Microfluidic single-cell cultivation chip with controllable immobilization and selective release of yeast cells†
Zhen
Zhu
*a,
Olivier
Frey
a,
Diana Silvia
Ottoz
b,
Fabian
Rudolf
b and
Andreas
Hierlemann
a
aETH Zurich, Department of Biosystems Science and Engineering (D-BSSE), Bio Engineering Laboratory (BEL), Mattenstrasse 26, CH-4058, Basel, Switzerland. E-mail: zhen.zhu@bsse.ethz.ch; Fax: +41 61 387 3989; Tel: +41 61 387 3296
bETH Zurich, Department of Biosystems Science and Engineering (D-BSSE), Computational Systems Biology (CSB), Mattenstrasse 26, CH-4058, Basel, Switzerland. E-mail: fabian.rudolf@bsse.ethz.ch; Fax: +41 61 387 3991; Tel: +41 61 387 3215
First published on 22nd December 2011
Abstract
We present a microfluidic cell-culture chip that enables trapping, cultivation and release of selected individual cells. The chip is fabricated by a simple hybrid glass-SU-8-PDMS approach, which produces a completely transparent microfluidic system amenable to optical inspection. Single cells are trapped in a microfluidic channel using mild suction at defined cell immobilization orifices, where they are cultivated under controlled environmental conditions. Cells of interest can be individually and independently released for further downstream analysis by applying a negative dielectrophoretic force via the respective electrodes located at each immobilization site. The combination of hydrodynamic cell-trapping and dielectrophoretic methods for cell releasing enables highly versatile single-cell manipulation in an array-based format. Computational fluid dynamics simulations were performed to estimate the properties of the system during cell trapping and releasing. Polystyrene beads and yeast cells have been used to investigate and characterize the different functions and to demonstrate biological compatibility and viability of the platform for single-cell applications in research areas such as systems biology.
1 Introduction
Cells in clonal populations can display profound variations at all levels for a variety of reasons. Yet, all of today's ‘omics’ measurement techniques require—mostly for sensitivity reasons—sample amounts consisting of a large number of cells, which consequently prevent the detection of cell-to-cell differences in the sampled population. Therefore, the analysis of single cells is necessary to obtain more precise information and, thus, reveal the properties of individual cells and cell-to-cell differences. In the past decades, single-cell analysis based on conventional technologies, such as capillary electrophoresis (CE)1–3 and flow cytometry (FC),4 was important in the fields of biology, medicine, and pharmacology. These technologies, however, analyze the resulting data from the instruments at an aggregate level, which restricts the further analysis of the intracellular information of individual cells and their intercellular communications. With the rapid development of MEMS and microfabrication technologies at the turn of the century, the concept called ‘Lab-on-a-Chip’ (LoC) or ‘Micro Total Analysis Systems’ (μTAS), based on microfluidic systems, has been increasingly attracting great interest of researchers for biological, chemical, and medical diagnostic applications. As a result of the micro-dimensional features of the microfluidic devices, these LoC or μTAS systems are appropriate for the micro-environmental mimesis, manipulation, reaction, separation and detection of single cells.In order to perform single-cell analysis on cell-based microfluidic chips, the first and most important step is to isolate the cells. One of the most popular methods is the microwell array,5–8 which enables a high-throughput cell-trapping platform, on which sedimentary cells just fit into tailored microwells where they are immobilized individually to undergo cultivation. A second, frequently used approach is based on microdam structures that are laid across a flow and mechanically retain cells at designated locations.9,10 Both of these single-cell culture systems are designed to cultivate a large number of isolated cells in a platform format, however, without any regime to sort out cells of interest for further analysis. Therefore, many researchers have been dedicated to developing single-cell culture systems incorporating cell isolation and cultivation functions as well as individual selection.
One of the proposed single-cell culture systems is associated with dielectrophoresis (DEP).11–18 When a dielectric particle is subjected to a non-uniform electric field, there is an induced force exerted on the particle, and this phenomenon is called DEP, whereas cells typically are dielectric particles.19–21 The DEP can exhibit a positive force (pDEP) that pushes the particle towards the region of the strong electric field or a negative one (nDEP) repelling the particle from regions of strong electric field. The force direction depends on the conductivity and permittivity of the particle relative to its surrounding medium, as well as on the frequency of the applied non-uniform electric field. Taff and Voldman14 designed a ‘ring–dot’ electrode geometry in a row/column array format on a microfluidic cell-sorting chip that can trap and retain individual cells above the dot by pDEP, and release the targeted single cell by simply switching off the AC signal corresponding to the relevant electrode. Retaining the immobilized cell in the strong electric field, however, can have an adverse effect on the cells, which can interfere with cell proliferation.22,23 To eliminate long-term exposure of cells to a strong electric field, microdam structures were integrated into microfluidic channels to trap and retain cells.15 A modified electrode-geometry was placed at each site to generate the nDEP force for the cell release. This allows for a high throughput cell culture platform with sorting capability. The retention of the trapped cells, however, is based on a continuous forward flow and is, therefore, sensitive to disturbance in the flow profile.
Another method is based on microwells, which can sedimentarily immobilize the cells by gravity and then selectively release them using optical scattering forces generated by a laser.24 Similarly, Tan and Takeuchi25,26 selectively release cells from trapping sites through an air bubble that is generated via laser heating behind the cell and pushes out the cell from its trapping site. The cells are previously encapsulated in alginate beads and hydrodynamically trapped in a specially designed channel geometry. Both methods require precise laser positioning equipment, and the exposure of the cells to intense coherent light sources or heat pulses that can influence the cell cycle and should therefore be thoroughly investigated.
