Imitation of drug metabolism in human liver and cytotoxicity assay using a microfluidic device coupled to mass spectrometric detection†
Sifeng
Mao
,
Dan
Gao
,
Wu
Liu
,
Huibin
Wei
and
Jin-Ming
Lin
*
Beijing Key Laboratory of Microanalytical Method and Instrumentation, Department of Chemistry, Tsinghua University, Beijing, 100084, China. E-mail: jmlin@mail.tsinghua.edu.cn; Fax: +86-10-62792343; Tel: +86-10-62792343
First published on 17th November 2011
Abstract
In this work, we developed a microfluidic device for the imitation of drug metabolism in human liver and its cytotoxicity on cells. The integrated microfluidic device consists of three sections: (1) bioreactors containing poly(ethylene) glycol (PEG) hydrogel encapsulated human liver microsomes (HLMs); (2) cell culture chambers for cytotoxicity assay; and (3) integrated micro solid-phase extraction (SPE) columns to desalt and concentrate the products of enzymatic reaction. To verify the feasibility of the integrated microchip, we studied uridine 5′-diphosphate-glucuronosyltransferase (UGT) metabolism of acetaminophen (AP) and the cytotoxicity of products on HepG2 cells. The products of the reaction in one region of the device were injected into the cell culture chamber for cytotoxicity assay, while those in another region were directly detected online with an electrospray ionization quadrupole time-of-flight mass spectrometer (ESI-Q-TOF MS) after micro-SPE pre-treatment. Semiquantitative analysis achieved in the experiments could be related to the drug-induced HepG2 cell cytotoxicity. Total analysis time for one product was about 30 min and only less than 4 μg HLM protein was required for one reaction region. The results demonstrated that the established platform could be used to imitate drug metabolism occurring in the human liver, thereby replacing animal experiments in the near future. In addition, the integrated microchip will be a useful tool for drug metabolism studies and cytotoxicity assays, which are pivotal in drug development.
Introduction
In the drug discovery process, it is necessary and important to screen candidate drugs during the preclinical stage.1 Toxicity, which includes both drug and metabolism toxicity, is a leading cause of failure at all stages of drug development.2 Most regrettably, only a few of the candidate drugs are effective, and most of these do not become marketed drugs partly due to undesired biological consequences of drug metabolism and induced toxicity.3 Thus, it is valuable to establish a rapid and effective method to characterize the toxicities of drugs and their metabolites. Animal experiments, a common method for drug toxicity studies, are inadequate to evaluate toxicity because of the species-specific differences between humans and animals.4 As an example, the common analgesic acetaminophen, which was demonstrated to be safe in animal experiments, resulted in human acute liver failure when applied to humans.5 Therefore, it is imperative to develop a more effective and reliable method for drug discovery. We believe that imitation of drug metabolism and cytotoxicity is a good strategy for solving the problem.In the last 20 years, researchers have paid growing attention to microfluidics. As a technology, microfluidics offers many advantages including reduced sample and reagent consumption, integration, and high throughput.6–8 Therefore, microfluidic technology has been widely used in separations coupled to mass spectroscopy,6,9 high-throughput screening in drug development,10,11 single cell analysis12–14 and cell to cell communications.15
Cytochrome P450 enzymes, which play a central role in drug metabolism, have been studied for more than 50 years,16 and more than 75% of the human drug enzymatic reactions are catalyzed by them.17–20 Currently, chip-based P450 has been proven to be a good strategy for drug metabolism and drug toxicity assays. Mostly, human liver microsomes (HLMs) are used as the carrier of P450. Uridine 5′-diphosphate-glucuronosyltransferase (UGT) is an significant enzyme belonging to P450, so we use HLMs to supply UGT in the experiment. Zguris et al.21 reported a method to entrap HLMs in poly(ethylene) glycol (PEG) hydrogels; the results showed that the entrapped HLMs retained the enzymatic activity during photopolymerization. Ma et al.3 reported an integrated microfluidic device containing three layers—a 1 mm thick quartz layer embedded with separation microchannels and a three-microwell array, sandwiched between two PDMS layers—for a cytotoxicity assay and metabolite detection by capillary electrophoresis (CE). However, CE cannot characterize the structure of the metabolites and fails to determine the quantity of the metabolites. Therefore, we aim to develop an integrated microfluidic device to provide the possibility of combining a drug metabolism bioreactor, cell culture for toxicity assay and integrated SPE columns for sample pre-treatment.
