 5, 622–631 CrossRef CAS.
- F. L. Dorman, J. J. Whiting, J. W. Cochran and J. Gardea-Torresdey, Anal. Chem., 2010, 82, 4775–4785 CrossRef CAS.
- M. C. Lonergan, E. J. Severin, B. J. Doleman, S. A. Beaber, R. H. Grubbs and N. S. Lewis, Chem. Mater., 1996, 8, 2298–2312 CrossRef CAS.
- E. Covington, F. I. Bohrer, C. Xu, E. T. Zellers and C. Kurdak, Lab Chip, 2010, 10, 3058–3060 RSC.
- C. K. Ho and R. C. Hughes, Sensors, 2002, 2, 23–34 CrossRef CAS.
- Q.-Y. Cai and E. T. Zellers, Anal. Chem., 2002, 74, 3533–3539 CrossRef CAS.
- T. Sukaew, H. Chang, G. Serrano and E. T. Zellers, Analyst, 2011, 136, 1664–1674 RSC.
- M. Penza and G. Cassano, Sens. Actuators, B, 2003, 89, 269–284 CrossRef.
- M. Rapp, J. Reibel, A. Voigt, M. Balzer and O. Bülow, Sens. Actuators, B, 2000, 65, 169–172 CrossRef.
- K.-T. Tang, C.-H. Li and S.-W. Chiu, Sensors, 2011, 11, 4609–4621 CrossRef.
- D. Matatagui, J. Martí, M. J. Fernández, J. L. Fontecha, J. Gutiérrez, I. Gràcia, C. Cané and M. C. Horrillo, Sens. Actuators, B, 2011, 154, 199–205 CrossRef.
- M. Fang, K. Vetelino, M. Rothery, J. Hines and G. C. Frye, Sens. Actuators, B, 1999, 56, 155–157 CrossRef.
- E. J. Staples, http://www.estcal.com/tech_papers/papers/Environmental/Phenol.pdf.
- E. J. Staples, T. Matsuda and S. Viswanathan, Environ. Strat. 21st Cent., Asia Pacific Conf., 1998.
- B. Sutapun, M. Tabib-Azar and A. Kazemi, Sens. Actuators, B, 1999, 60, 27–34 CrossRef.
- B. Michael, P. C. Kevin, B. Matrika, R. S. Philip and M. Mokhtar, IEEE Photonics Technol. Lett., 2007, 19, 255–257 Search PubMed.
- K. Schroeder, W. Ecke and R. Willsch, Opt. Lasers Eng., 2009, 47, 1018–1022 CrossRef.
- H. J. Patrick, A. D. Kersey and F. Bucholtz, J. Lightwave Technol., 1998, 16, 1606–1612 CrossRef CAS.
- A. Cusano, P. Pilla, L. Contessa, A. Iadicicco, S. Campopiano, A. Cutolo, M. Giordano and G. Guerra, Appl. Phys. Lett., 2005, 87, 234105 CrossRef.
- J. Zhang, X. Tang, J. Dong, T. Wei and H. Xiao, Sens. Actuators, B, 2009, 135, 420–425 CrossRef.
- B. Liedberg, C. Nylander and I. Lunström, Sens. Actuators, 1983, 4, 299–304 CrossRef CAS.
- C. de Julián Fernández, M. G. Manera, G. Pellegrini, M. Bersani, G. Mattei, R. Rella, L. Vasanelli and P. Mazzoldi, Sens. Actuators, B, 2008, 130, 531–537 CrossRef.
- K.-J. Chen and C.-J. Lu, Talanta, 2010, 81, 1670–1675 CrossRef CAS.
- T. Karakouz, A. Vaskevich and I. Rubinstein, J. Phys. Chem. B, 2008, 112, 14530–14538 CrossRef CAS.
- A. Ksendzov, M. L. Homer and A. M. Manfreda, Electron. Lett., 2004, 40, 63–65 CrossRef.
- S. I. Shopova, I. M. White, Y. Sun, H. Zhu, X. Fan, G. Frye-Mason, A. Thompson and S.-J. Ja, Anal. Chem., 2008, 80, 2232–2238 CrossRef CAS.
- Y. Sun, J. Liu, D. J. Howard, G. Frye-Mason, A. K. Thompson, S.-J. Ja and X. Fan, Analyst, 2010, 135, 165–171 RSC.
- N. A. Yebo, P. Lommens, Z. Hens and R. Baets, Opt. Express, 2010, 18, 11859–11866 CrossRef CAS.
- J. Villatoro, M. P. Kreuzer, R. Jha, V. P. Minkovich, V. Finazzi, G. Badenes and V. Pruneri, Opt. Express, 2009, 17, 1447–1453 CrossRef CAS.
- G. Gauglitz, A. Brecht, G. Kraus and W. Nahm, Sens. Actuators, B, 1993, 11, 21–27 CrossRef.
- R. K. D. Reichl, C. Krumme and G. Gauglitz, Appl. Spectrosc., 2000, 54, 583–586 CrossRef.
- J. Zhang, M. Luo, H. Xiao and J. Dong, Chem. Mater., 2006, 18, 4–6 CrossRef CAS.
- J. Liu, Y. Sun and X. Fan, Opt. Express, 2009, 17, 2731–2738 CrossRef CAS.
- J. Liu, Y. Sun, D. J. Howard, G. Frye-Mason, A. K. Thompson, S.-J. Ja, S.-K. Wang, M. Bai, H. Taub, M. Almasri and X. Fan, Anal. Chem., 2010, 82, 4370–4375 CrossRef CAS.
- C. Martínez-Hipatl, S. Muñoz-Aguirre, G. Beltrán-Pérez, J. Castillo-Mixcóatl and J. Rivera-De la Rosa, Sens. Actuators, B, 2010, 147, 37–42 CrossRef.
- K. Reddy, Y. Guo, J. Liu, W. Lee, M. K. Khaing Oo and X. Fan, Sens. Actuators, B, 2011, 159, 60–65 CrossRef CAS.
- M. Agah, J. A. Potkay, G. Lambertus, R. Sacks and K. D. Wise, J. Microelectromech. Syst., 2005, 14, 1039–1050 CrossRef.
- J. Liu, N. K. Gupta, K. D. Wise, Y. B. Gianchandani and X. Fan, Lab Chip, 2011, 11, 3487–3492 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2lc20922e |
| This journal is © The Royal Society of Chemistry 2012 |