Greve et al.27 proposed a method using hydrodynamic forces exerted by a common negative pressure on small holes in the bottom substrate to capture hundreds of cells in an array. Laminar flow conditions are then used to expose the cells to different drug concentrations. The fabrication requires silicon micromachining, and cell release has not been implemented. A similar retention method is used by Valero et al.28 Single cells are aspirated at the entrance of small side channels of a larger microfluidic channel. The chip, fabricated from a silicon substrate with etched channels and anodically bonded to glass, is used for electroporation. Only a small number of sites are integrated, and selective release of cells is not possible. However, this design has the potential to integrate more trapping sites and achieve a stable immobilization of single cells without any restrictions regarding the cell type and size. In both variations, a controlled negative pressure minimizes the influence on the cell metabolism.
In this article we combine trapping of single cellsvia mild suction in an array-format with selective single-cell release using a superimposed nDEP force at the specific trapping site. A main perfusion channel comprises several sub-cellular-sized side channels, where a single cell can be immobilized and retained for cultivation. Each site is equipped with an individually addressable microelectrode that allows generation of a non-uniform field and release of the cell of interest by the induced nDEP force pulse. The released cell is dragged by the passing fluid flow towards the outlet or subsequent units for further analysis.
The design, fabrication and operation of the microfluidic chip have been kept as simple as possible. A hybrid glass-SU-8-PDMS approach is used that simplifies critical fabrication steps, such as sealing, insures compatibility with inverted optical transmission light microscopes as well as fluorescent microscopes for biological applications, and allows straightforward fluidic and electrical connections.
To demonstrate the biological compatibility and application of the device, budding yeast (S. cerevisiae) has been used in various experiments. We successfully demonstrate that our microfluidic single-cell culture chip enables the single cells to be individually trapped and selectively released. Furthermore, the trapped cells are able to undergo proliferation successfully as demonstrated by long-term time-lapse monitoring of the budding process of yeast.
2 Materials and methods
2.1 Idea and design of the microfluidic device
The microfluidic cell culture device and the principle for individual trapping, cultivation and selective release of single cells are schematically illustrated in Fig. 1. The microfluidic chip consists of a cell culture channel with a width of 150 μm and two suction channels, each with a width of 300 μm, that are situated besides it. Several bottleneck orifices of 5 μm width are placed along the flow between the cell culture and suction channels, which results in an array-format of cell-immobilization sites in the cell culture channel (see 3D close-ups in Fig. 1a). The height of all channels is 30 μm. In this chip type, 10 trapping sites are located on each side of the culture channel at a pitch of 200 μm. The cells are loaded by introducing the cell suspension at the inlets of the culture channel using a conventional syringe pump, and are focused to flow along the channel wall by a side sheath-flow generated by cell culture medium applied through the medium inlet. To generate a sufficiently high suction force in order to achieve reliable cell immobilization, a controlled negative pressure (relative to atmospheric pressure, similarly hereinafter) is applied through these bottleneck orifices via the suction channel. | ||
| Fig. 1 Working principle of the microfluidic single-cell culture chip. (a) Picture of the whole device and schematic top view of a functional part without the PDMS cover for better visibility. The two close-ups show the 3D profile of one orifice with the immobilized cell. (b) Re(K(ω)) is the real part of the Clausius–Mossotti factor,35 which can be expressed in terms of complex permittivities of the cell and medium. A pDEP force FpDEP attracts the cell towards the region of stronger electric field and the orifice when Re(K(ω)) > 0; while an nDEP force FnDEP overcomes the cell trapping force Ftrap to repel the cell towards the region of low electric field when Re(K(ω)) < 0. | ||
The magnitude of the pressure is accurately controlled by a pressure controller, allowing for precise trapping of single cells at each orifice. After immobilization of single cells, they are cultivated under constant perfusion. Using a laminar flow regime, each side of the channel—and the respective resident cells—can be exposed to different/modulated culture media or reagents, which enables conducting different experiments on the same chip simultaneously.
Each orifice is further equipped with a 10 μm wide microelectrode, which is situated 2 μm into the orifices opposite to the cell culture channel wall. Thus, the microelectrodes never obstruct optical observation when using the inverted microscope. In the center of the cell culture channel, there is a long common electrode with a width of 50 μm.
The microelectrodes are used to generate a local nDEP force that repels the respective cell from its immobilization site. Therefore an AC voltage is applied between the electrode at the corresponding orifice and the long common electrode. As schematically illustrated in Fig. 1b, due to the specific geometry of the design, a strong electric field is generated by the electrode under the trapping orifice, which spreads out toward the larger electrode, creating the non-uniform field to polarize the cell. This asymmetric electrical field is required for DEP. The DEP force can be either attractive (pDEP) or repulsive (nDEP), depending on the frequency of the voltage and the relative polarizability of the cells and culture medium.21 When the cells are more polarizable than the medium, a pDEP force is generated that attracts the cells towards the region of the strong electric field. On the other hand, when the effective permittivity and/or conductivity of the cells are/is smaller than that of the medium, an nDEP force results that repels the cells from the region of the strong electric field around the trapping sites (Fig. 1b). It is notable that the immobilized cell can be released only when the nDEP force is high enough to overcome the suction force for the cell retention.