Acetaminophen (AP), the most common cause of drug-induced liver disease,5 has been frequently selected as a model drug to verify the feasibility of the integrated microchip for drug metabolism. In phase I, AP was metabolized by cytochrome P450 enzymes in human liver. Then in phase II, AP was mainly conjugated with sulfate, or glucuronide (UGT pathway) to produce water-soluble compounds, while a small amount of AP was metabolized by CYP2E1 (which also belongs to P450 enzymes) to a highly electrophilic metabolite, N-acetyl-p-benzoquinone-imine (NAPQI). Normally, hepatic glutathione (GSH) subsequently detoxified NAPQI into nontoxic mercapturic acid and cysteine derivatives. When AP was excessive, the sulfation and UGT pathways would be saturated, which would lead to NAPQI increase through the P450 pathway. After hepatic GSH depletion, NAPQI would transfer into mitochondria to conjugate with mitochondria GSH, and the mitochondrial GSH covalent binding P-Bcl-Xl (GSH-P-Bcl-Xl) decreased, which caused the activation of c-Jun-N-terminal kinase (JNK). JNK then promoted translocation of death-inducing proteins, which led to mitochondrial permeability transition (MPT) and necrotic cell death (Scheme 1).5
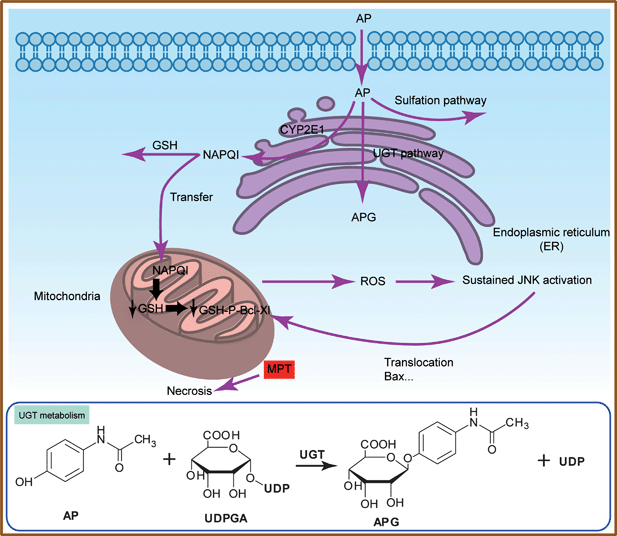 | ||
| Scheme 1 Acetaminophen metabolism pathway in human liver and toxicity on human liver cells. | ||
Herein, we have developed an integrated microfluidic device to imitate drug metabolism in human liver and toxicity in HepG2 cells, in which the drug metabolites were detected with online ESI-Q-TOF MS. After a bioreactor for in vitrodrug metabolism was fabricated, cell culture was realized in the modified microchannels and the microfluidic chip was directly connected to ESI-Q-TOF MS by a fused-silica capillary. We found that an AP solution appeared to exhibit less toxicity after UGT metabolism by encapsulated HLMs on the integrated microchip. The cell cytotoxicity is relevant to the amount of metabolite detected by MS: the higher the AP concentration, the lower the viability of the cells. This established platform is robust for the characterization of low levels of compounds and shows potential for new drug development and metabolism studies.
Experimental
Microfluidic device fabrication
Three different functional parts were designed on the microfluidic device to perform drug metabolism, a drug or metabolite cytotoxicity assay and ESI-Q-TOF MS analysis of metabolites (Fig. 1a). For drug metabolism, zigzag channels (Fig. 1b, 0.5 mm width × 16 cm length × 60 μm depth) were designed to form the bioreactor where HLMs (HMMCPL-PL050, Invitrogen, Austin, TX, USA) were encapsulated by PEG hydrogels. For metabolite analysis, straight channels (Fig. 1c, 1.0 mm width × 1.5 cm length × 60 μm depth) with triangular ends (40 μm width) were designed. Then, commercial SPE beads with diameters larger than the width of the triangular ends were immobilized to form on-chip micro-SPE columns. To carry out cytotoxicity assays, simple straight channels (Fig. 1d, 1.0 mm width × 1.5 cm length × 60 μm depth) were designed for cell culture and drug stimulation. Microfluidic devices were fabricated from PDMS (Sylgard 184, Dow Corning) using soft lithography and rapid phototyping, as reported elsewhere.22–24 Briefly, an SU-8 2050 negative photoresist (Microchem) was spin-coated on a clean silicon wafer. After spinning, the wafer was soft-baked (6 min at 65 °C and then 3 min at 95 °C) and then exposed to UV light through a photomask. After post-exposure bake and development, the master was hard-baked for 5 min at 65 °C. A 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight mixture of PDMS prepolymer and curing agent (Sylgard 184, Dow Corning) was stirred thoroughly and then degassed under vacuum for 1 h. Then, the mixture was poured onto the silicon master and baked in the oven at 80 °C for 2 h. After curing, the PDMS replicas were peeled off the master and the connection holes were punched with a stainless needle before sealing. The PDMS replicas were irreversibly sealed with glass slides after oxygen plasma (PDC-32G, Harrick Plasma, Ithaca, NY) treatment for 90 s. Finally, the devices were cured at 60 °C for 2 h to reinforce the bonding.
:1 weight mixture of PDMS prepolymer and curing agent (Sylgard 184, Dow Corning) was stirred thoroughly and then degassed under vacuum for 1 h. Then, the mixture was poured onto the silicon master and baked in the oven at 80 °C for 2 h. After curing, the PDMS replicas were peeled off the master and the connection holes were punched with a stainless needle before sealing. The PDMS replicas were irreversibly sealed with glass slides after oxygen plasma (PDC-32G, Harrick Plasma, Ithaca, NY) treatment for 90 s. Finally, the devices were cured at 60 °C for 2 h to reinforce the bonding.
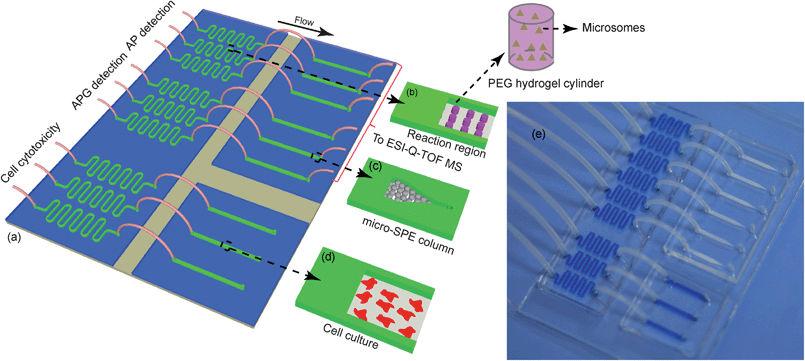 | ||
| Fig. 1 Microfluidic device for cell culture, metabolite analysis and cytotoxicity assay. (a) The integrated microfluidic device. (b) Microchannels for HLM encapsulation by PEG hydrogels. (c) Design of the on-chip micro-SPE column. (d) Cell culture channel. (e) An image of the microfluidic device filled with a blue dye in the bioreactor part cell culture part. | ||
Preparation of PEG hydrogels inside microchannels for entrapping HLMs
The hydrogel microstructures were generated using a simple method, controlled photopolymerization, as reported in our earlier work.25 It is a common method used to fabricate hydrogels with different shapes inside microchannels; moreover, bioactive reagents could be encapsulated inside the hydrogel where bioreactions could take place.21 In short, the PEG-DA (MW 700, Sigma, St. Louis, MO) precursor containing PEG-DA, phosphate buffered saline (PBS, 0.01 M, pH 7.4) and a photoinitiator, 2-hydroxy-2-methylpropiophenone (Sigma, St. Louis, MO) was polymerized inside microchannels under a UV light beam (wavelength 340–380 nm) ejected from an inverted fluorescence microscope (Leica DMI 4000 B, Wetzlar, Germany). For visualization, the microsome membrane was stained with Vybrant DiO (ex/em: 484/501 nm) cell-labeling solution (Invitrogen, Carlsbad, CA).For hydrogel microstructures entrapping HLMs, the composition of components was 70 μL of PEG-DA, 1 μL of 2-hydroxy-2-methylpropiophenone, 26 μL of PBS, 2 μL of HLMs at 20 mg protein mL−1 and 1 μL Vybrant DiO cell-labeling solution (1 mM). After the mixture was incubated at 37 °C for 20 min, the HLMs were stained with Vybrant DiO cell-labeling solution. Then UV light beam was manually controlled to induce the photopolymerization of the PEG-DA precursor to generate hydrogel microstructure arrays with cylindrical shapes, and the remaining unreacted hydrogel precursor was flushed away with PBS. A 300 ms exposure of UV light (700 mW) for photopolymerization was controlled by an automatic shutter on the microscope. Micrographs were captured by a cooled CCD camera (Leica DFC300 FX, Wetzlar, Germany) coupled with the microscope.