2.2 Device fabrication
The microfluidic chip is fabricated using a simple hybrid glass-SU-8-PDMS process, as shown in Fig. 2. First, 200 nm Pt electrodes with a 20 nm thick TiW adhesion layer beneath are patterned on a Pyrex wafer according to a common lift-off metallization process (Fig. 2a). The microfluidic structure with a thickness of 30 μm is fabricated in SU-8 3025 (MicroChem Co., USA) directly on top of the metal layer (Fig. 2b). With the mask aligner (MA/BA8 Gen3, SUSS Microtec AG, Germany), SU-8 patterns can be precisely aligned with Pt electrodes on the substrate to make sure that the cell immobilization orifices are accurately located at the right positions on the chip, which is needed for the proper functioning of cell-immobilization and cell-release of this chip. Both main fluidic channels and the cell-immobilization orifices are realized using SU-8. The wafer is then diced into single chips (Dicing Saw 8003, Esec AG, Switzerland) and irreversibly bonded to an unstructured PDMS (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w, Sylgard® 184, Dow Corning Co., USA) cover, which seals the microfluidic channels and completes the chip fabrication (Fig. 2d). For a tight seal, the SU-8 surface of each chip has to be modified with 3-aminopropyltriethoxysilane (APTES) (Sigma-Aldrich, USA) using a vapor phase silanization (Fig. 2c).29 The unstructured PDMS layer comprises punched fluidic inlets and outlets, and its surface has to be activated by oxygen plasma (200 Plasma System, TePla AG, Germany) before assembly. It is important to notice that for the final channel sealing, no precise alignment is required so that it can be performed under a conventional stereomicroscope. This substantially simplifies the final bonding procedure (Fig. 2d). All used materials have excellent light transmittance, so that a completely transparent microfluidic system for optical observation of samples in the microchannels results.
:1 w/w, Sylgard® 184, Dow Corning Co., USA) cover, which seals the microfluidic channels and completes the chip fabrication (Fig. 2d). For a tight seal, the SU-8 surface of each chip has to be modified with 3-aminopropyltriethoxysilane (APTES) (Sigma-Aldrich, USA) using a vapor phase silanization (Fig. 2c).29 The unstructured PDMS layer comprises punched fluidic inlets and outlets, and its surface has to be activated by oxygen plasma (200 Plasma System, TePla AG, Germany) before assembly. It is important to notice that for the final channel sealing, no precise alignment is required so that it can be performed under a conventional stereomicroscope. This substantially simplifies the final bonding procedure (Fig. 2d). All used materials have excellent light transmittance, so that a completely transparent microfluidic system for optical observation of samples in the microchannels results.
 | ||
| Fig. 2 Fabrication process illustrated in cross-sectional views along AA' and BB′ in Fig. 1. (a) Pt electrodes are fabricated by a common lift-off process; (b) the SU-8 structure is precisely aligned with the electrodes to create the fluidic components; (c) a monolayer of APTES is applied to the SU-8 surface to increase the bond strength to the unstructured PDMS cover; (d) PDMS cover is irreversibly bonded to SU-8 for sealing the microfluidic channels. | ||
2.3 Experimental setup
The bonded microfluidic chip is placed on an aluminium holder, which properly fits the inverted microscope stage (Zeiss Jena GmbH, Germany). Then the chip is screwed down tightly on the aluminium holder by a PMMA cover-flat with holes, through which PTFE tubings (Bohlender GmbH, Germany) connect the inlets and outlets of the chip to the corresponding fluidic control units. The bead samples, cell suspensions or media are first loaded into syringes (ILS Microsyringes AG, Germany) and then injected into the cell culture channel with a controllable continuous-flow provided by dedicated syringe pumps (neMESYS, Cetoni GmbH, Germany). The suction for cell trapping is exerted on the pressure ports of suction channels by a pressure controller (DPI 520, Druck Ltd., UK), which is connected to the in-house pressure and vacuum supply. To implement the release of selected cells by the nDEP force, the electrode pads are electrically connected to a printed circuit board (PCB) with switches that enable the activation of the AC voltage from a signal generator (8116A Pulse/Function generator 50 MHz, HP, USA). During the experiment, continuous imaging of the cell trapping, budding and selective release is recorded by either a digital CCD camera (Foculus 124TC, NET New Electronic Technology GmbH, Germany) or a monochrome CCD camera (F-View II, Soft Imaging System GmbH, Germany).2.4 Bead preparation
In the first stage, commercial polystyrene beads (Fluka, Sigma-Aldrich Production GmbH, Germany) with a standard size of 8 μm diameter are employed as a test model for cellular experiments. They are used to evaluate the functionalities of this microfluidic single-cell culture system, such as cell trapping and selective release by the nDEP force. Before loading the bead suspension into the syringe, we mix polystyrene beads into the medium, which is composed of a 0.01 M phosphate buffered saline (PBS) solution (Sigma-Aldrich Co., USA) supplemented with 1% w/v bovine serum albumin (BSA, Sigma-Aldrich Co., USA) and 1% v/v Triton X-100 surfactant (AppliChem GmbH, Germany), and then mechanically separate bead-clusters into individual beads using the ultrasonic bath (Bioblock® Scientific 86480, Fisher Scientific GmbH, Germany) at 350 W for 3 minutes at room temperature.2.5 Yeast cell preparation
Standard methods are used to grow liquid cultures of S. cerevisiae.30 The used yeast cells are BY4743, a diploid strain with a homozygous cln3 deletion as the relevant genotype. Cells are grown in complete synthetic medium made of 0.17% Yeast Nitrogen Base (YNB) (Difco™, BD GmbH, Germany), 0.5% ammonium sulfate (Sigma-Aldrich Co., Germany) and 2% glucose sulfate (Sigma-Aldrich Co., Germany) at 30 °C.The prepared yeast cell suspension is first diluted to reach a concentration of ∼1 × 105 to 1 × 106cells per ml in the cell culture medium. It is then carefully loaded into the syringe without inducing any bubbles and mounted in the syringe pump. Cell culture medium without cells is loaded into another syringe. Before delivering the cell suspension and cell culture medium into the chip, the fluidic channels are flushed with 1% BSA solution. The main purpose of the last steps is to attain a bubble-free filling of the channel system and a protein-surface coating of the channel surfaces to reduce cell stiction. Afterwards, the cell suspension is delivered into the cell culture channel by a continuous flow focused toward the sidewalls of the channel by means of a sheath-flow generated via a cell culture medium influx from the medium inlet (see Fig. 1a). When only one side of the cell-immobilization sites is going to be used in the experiments, the other inlet for the cell loading can be connected to an individual syringe with cell culture medium. The pressure ports of the suction channel are always connected to the pressure controller by conventional tubing. All flow rates stated in the following sections indicate the sum of the flow rates of all incoming fluids to the chip. The cell suspension growth curves were determined at room temperature using a Tecan M200 reader (Tecan Group Ltd., Switzerland).