Microfluidic cell culture
HepG2 cells (Cancer Institute & Hospital of the Chinese Academy of Medical Science, Beijing, China) were maintained in Dulbecco's modified Eagle's minimal essential medium (DMEM) with high glucose supplemented with 10% fetal bovine serum (FBS), 100 U mL−1penicillin and 100 U mL−1streptomycin in a humidified atmosphere of 95% air, 5% CO2 at 37 °C. The HepG2 cells were maintained in petri dishes for 2–3 days prior to microfluidic experiments. All the experiments were carried out when the cells were in the exponential growth phase. After assembly, the cell culture channels were filled with 0.1% poly-L-lysine (PLL) solution for 1 h at room temperature and then washed with PBS. Before cell loading, the modified channels were first sterilized under UV light for 5 min, and then rinsed with 75% ethanol and sterile PBS. HepG2 cells were then detached from the petri dishes with 0.25% trypsin, resuspended in DMEM containing 10% FBS, counted with a hemocytometer and injected into the cell culture channel at a density of ∼2 × 106cells mL−1. To decrease liquid evaporation and supply medium, the top surface of the chip was covered with cell culture medium. The seeded microfluidic device was then placed into a humidified atmosphere of 95% air, 5% CO2 at 37 °C for more than 6 h, and cells became attached to the bottom of the channels.Fabrication of on-chip micro-SPE column and integration with ESI-Q-TOF MS detection
The reactions catalyzed by P450 enzymes took place in a complex environment (two membrane-bound protein components, with cofactor, MgCl2).6 In the experiments, it is difficult to avoid introducing PEG-DA and other salt ions if the metabolites are directly injected into the ESI source without sample pre-treatment, so integrated on-chip micro-SPE columns for sample pre-treatment before ESI-Q-TOF MS analysis are indispensable.To fabricate micro-SPE columns, the polymeric SPE beads were suspended in methanol and then injected into the channel, where they would be intercepted by the triangular end while methanol could flow out. Then the channel was conditioned with methanol and water at a flow rate of 10 μL min−1 for 5 min. The extraction procedure consisted of pressure-driven loading, washing, and elution steps. To characterize AP, reaction products in one region were introduced into the micro-SPE column filled with Bond Elut Plexa SPE beads (Varian, Moscow, Russian Federation, diameter 45 μm), a special commercial SPE material with different size hollow cavities which adhere to size exclusive separation schemes, by water at a flow rate of 2 μL min−1 for 5 min using a 500 μL Hamilton gastight syringe (Hamilton, Las Vegas, NV). Then the micro-SPE column was washed with 5% (v/v) methanol in water at a flow rate of 5 μL min−1 for 10 min to remove any unbound proteins, salts, and then connected directly to the ESI source utilizing a fused silica capillary with an inner diameter of 50 μm and outer diameter of 365 μm. The target was eluted from the micro-SPE column with 5% ammonia in methanol and analyzed by online ESI-Q-TOF MS. To characterize acetaminophen-glucuronides (APG) which is a product of AP metabolism, the micro-SPE column was filled with Cleanert PAX SPE microspheres (Agela Technologies, Tianjin, China, diameter 55 μm) instead, and the solutions for sample introduction, washing and elution were 1% ammonia in methanol, methanol and water, and 2% formic acid in methanol, respectively.
To quantify the target compounds in the metabolites, a series of drug concentrations were prepared to construct a calibration curve. The drug solutions were incubated in the reaction region with PEG hydrogel microstructures for 12 h at 37 °C, then extracted with the on-chip micro-SPE columns and detected by ESI-Q-TOF MS. ESI-Q-TOF MS, which has a high sensitivity and high resolution, is an elegant tool for substance identification and structure characterization.
In this work, we combined different channels on two chips with polytetrafluoroethylene (PTFE) tubes. The syringe for eluting was connected to the inlet of the on chip micro-SPE column by a fused-silica capillary (20 cm length), and the on-chip micro-SPE column was connected directly to the ESI source by a fused-silica capillary (15 cm length) with a PTFE cannula at the end.
Microfluidic device operation
The operation procedures included drug metabolism, cell culture and cytotoxicity assays, reaction product pre-treatment and ESI-Q-TOF MS analysis. Briefly, a suspension of HepG2 cells at a density of ∼2 × 106cells mL−1 was loaded into the cell culture channel, which was then maintained in a humidified atmosphere of 95% air, 5% CO2 at 37 °C for cell adhesion. To reduce liquid evaporation and maintain cell culture medium supply, the top surface of the chip was covered with culture medium. Sample mixtures designed as shown in Table 1 were introduced into different reaction regions (5 μL min−1), where PEG hydrogels had been encapsulated with or without HLMs. To reduce evaporation, the inlets and outlets were plugged with pipet tips filled with PBS. AP, APG, uridine 5′-diphosphate glucuronic acid (UDPGA) and MgCl2 were purchased from Sigma (St. Louis, MO). After incubation at 37 °C for 12 h, reaction products were injected into micro-SPE columns for desalting and concentration prior to ESI-Q-TOF MS detection at a flow rate of 10 μL min−1 for 1 min. The washing step (5 μL min−1) and elution step (3 μL min−1) for AP and APG detection are the same. Meanwhile, for the cytotoxicity assay, reaction products were injected into cell culture channel at a flow rate of 10 μL min−1 for 1 min. Also, we used the same scheme to alleviate water evaporation. After metabolite exposure at 37 °C for 12 h, the channels were washed with PBS (1 μL min−1) and then cells were stained with a 5 μg mL−1 Hoechst 33342 (ex/em: 350/461 nm) solution at 37 °C for 30 min. Subsequent visualization and fluorescence analysis of the stained cells was carried out using an inverted fluorescence microscope. The fluorescence images were analyzed by image processing and analysis software (Image-Pro 6.0, Media Cybernetics, Silver Spring, MD).| MgCl2 (mM) | UDPGA (mM) | AP (mM) | HLM (mg mL−1) | |
|---|---|---|---|---|
| AP control | 20 | 10 | 0 | 0.4 |
| HLM control | 20 | 10 | 10 | 0 |
| Experiment | 20 | 10 | 10 | 0.4 |
Results and discussion
Fabrication of PEG hydrogels containing HLMs
HLMs were stained with Vybrant DiO as detailed in the experimental part, green fluorescence could be detected using the inverted fluorescence microscope when the stained HLMs were stimulated by a blue light beam (wavelength 450–490 nm) (Fig. 2a, microscope objective 10×, microscope eyepiece 10×). After the PEG precursor was irradiated for 300 ms under UV light beam (7 mW), an array of PEG hydrogel cylinders (height, ∼60 μm) were fabricated as shown in Fig. 2b. After PBS washing, the fluorescence image of an individual cylinder (diameter, ∼120 μm) was shown in Fig. 2c (microscope objective 20×, microscope eyepiece 10×). When the PEG cylinders containing stained HLMs were washed with PBS, no substantial decrease in fluorescent intensity was observed (Fig. S1†), indicating that no HLMs escaped from the hydrogels. ESI-Q-TOF MS analysis showed that the metabolite APG was detected after incubation, which verified that the drug AP could reach HLMs in PEG hydrogels and the metabolite APG could get out of the hydrogels. Many experiments have reported that the mesh size of the PEG hydrogels was about 100 nm, calculated using the Peppas–Merrill equation,21,26 so molecules (like AP and APG) which are smaller than the mesh size can get in and out of the PEG hydrogels freely. | ||
| Fig. 2 Micrographs of HLMs and hydrogel cylinder. (a) HLMs stained with Vybrant DiO. (b) Controlled encapsulation of HLMs inside hydrogel microstructures. (c) Fluorescence image of HLMs entrapped inside PEG hydrogel microstructures. The edge of the microstructure was labelled with dash lines. | ||
Integration of on-chip micro-SPE column and ESI-Q-TOF MS detection
The integrated micro-SPE columns were shown in Fig. 3a, where packing materials were kept in the microchannels evenly and tightly, as shown in Fig. 3b and 3c. The triangular ends of the microchannels were not as narrow as designed (∼60 μm width instead of 40 μm) and were larger than that of the SPE particles (diameter 45 μm), mainly resulting from unavoidable expansion in chip fabrication process. However, the SPE particles could still be intercepted firmly in the end, stopped by two adjacent parallel particles. | ||
| Fig. 3 Integrated on-chip micro-SPE columns. (a) Photograph of the on-chip micro-SPE column. (b) The triangular end of a micro-SPE column. (c) Particles tightly accumulated in the channel. | ||
For MS detection, we optimized the collision energy of ESI-Q-TOF MS (data not shown) and the pH of the eluates (data not shown) for APG detection. Finally, 2% formic acid in methanol was selected as the eluate, and 6 eV was the best collision energy. For AP, 5% ammonia in methanol was selected as the eluate and 10 eV selected as the collision energy after optimization (data not shown). When characterizing AP, the identity was confirmed by the mass spectra obtained under the mass range m/z 50–300 in negative ion mode. As shown in Fig. 4a, AP (1.00 mM) with [M–H]− = 150.0 was detected after the corresponding extraction procedure, and its structure was further identified by an MS/MS spectrum. After an extra energy of 10 eV was applied on the extracted ion m/z 150.0, we obtained the corresponding fragments, m/z 107.0 (Fig. 4b). For APG, the identity was confirmed by the mass spectrum obtained under the mass range m/z 50–500 in the negative ion mode. As shown in Fig. 4c, APG (1.00 mM) with [M–H]− = 326.0 was detected after corresponding extraction procedure, and its structure was further identified by the MS/MS spectrum. The corresponding fragments, m/z 150.0 and m/z 175.0 were obtained by adding an extra energy of 10 eV on the extracted ion m/z 326.0 (Fig. 4d). All the results above showed that AP and APG could be well extracted by an on-chip micro-SPE column (Plexa) and an on-chip micro-SPE column (PAX), respectively. In addition, they could be detected by ESI-Q-TOF MS and their structures were further identified by MS/MS spectra.