2.6 Simulations
2D computational fluid dynamics (CFD) simulations are performed in COMSOL Multiphysics 3.5a using ‘Incompressible Navier–Strokes’ physics from the MEMS Module. If not otherwise stated, all subdomains are assigned with a density of ρ = 1000 kg m−3 and a dynamic viscosity of η = 0.001 Pa s (for water). Further, no-slip boundary conditions are applied to the walls of channels and orifices. The height of all channels and orifices is 30 μm and considered as a shallow channel approximation in the simulation.313D CFD simulations are performed with the same parameters and boundary conditions as for 2D simulations. Due to time and memory reasons, the simulated geometry is reduced to the critical section, hence, the orifice. The boundaries are chosen in uncritical regions and their values are taken from the 2D simulation results.
Simulations of the nDEP force are based on a multi-shell yeast cell model,32 where the physical parameters such as the thickness of each membrane, as well as the electrical parameters including the permittivity and conductivity of the cytoplasm, the cell membrane, and the cell wall of yeast cell, have been simplified and integrated into a simple dielectric particle with an effective complex permittivity. These aforementioned parameters are derived from the work of Talary et al.33 For the yeast cells, the relative permittivities of cell cytoplasm, membrane and wall are 50, 6, and 60 respectively. The respective conductivities are 0.3 S m−1, 0.25 μS m−1, and 24 mS m−1. The yeast cell cytoplasm has a diameter of 8 μm with an 8 nm membrane and 0.22 μm cell wall. The medium conductivity is 0.53 S m−1 and the relative permittivity is 81 referring to the yeast cell culture medium used in the study.
3 Results and discussion
3.1 Fluidic simulation and bead experimental results
To estimate the efficiency of the single-cell trapping process, we first carried out CFD simulations, based on the geometric design of this microfluidic chip. Fig. 3a illustrates the 2D simulation result of the flow-velocity field in the region of cell immobilization sites. A laminar inflow boundary condition with a flow rate of 4 μl min−1 is applied to the inlet of the cell culture channel, and the boundary conditions at the outlets of the cell culture channel and the suction channel are assigned with a pressure of 0 Pa. As observed from the simulation result, the maximum of the flow-velocity is located in the cell immobilization orifices, while there is a decrease of the flow-velocity from the leftmost orifice to the rightmost one. This phenomenon is a result of the pressure drop along the suction channel. A close-up view of the pressure distribution and velocity streamlines around the leftmost site (S1) is shown in Fig. 3b. The velocity streamlines indicate that a part of the liquid is diverted into the orifice due to the pressure drop and accelerated inside S1. This generates the hydrodynamic drag force on the cells in the culture channel and causes them to flow towards the immobilization orifice. An important fact here is that cells flowing approximately 20 μm away from the channel wall prefer flowing downstream instead of being trapped by the orifices. | ||
| Fig. 3 CFD analysis with resulting flow-velocity, pressure and force distributions before and after cell trapping. 2D simulation without cell immobilization: (a) flow-velocity field distribution in the area of the cell immobilization sites; (b) close-up of the first site in (a) with the pressure distribution and velocity streamlines. 3D CFD simulation with an immobilized cell: (c) flow-velocity field distribution in the horizontal cross-section at 4 μm above the bottom of the channel after a single cell has been trapped at the bottom of the orifice; (d) vertical cross-section through the center of the cell and immobilization orifice. The red arrows in (c) and (d) proportionally represent the calculated x-, y- and z-component of the net force exerted on the 8 μm diameter sphere. (e) Calculated forces in the x-, y-, and z-direction on the 8 μm diameter sphere in relation to different flow rates at a constant applied pressure of −2000 Pa at the suction channel. The boundary parameters in the 3D simulation of (c) and (d) are derived from the pressure values at the same boundary positions in the 2D simulation results depicted in (b). | ||
For comparison, we placed an 8 μm diameter spherical particle in front of the orifice serving as an immobilized single cell in 3D CFD simulation. From observations in real experiments using beads or yeast cells, they, indeed, are prone to be immobilized at the bottom of the channel. Therefore the sphere in the 3D simulation is placed at the bottom. With the boundary parameters derived from Fig. 3b, we obtained the 3D simulation results illustrated by the cross-sectional views in Fig. 3c and d. The maximal velocity in the orifice in Fig. 3c substantially decreases from 0.0657 m s−1 for the case without an immobilized sphere in a 2D simulation to 0.0324 m s−1 with the immobilized sphere in a 3D simulation. This prevents additional particles being trapped in the same plane around the already occupied orifice. Fig. 3d shows the vertical cross-section of the velocity profile, where the velocity in the orifice is still high—the maximal velocity is even slightly higher than that without the immobilized sphere in the 2D simulation, which could suggest that a second particle can be trapped more easily. But as already mentioned, trapping of two particles above each other was rarely observed in practice supporting the assumption that they tend to flow at the bottom of the channel and are trapped at the lower part of orifices.