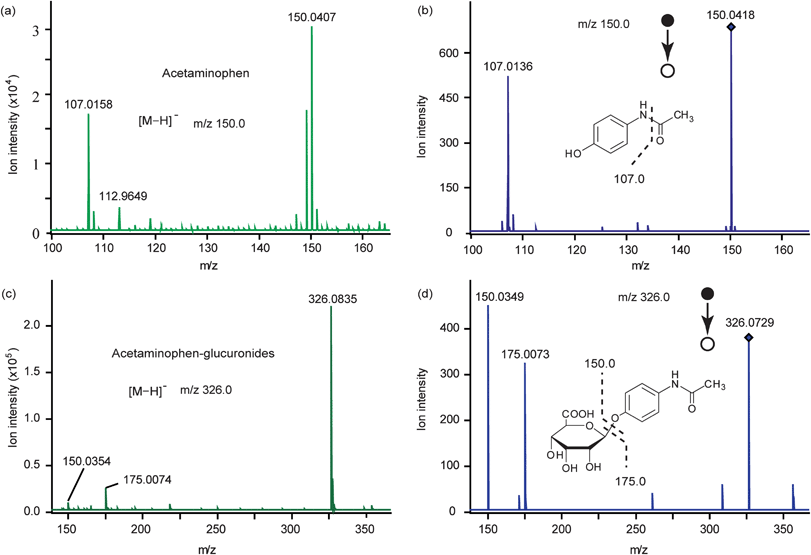 | ||
| Fig. 4 Mass spectra of standard acetaminophen (AP) (a) and acetaminophen-glucuronides (APG) solution (c) and corresponding tandem mass spectra (b, d). Spectra were obtained in the negative ion mode. | ||
The drugs, AP and APG, were respectively dissolved in 50% (v/v) ethanol–water at a concentration of 1 M, and diluted to the required concentrations using cell culture medium. Then the solutions were incubated and extracted following the extraction procedure, and eluted for direct MS detection to obtain the relationship between intensity and drug concentration. The molecular ion peak intensity (Y) increased linearly with AP concentration (X) in the range of 0.20–2.00 mM, and the fitting formula was Y = 3.70 × 103 + 2.52 × 104X (R2 = 0.989) (Fig. 5a); while the relationship between the molecular ion peak intensity (Y) and APG concentration (X) was linear in the range of 0.01–1.00 mM, and the fitting formula was Y = 6.55 × 102 + 1.80 × 104X (R2 = 0.968), as shown in Fig. 5b.
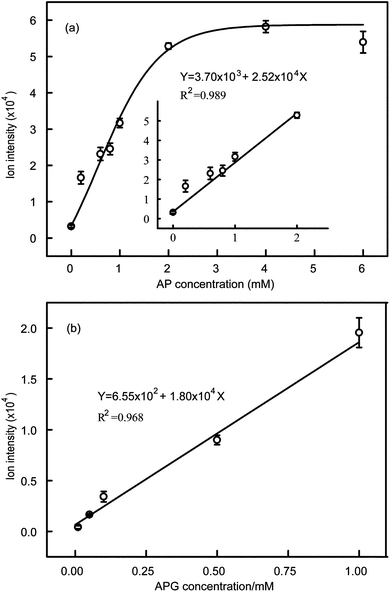 | ||
| Fig. 5 (a) Relationship between molecular ion intensity and AP concentration. (b) Relationship between molecular ion intensity and APG concentration. The standard error bars are the variation of three individual experiments. | ||
Metabolite detection and its cytotoxicity on HepG2 cells
According to the relationship between cell viability and AP concentration shown in Fig. S2†, a 10 mM concentration close to the IC50 (8 mM) was chosen for the experiment. In contrast, the metabolite APG is nontoxic to HepG2 cells (Fig. S2†). Three solutions (as detailed in Table 1) were injected into the reaction regions with or without encapsulated HLMs, respectively, for AP detection, APG detection and cell cytotoxicity. After metabolism and drug exposure, the cells were stained with Hoechst 33342. Hoechst 33342 is a blue fluorescent dye which penetrates the cell membrane and links to DNA, it is usually selected to detect apoptosis through nucleus dyeing. The permeability of cell membrane will increase when cells are at the apoptotic stage, which allows the dye to go through the cell membrane more easily. Therefore cells which have started the death process or are dead appear brighter than viable cells after staining by Hoechst 33342. We can consider that the drug concentration is toxic to cells, no matter whether the cells are dead or have begun the death process. The live/dead viability kit only classifies cells as live (green fluorescence) or dead (red fluorescence), and will classify cells between live and dead states as live cells, so it cannot show us all the abnormal cells. Therefore, we used Hoechst 33342 in the experiments. Fluorescence images obtained with the inverted fluorescence microscope are shown in Fig. 6a, while survival rate of cells are shown in Fig. 6b. Cell viabilities of AP control, HLM control and experiment groups (as detailed in Table 1) were 99.3%, 56.2% and 79.3%, respectively. For AP control group, it is easy to understand that cell viability is higher without drug AP cytotoxicity. In HLM control group, AP could not be metabolized into non-toxic compounds, so a high concentration AP solution would cause cell cytotoxicity and results in low cell viability, 56.2%. When the AP was converted to APG by UGT metabolism in the experiment group, the cell viability raised to 79.3% compared to the HLM control group 56.2%. These results demonstrated that while most AP was detoxified by UGT metabolism, the residual still exhibited cytotoxicity on HepG2 cells; as a result the cell viability of the experiment group was lower than that of the AP control group. To ensure the results of the HLM control and the experiment are the statistically different, we performed a t-test. When we set the significant level α = 0.05, the results showed us that the P value = 0.02 < 0.05 and t value = 2.92 > tα (tα = 2.13). Therefore, there were significant differences.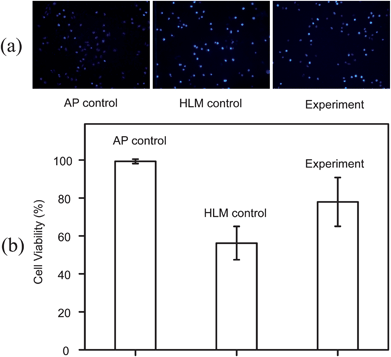 | ||