As already observed and illustrated in Fig. 3a, there is a critical velocity decline along the 10 cell immobilization sites. Therefore a thorough characterization of the suction pressure affecting the cell-immobilization becomes important in this single-cell culture system. On the basis of CFD simulation results shown in Fig. 3, Fig. 4a shows the velocity variation of each immobilization site along the channel with the applied suction of 0 Pa, −500 Pa and −1000 Pa, respectively. This specific feature of the velocity variation, which creates the inhomogeneous immobilization of single cells, however, offers the advantage to control the orifice filling by adjusting the overall pressure applied through the suction channel. We hypothesize that there is a velocity threshold, dependent on the flow rate of cell loading and the suction pressure. The cells are immobilized at those sites, where the velocity is above this threshold, while other sites cannot capture any cells because of a subcritical velocity. In Fig. 4a, the velocity threshold is assumed to be in the range of 0.05 m s−1. In the case of 0 Pa sucking pressure, the velocities at S1, S2 and S3 are above the threshold, which creates the capability of cell immobilization at those three sites. As the suction pressure is lowered to −500 Pa, another site, S4, is able to capture a cell due to the higher velocity. Compared with the former two cases, the velocities at 10 sites are all above the threshold when a pressure of −1000 Pa is applied, which leads to the full immobilization at 10 sites.
 | ||
| Fig. 4 Controllable bead trapping at different sites by varying the suction pressure. (a) The velocity in the center of 10 cell immobilization orifices derived from the CFD simulation with the geometry shown in Fig. 3. The flow rate is 4 μl min−1, the pressure at the outlet of the cell culture channel is 0 Pa, and the pressure at the suction port is 0 Pa, −500 Pa and −1000 Pa, respectively. (b) Sites trapping single beads (8 μm diameter) versus the pressure variation at the suction port; pressure difference Δp = p(cell culture channel) − p(pressure port of suction channel); the black dot represents an occupied site with a single bead while the white dot represents an empty site. | ||
This effect is experimentally confirmed in Fig. 4b, which shows the results of single-bead trapping at different values of suction pressure. It should be noted that in these bead experiments, the microfluidic chip layout corresponds exactly to the simulation geometry: immobilization sites are placed only on one side of the cell culture channel. Since a syringe pump is employed to drive the fluid flow into the cell culture channel, it is difficult to estimate the pressure difference, Δp, between the cell culture channel and the pressure port of the suction channel. We therefore define the pressure difference as Δp12, when the cell immobilization sites, S1 and S2, are occupied by single beads. As the pressure difference is increased from Δp12 to Δp12 + 2500 Pa by lowering the pressure in the suction channel, single beads are “filling up” the cell-immobilization orifices until all 10 sites are occupied. Using intermediate pressure steps, it is, therefore, possible to reproducibly control the number of sites that will be occupied by a single bead. Higher values are required in practice, assumedly due to connection tubes that have not been considered in the simulations.
It is clear that the applied flow rate of fluid—that has been set to 4 μl min−1 in these experiments—has a significant influence on the process of cell-trapping and the trapping pressure has to be adjusted accordingly. At very high flow rates, Δp with 0 Pa suction pressure is already high enough to trap cells. In some cases the suction pressure has to be shifted towards positive pressures in order to avoid multi-cell trapping. Further, shear-forces and interactions with successive cells can cause trapped cells to be dragged or kicked away from their sites. Therefore the suction pressure must be high enough to retain the immobilized cells at the orifices. Consequently, the number of sites that are occupied by single cells can be controlled by the independent modulation of the magnitude of suction, which is in conformity with the velocity variation of each site along the channel. (Real images corresponding to Fig. 4b are illustrated in Fig. S1†, and Movie S1† shows the trapping status of each site in the case of Δp12 + 2000 Pa pressure difference, where S7 cannot stably trap a single bead, hence, being at a pressure difference just around the threshold.)
In order to release the immobilized single cell at the orifice, the applied nDEP force has to overcome the suction force, used to drag a cell flowing in the culture channel towards the immobilization orifice. The CFD model shown in Fig. 3 was used to calculate the suction force on the particle during immobilization at different flow rates. The net force is obtained by integration of the force acting on the overall surface of the sphere. For a flow rate of 4 μl min−1 with an applied pressure of 0 Pa, the x-, y- and z-components are 0.4 nN, 11 nN, and −1.9 nN, respectively. These values are visualized in Fig. 3c and d. For biological experiments with yeast cells, the flow rate is set to a much lower value of 0.1 μl min−1 with an applied pressure of −2000 Pa, resulting in a suction force with x-, y- and z-components of 0.01 nN, 0.4 nN, and −0.058 nN, respectively. The relationship between different applied flow rates at a constant applied pressure of −2000 Pa at the suction channel and the calculated force exerted on the 8 μm diameter sphere is shown in Fig. 3e. These simulation results point out that the major part of the net force is in the y-direction immobilizing the sphere at the respective orifice. Further, a negative z-component keeps the sphere at the bottom of the channel, whereas the drag force in the direction of the main channel (x-component) is relatively small.