| Fig. 6 The metabolite cytotoxicity on HepG2 after UGT metabolism of AP. (a) Fluorescence images of HepG2 cells stained with Hoechst 33342, after incubation with AP control, HLM control and experiment groups. (b) Cell viabilities of three different designed groups. The standard error bars show the variation of three individual experiments. | ||
To investigate the relationship between the cell cytotoxicity assay and the products of the reaction, it is necessary to determine the concentrations of AP and APG. As shown in Fig. 7a, AP with [M–H]− = 150.0 was detected after extraction by an on-chip micro-SPE column (Plexa), while APG with [M–H]− = 326.0 was detected after extraction by an on-chip micro-SPE column (PAX) (Fig. 7b). The results showed that no AP or APG was detected in the AP control group, only AP was detected in the HLM control group and both AP (the ion intensity of the peak m/z 150.0 = 44247) and APG (the ion intensity of the peak m/z 326.0 = 11738) were detected in the experiment group. According to the calibration curves shown in Fig. 5a and 5b, AP and APG concentrations in experiment group were ∼1.61 mM and ∼0.62 mM, respectively, which could be related to cell cytotoxicity on HepG2 cells. AP concentration became lower when AP was metabolized to APG and other metabolites, so cytotoxicity became lower accompanied by high cell viability 79.3%, shown in Fig. 6b. Therefore, AP concentrations detected by ESI-Q-TOF MS could be related to cell cytotoxicity.
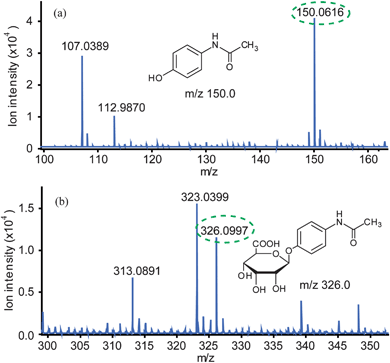 | ||
| Fig. 7 Mass spectra of metabolites in the experiment group. (a) Mass spectrum of AP in metabolites. (b) Mass spectrum of APG in metabolites. | ||
Conclusions
An integrated microfluidic device coupled with ESI-Q-TOF MS was developed for drug metabolism research and cytotoxicity assay. Besides low reagents consumption, high efficiency and high through-put capability, the platform offered several vital advantages: (1) on-chip drug-induced cytotoxicity assay and ESI-Q-TOF MS analysis, which allowed the determination of the relationship between drug-induced cytotoxicity and drug/metabolite concentration; (2) integrated on-chip micro-SPE columns for sample pre-treatment, which is convenient for online detection by ESI-Q-TOF MS; (3) an encapsulated HLMs bioreactor for imitating drug metabolismin vitro. Because of the successful imitation of drug metabolism in human liver, we demonstrate that human liver has the ability to detoxify AP and excessive AP could induce HepG2 cell death, which are consistent with reported work.5 In the future, the established platform is expected to be a useful tool for assaying drug-induced cytotoxicity caused by P450 metabolism and for metabolite screening studies.Acknowledgements
This work was supported by National Natural Science Foundation of China (Nos. 20935002 and 90813015) and National Basic Research Program of China (2007CB714507). Prof. Richard N. Zare (Department of Chemistry, Stanford University) is thanked for critical appraisal of the manuscript.Notes and references
- E. F. A. Brandon, C. D. Raap, I. Meijerman, J. H. Beijnen and J. H. M. Schellens, Toxicol. Appl. Pharmacol., 2003, 189, 233–246 CrossRef CAS.