At the same time, shear stress exerted on the cells by a liquid flow can have a significant influence on cell proliferation and viability.34 For this reason, the same CFD model shown in Fig. 3 was used to calculate the shear stress on the cells at the two different flow rates used in the experiments. The maximum value of the shear stress at a flow rate of 0.1 μl min−1 is on the order of 4 Pa, and it is on the order of 120 Pa in the case of a flow rate of 4 μl min−1. In both cases, the maximum values of shear stress are much lower than values reported to affect cell viability (>1000 Pa).34
3.2 DEP force simulation
In the simulation of DEP force, an AC voltage with a peak-to-peak amplitude of 20 V at a frequency of 5 MHz was used as stimulus for generating the non-uniform electric field. Fig. 5a shows the simulation results of the y-component of the nDEP force in the xy-plane (z = 0), in the region of the orifice. The force distribution shows values above 10 nN in an area of about 3 μm around the cell immobilization site. Further, nDEP force distribution curves are shown in Fig. 5b. They represent the absolute values in the y-direction along five lines parallel to the x-axis at a distance of 1 to 5 μm from the channel wall (see the black lines in Fig. 5a). At a distance of 1 μm in front of the trapping orifice, the nDEP force has a peak value of 50 nN with a valley of 25 nN in the center; with increasing distance from the trapping orifice—between 1 μm and 5 μm—the nDEP force decreases to 3 nN at y = 5 μm. Additionally, Fig. 5c shows the distribution of the y-component of the nDEP force in the yz-plane at x = 0. The nDEP force in the center of an immobilized cell (x = 0, y = 3 μm, z = 4 μm) is 5 nN. This corresponds to a decrease of approximately 50% with respect to the value of 10 nN at the bottom of the channel (z = 0). When comparing these values with the calculated suction force for cell immobilization (cf.Fig. 3e), the expected nDEP force of 5 nN is theoretically sufficient to repel a single yeast cell from its immobilization site at a flow rate of up to 2 μl min−1 with an applied pressure of −2000 Pa. It has to be mentioned that the flow rate used in bead/cell release experiments was only 0.1 μl min−1, which generates a much smaller trapping force on the bead/cell, as discussed in Section 3.1. Further, for higher flow-rates, the applied suction pressure has to be adjusted and is in most scenarios less negative, which further reduces the suction force. We can, therefore, conclude that the nDEP derived from the applied AC voltage (20 Vpp, 5 MHz) is sufficient for releasing single cells that are trapped at the orifices. On the other hand it has to be pointed out that yeast cells are not ideal spheres but ellipsoids, with even more irregular shapes caused by buds. Moreover, they can be clamped at the edges of immobilization orifices due to their flexible cell membranes. This can result in frictional forces increasing the required release force in some cases.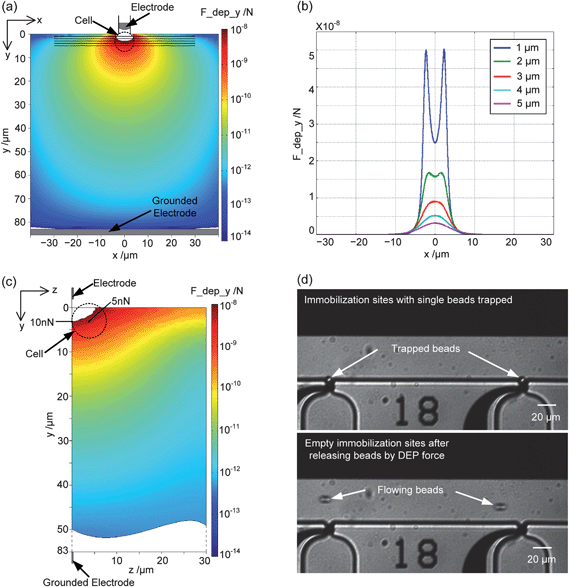 | ||
| Fig. 5 Characterization of single-bead releasing by the nDEP force. Simulation results of the nDEP force distribution (AC voltage: 20 Vpp at 5 MHz): (a) y-component of the nDEP force distribution around the cell orifice in the xy-plane (z = 0); (b) nDEP force along the five black lines in (a), which refers to the distance from the entrance of the trapping orifice; (c) y-component of the nDEP force distribution around the cell orifice in the yz-plane (x = 0). The dotted ring is the assumed 8 μm bead immobilized at the orifice. Experimental results of trapping and releasing of single beads: (d) Single beads are immobilized at each immobilization site (top picture) by an applied pressure of −2000 Pa with the flow rate of fluid at the cell culture channel of 0.1 μl min−1; and single beads are released (bottom picture) by the nDEP force activated by 15 Vpp AC voltage at 7.5 MHz. | ||
Fig. 5d shows the experimental result with polystyrene beads, which are trapped by suction and subsequently released by the nDEP force. The lower stimulus voltage results in a lower nDEP force value but is still sufficient because of the lower flow rate chosen that produces less suction force on the beads. The beads are released within the same second of the applied pulse resulting in a very fast response time. As soon as the voltage is switched off, new beads that have been introduced into the chip are attracted to the orifice. In the situation when the AC voltage is kept on, these new beads are repelled from the trapping sites and flow away from the channel wall (see bottom picture in Fig. 5d). This provides the possibility to protect immobilized cells from being hit by subsequent cells travelling along the channel wall and to define a specific time period, in which cells of interest can be captured. A specially designed repulsion electrode can, therefore, be structured upstream of the trapping sites in future device designs.
3.3 Trapping and releasing experiment of yeast cells
The intention of first experiments with yeast cells in the microfluidic single-cell culture chip was to provide evidence of its biological functionality. In many cases, the appropriate negative pressure for trapping single cells has to be optimized manually during the experiment, since it highly depends on the flow rate of cell suspension delivery and the location of cell immobilization orifices (see Section 3.1). Fig. 6a shows the experimental result of trapping and selectively releasing single yeast cells. As a result of the fluidic conditions, both of the immobilization sites successfully capture single yeast cells, and the cell trapped at the left site is a budding yeast cell. Afterwards, we stimulate the electrode under the right immobilization site by applying an AC voltage, which generates a sufficient nDEP force to repel the cell away from the original position so that it flows downstream with the fluid (Fig. 6a, middle left). The stimulus has no influence on the neighboring cell on the left side. Then, the left electrode is stimulated by the same signal as the right one to release the budding yeast cell from its immobilization site (Fig. 6a, bottom left). These simple experiments demonstrate the whole process of trapping and selectively releasing individual yeast cells—the intended functions of this microfluidic single-cell culture chip. | ||
| Fig. 6 Experimental results of yeast cells. (a) Single yeast cells (budding yeast cell at left site) are immobilized by the suction pressure of −2000 Pa with 0.1 μl min−1 flow rate, and immobilized cells are selectively released by the nDEP force, activated by a 5 MHz, 20 Vpp AC voltage. The long common electrode, located in the cell culture channel at 85 μm distance from the targeted electrode, is grounded and not shown in the figures for the reason of space. (b) 70 min real-time imaging of the budding process of an immobilized single yeast cell with the same flow conditions as in (a). | ||
With this approach, cells are not exposed to electric fields for long periods of time. The AC voltage stimulus is normally switched off after the releasing process, but it can be kept on to maintain an empty orifice. After cell loading, further cell introduction into the chip can be interrupted immediately. Continuous cell culture medium supply, however, is ensured by one of the other inlet channels.