- J. A. Kramer, J. E. Sagartz and D. L. Morris, Nat. Rev. Drug Discovery, 2007, 6, 636–649 CrossRef CAS.
- B. Ma, G. H. Zhang, J. H. Qin and B. C. Lin, Lab Chip, 2009, 9, 232–238 RSC.
- S. R. Khetani and S. N. Bhatia, Nat. Biotechnol., 2008, 26, 120–126 CrossRef CAS.
- B. K. Gunawan and N. Kaplowitz, Clin. Liver Dis., 2007, 11, 459–475 CrossRef.
- S. Benetton, J. Kameoka, A. M. Tan, T. Wachs, H. Craighead and J. D. Henion, Anal. Chem., 2003, 75, 6430–6436 CrossRef CAS.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- P. J. Resto, B. J. Mogen, E. Berthier and J. C. Williams, Lab Chip, 2010, 10, 23–26 RSC.
- B. Bajrami, L. L. Zhao, J. B. Schenkman and J. F. Rusling, Anal. Chem., 2009, 81, 9921–9929 CrossRef CAS.
- N. N. Ye, J. H. Qin, W. W. Shi, X. Liu and B. C. Lin, Lab Chip, 2007, 7, 1696–1704 RSC.
- R. Wei, G. D. Li and A. B. Seymour, Anal. Chem., 2010, 82, 5527–5533 CrossRef CAS.
- B. Huang, H. K. Wu, D. Bhaya, A. Grossman, S. Granier, B. K. Kobilka and R. N. Zare, Science, 2007, 315, 81–84 CrossRef CAS.
- X. J. Li, V. Ling and P. C. H. Li, Anal. Chem., 2008, 80, 4095–4102 CrossRef CAS.
- D. Wlodkowic, S. Faley, M. Zagnoni, J. P. Wikswo and J. M. Cooper, Anal. Chem., 2009, 81, 5517–5523 CrossRef CAS.
- E. S. Park, A. C. Brown, M. A. DiFeo, T. H. Barker and H. Lu, Lab Chip, 2010, 10, 571–580 RSC.
- F. P. Guengerich, M. V. Martin, C. D. Sohl and Q. Cheng, Nat. Protoc., 2009, 4, 1245–1251 CrossRef CAS.
- U. A. Meyer, J. Pharmacokinet. Biopharm., 1996, 24, 449–459 CrossRef CAS.
- L. Vignati, E. Turlizzi, S. Monaci, P. Grossi, R. de Kanter and M. Monshouwer, Toxicology, 2005, 216, 154–167 CrossRef CAS.
- R. F. Staack and G. Hopfgartner, Anal. Bioanal. Chem., 2007, 388, 1365–1380 CrossRef CAS.
- J. A. Williams, R. Hyland, B. C. Jones, D. A. Smith, S. Hurst, T. C. Goosen, V. Peterkin, J. R. Koup and S. E. Ball, Drug Metab. Dispos., 2004, 32, 1201–1208 CrossRef CAS.
- J. C. Zguris, L. J. Itle, D. Hayes and M. V. Pishko, Biomed. Microdevices, 2005, 7, 117–125 CrossRef CAS.
- D. Qin, Y. N. Xia and G. M. Whitesides, Adv. Mater., 1996, 8, 917 CrossRef CAS.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- A. Rosenthal, A. Macdonald and J. Voldman, Biomaterials, 2007, 28, 3208–3216 CrossRef CAS.
- J. J. Liu, D. Gao, H. F. Li and J. M. Lin, Lab Chip, 2009, 9, 1301–1305 RSC.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Mass transfer to HLMs in PEG hydrogel microstructures, drug and metabolite APG cytotoxicity on HepG2 cells. See DOI: 10.1039/c1lc20678h |
| This journal is © The Royal Society of Chemistry 2012 |