One of the main advantages of the presented system becomes visible during the trapping process of single cells and is associated with the cell-releasing function, which can serve as a custom-defined cell-sorting approach before cell culturing and further analysis: if the trapped cell is not the right one (this can be determined by the appearance of cells under microscopy), then it can be released by the nDEP force until a favored cell is trapped. This procedure can be repeated independently with every site over the whole array until some or all trapping sites are loaded with specifically chosen cells that are of interest for further cultivation studies. As soon as one of the cells shows an interesting behavior it can be instantaneously repelled away from its original immobilization orifice by the applied electric stimulus for collection or further downstream analysis.
3.4 Long-term and real-time monitoring of the budding process of immobilized single yeast cells
To access whether the trapped yeast cells on the chip inhibit their ability to grow, we compared yeast cell growth in suspension culture and on the chip under identical environmental conditions. To this end, single yeast cells are trapped individually using the same conditions as in Section 3.3. The flow rate is kept constant during the loading of the cell suspension and the real-time recording of the budding process. An individual cell is selected when it is trapped in a stable position and is observed with time-lapse imaging at intervals of 30 s. Budding yeast morphology changes during the cell cycle and thereby provides an easy tool to monitor growth. In short, cells grow in an unbudded state (variable time) and start budding once they commit to a new round of the cell cycle. Budding starts at the G1/S boundary and proceeds until the completion of M phase which subsequently leads to cell separation (constant time; Fig. S2† for illustration). Fig. 6b illustrates the budding process of a trapped yeast cell for 70 min. In the first image, a tiny bud is visible on the mother cell. During the recording process of 70 min, the growth of the bud is continuously observed. This compares well with the reported literature values and is smaller than the ∼215 min generation time measured for this strain in suspension culture. However, at the end, the final splitting of the bud from its mother cell appears to be difficult. This may be due to the fact that shear-forces on the bud are not equal to shaking or ultrasonic mixing in standard incubators for yeast cells. Importantly, even though cells are not able to completely separate, the mother cell is able to re-enter the cell cycle by the outgrowth of another bud. This real-time and long-term monitoring of the budding process of an immobilized single yeast cell demonstrates that the force acting on the immobilized yeast cells by mild suction has no observable effect on the proliferation of yeast cells. The device allows for continuous monitoring of the growth of a single cell on an array-based platform over at least one cell cycle period.4 Discussion and conclusion
In this article, we presented a microfluidic cell-culture system that integrates the functions of immobilization, cultivation, and selective release of single cells. The fabrication of this microfluidic chip is based on a simple hybrid glass-SU-8-PDMS approach. Planar electrodes are patterned by a standard metallization process, on top of which a microfluidic network is constructed using SU-8 photoresist. The critical alignment of electrodes at the orifice is performed directly during exposure of SU-8 in the mask aligner, allowing submicron precision. A flexible PDMS flat cover, which does not require precise alignment, is used to conformally seal the microchannels, and standard tubing is used to make external connections. This simple chip construction allows for fast iteration cycles of chip design and fabrication, and provides single-use chips, thus substantially reducing the risk of cross-contamination between critical experiments. Further, all materials are transparent and compatible with light and fluorescence microscopy.Besides these features of the chip fabrication, the approach also provides superior handling and operation of the device. The immobilization strategy of the system, which is adapted from the previous work,27 employs a trapping force in the form of mild suction through the small orifices to reproducibly capture and stably retain the cells at defined positions. The applied mild suction minimizes the influence on the cell's behavior, such as the adverse effect of continuous exposure to an electric field during cell proliferation.14 The loading site number is controlled by modulation of the suction, which is precisely optimized by a pressure controller. The suction pressure can be controlled independently and allows for modification to different flow rates of cell loading and variable cell types. The immobilized cells can be selectively released by the stimulus of nDEP force at sub-second response time.
There are other impacts of the operation of this device. The combination of trapping and releasing regimes means that single cells can be selectively trapped on the chip. If the immobilized single cell is not the one of interest, it can be easily released by the nDEP force, and another interesting cell can be found. The cells on the chip undergo a continuous observation by an inverted microscope at any time as a consequence of the chip transparency. Cells are individually cultivated in a continuous flow without any cross-talk to their neighbors, even during long-term recording.
Some functions of the chip will be improved in future work. More cell immobilization sites will be integrated on the chip and, therefore, achieve a higher throughput in cell culturing. A serpentine-channel geometry, which enhances the channel length on chip, could be an option to situate more trapping sites. The DEP force can be employed to direct the cell flow along the channel wall during cell loading, or away from the channel wall during cell culturing.
Finally, the biological experiments on this chip have been performed with budding yeast cells (S. cerevisiae). The experimental results of individual trapping and selective release of yeast cells successfully demonstrate the expected functionalities of the microfluidic single-cell culture system. The 70 min long-term monitoring of the budding process of the immobilized single-yeast-cell demonstrates the biological compatibility of this system. Therefore, this microfluidic single-cell culture system provides a promising platform for single cell manipulation, cultivation, and analysis.
Acknowledgements
The authors acknowledge financial support through the Swiss SystemX.ch program in Systems Biology within the RTD project “CINA”, as well as the individual funding of Zhen Zhu from the Chinese Scholarship Council.References
- R. T. Kennedy, M. D. Oates, B. R. Cooper, B. Nickerson and J. W. Jorgenson, Science, 1989, 246, 57–63 CrossRef CAS.
- L. A. Woods and A. G. Ewing, Anal. Bioanal. Chem., 2003, 376, 281–283 CAS.
- L. A. Woods, T. P. Roddy and A. G. Ewing, Electrophoresis, 2004, 25, 1181–1187 CrossRef CAS.
- J. P. Nolan and L. A. Sklar, Nat. Biotechnol., 1998, 16, 633–638 CAS.
- J. R. Rettig and A. Folch, Anal. Chem., 2005, 77, 5628–5634 CrossRef CAS.
- D. K. Wood, D. M. Weingeist, S. N. Bhatia and B. P. Engelward, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10008–10013 CrossRef CAS.
- X. A. Figueroa, G. A. Cooksey, S. V. Votaw, L. F. Horowitz and A. Folch, Lab Chip, 2010, 10, 1120–1127 RSC.
- M. C. Park, J. Y. Hur, H. S. Cho, S.-H. Park and K. Y. Suh, Lab Chip, 2011, 11, 79–86 RSC.
- D. Di Carlo, N. Aghdam and L. P. Lee, Anal. Chem., 2006, 78, 4925–4930 CrossRef.
- D. Di Carlo, L. Y. Wu and L. P. Lee, Lab Chip, 2006, 6, 1445–1449 RSC.
- S. Fiedler, S. G. Shirley, T. Schnelle and G. Fuhr, Anal. Chem., 1998, 70, 1909–1915 CrossRef CAS.
- Y. Huang, S. Joo, M. Duhon, M. Heller, B. Wallace and X. Xu, Anal. Chem., 2002, 74, 3362–3371 CrossRef CAS.
- T. Muller, A. Pfennig, P. Klein, G. Gradl, M. Jager and T. Schnelle, IEEE Eng. Med. Biol. Mag., 2003, 22, 51–61 CrossRef.
- B. M. Taff and J. Voldman, Anal. Chem., 2005, 77, 7976–7983 CrossRef CAS.
- B. M. Taff, S. P. Desai and J. Voldman, Appl. Phys. Lett., 2009, 94, 084102 Search PubMed.
- M. S. Jaeger, K. Uhlig, T. Schnelle and T. Mueller, J. Phys. D: Appl. Phys., 2008, 41, 175502 CrossRef.
- K. Khoshmanesh, S. Nahavandi, S. Baratchi, A. Mitchell and K. Kalantar-zadeh, Biosens. Bioelectron., 2011, 26, 1800–1814 CrossRef CAS.
- K. Khoshmanesh, J. Akagi, S. Nahavandi, J. Skommer, S. Baratchi, J. M. Cooper, K. Kalantar-Zadeh, D. E. Williams and D. Wlodkowic, Anal. Chem., 2011, 83, 2133–2144 Search PubMed.
- H. A. Pohl and I. Hawk, Science, 1966, 152, 647–649 CrossRef.
- R. Pethig, Y. Huang, X. B. Wang and J. P. H. Burt, J. Phys. D: Appl. Phys., 1992, 25, 881–888 CrossRef CAS.
- R. Pethig, Biomicrofluidics, 2010, 4, 022811 CrossRef.
- R. Pethig, M. S. Talary and R. S. Lee, IEEE Eng. Med. Biol. Mag., 2003, 22, 43–50 CrossRef.
- A. Menachery and R. Pethig, IEE Proc.: Nanobiotechnol., 2005, 152, 145–149 CrossRef CAS.
- J. R. Kovac and J. Voldman, Anal. Chem., 2007, 79, 9321–9330 CrossRef CAS.
- W. H. Tan and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1146–1151 CrossRef CAS.
- W. H. Tan and S. Takeuchi, Lab Chip, 2008, 8, 259–266 RSC.
- F. Greve, L. Seemann, A. Hierlemann and J. Lichtenberg, J. Micromech. Microeng., 2007, 17, 1721–1730 Search PubMed.
- A. Valero, J. N. Post, J. W. van Nieuwkasteele, P. M. ter Braak, W. Kruijer and A. van den Berg, Lab Chip, 2008, 8, 62–67 RSC.
- S. Talaei, O. Frey, P. D. van der Wal, N. F. de Rooij and M. Koudelka-Hep, Hybrid Microfluidic Cartridge Formed by Irreversible Bonding of SU-8 and PDMS for Multi-Layer Flow Applications, Switzerland, Lausanne, 2009 Search PubMed.
- M. D. Rose, F. M. Winston and P. Hieter, Methods in Yeast Genetics: a Laboratory Course Manual, Cold Spring Harbor Laboratory Press, New York, 1990 Search PubMed.
- COMSOL, Comsol Multiphysics Version3.3: User's Guide, COMSOL AB., Los Angeles, 2006 Search PubMed.
- Y. Huang, R. Holzel, R. Pethig and X. B. Wang, Phys. Med. Biol., 1992, 37, 1499–1517 CrossRef CAS.
- M. S. Talary, J. P. H. Burt, J. A. Tame and R. Pethig, J. Phys. D: Appl. Phys., 1996, 29, 2198–2203 CrossRef CAS.
- H. Lange, P. Taillandier and J. P. Riba, J. Chem. Technol. Biotechnol., 2001, 76, 501–505 Search PubMed.
- J. Nilsson, M. Evander, B. Hammarstrom and T. Laurell, Anal. Chim. Acta, 2009, 649, 141–157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 illustrates the real images corresponding to Fig. 4b, Fig. S2 shows an illustration of the yeast morphology changes during the cell cycle and growth curves recorded in suspension culture for comparison, and Movie S1 shows the bead-trapping status of each site in the case of Δp12 + 2000 Pa pressure difference. See DOI: 10.1039/c2lc20911j |
| This journal is © The Royal Society of Chemistry 2012 |