Dynamically bonded layer-by-layer films for self-regulated insulin release†
Xi
Zhang
,
Ying
Guan
and
Yongjun
Zhang
*
State Key Laboratory of Medicinal Chemical Biology and Key Laboratory of Functional Polymer Materials, Institute of Polymer Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: yongjunzhang@nankai.edu.cn; Tel: +86-22-23501657
First published on 3rd July 2012
Abstract
Layer-by-layer assembled films using reversible/dynamic bonds between the polymer pairs as driving forces are dynamic in nature. They may disintegrate under certain conditions as a result of the breakage of the dynamic bonds. More importantly they disintegrate gradually under conditions of equilibrium control, making them suitable for sustained drug release. Here insulin release from dynamic LbL films was demonstrated as an example. The films were fabricated from a fluorescently-labeled insulin–PVA (PVA: polyvinyl alcohol) conjugate and poly[acrylamide-co-3-(acrylamido)-phenylboronic acid], using a reversible covalent bond, i.e., phenylboronate ester bond, as the driving force. Film fabrication was followed by UV-vis and fluorescence spectroscopy. In all cases linear film growth was observed. The film growth rate increases with decreasing pH and increasing ionic strength of the assembly solutions. Successful integration of insulin into the films was achieved by covalent conjugation of insulin with PVA. When immersed in an aqueous solution, the films disintegrate gradually, thus releasing insulin into the media. Insulin release rate increases with increasing pH and decreasing ionic strength of the media. More importantly, it increases with increasing glucose concentration in the media. The glucose response of the film was attributed to the conversion of PBA (PBA: phenylboronic acid) groups in the film from a neutral form to a negatively charged one as a result of the formation of glucose–PBA complexes, thus increasing the rate of the film disintegration.
Introduction
A bond that can selectively undergo reversible breaking and reformation, usually under equilibrium conditions, is defined as a “dynamic bond”.1 There are two categories of dynamic bond: non-covalent and covalent ones. Non-covalent dynamic bonds are supramolecular interactions, such as π–π stacking, hydrogen bonding, and so on, while dynamic covalent bonds are covalent bonds that can break and reform under appropriate conditions without irreversible side reactions.1 By introduction of these reversible/dynamic bonds into polymer structures, novel polymers with dynamic properties, such as a changeable and tunable constitution even after polymerization, were designed.2 These polymers were named “dynamers” or dynamic polymers.2 The constitution and structure of the polymer were tuned by manipulation of the equilibrium in reversible bonding in response to environmental conditions under thermodynamic control.1,3Similarly dynamic bonds can be introduced into layer-by-layer (LbL) assembled films. LbL assembly is a method to fabricate thin films by alternately dipping a substrate into two polymer solutions with complementary functional groups.4 Up to now, numerous films have been fabricated using this method; these nanostructured films have found a wide range of applications.5,6 Because of their extraordinary advantages, such as ease of preparation, versatility and fine control over the film structure and properties, these films are highly desirable for biomedical applications.7 A lot of applications of LbL films in biomimetics, biosensors, drug delivery, protein and cell adhesion, mediation of cellular functions, and implantable materials have been reported.7
The driving force for the buildup of an LbL film, i.e., the interaction between the polymer pairs, can be electrostatic interaction,8,9 covalent bonding,10,11 hydrogen bonding,12–15 charge transfer interaction,16,17 and so on.18 Many of them are reversible, i.e., they fall into the category of dynamic bonds, as they can undergo reversible breaking and reformation, usually under equilibrium conditions.1–3 Just like dynamic polymers, LbL films linked with dynamic bonds, or dynamic LbL films, will have a changeable and tunable constitution. Their properties can be tuned by shifting the equilibrium of the reversible bonding in response to environmental conditions under thermodynamic control.
A unique property of the dynamically bonded films is that they can disintegrate under certain conditions. It has long been observed that hydrogen-bonded LbL films disintegrate immediately when immersed in alkali solutions.19,20 In these cases the film disintegrates as a result of the direct disruption of the bonds. Film disintegration can also be achieved under conditions of equilibrium control. We previously observed the gradual erosion of hydrogen-bonded LbL films when immersed in water.21–23 Films fabricated using reversible phenylboronate ester bonds as driving forces also disintegrate gradually in aqueous solutions.24 Here we exploited the gradual disintegration of dynamic LbL films under conditions of equilibrium control for sustained drug release.
LbL films are extraordinarily suitable to be used as drug delivery vehicles.25 For this purpose, two strategies have been used. In the first strategy, drugs are encapsulated in LbL microcapsules and the LbL films act as diffusion barriers.5,26,27 As the permeability of the capsule wall can be tuned by a lot of factors, such as the bilayer number of the coatings,28 ionic strength,29 pH and temperature,30 drug release rate can be controlled by fine control of these factors.31 In the second strategy, drugs are integrated into macroscopic planar LbL films during or after the film fabrication. They are released by diffusion32,33 or the degradation of the films. For example, polyionic drugs as one of the building blocks were used to fabricate LbL films with a degradable polymer.34–36 The drugs are released as the films are eroded in water. In these cases the release rate is determined by the degradation rate of the degradable polymer.34–36
The strategy we used here is different from the above-mentioned ones. Similar to the second strategy mentioned above, the drugs are loaded in the film as one of the building blocks and they are released as a result of the film disintegration. However, the film disintegration is not achieved via the degradation of the polymer, but via the breakage of the bonds between the two polymers. Here dynamic LbL films were fabricated from an insulin–PVA (PVA: polyvinyl alcohol) conjugate and a polymer with phenylboronic acid (PBA) groups using reversible phenylboronate ester bonding as the driving force. Insulin release was achieved as a result of the film disintegration. In addition, the release rate can be tuned by glucose, making this system potentially useful for self-regulated insulin release.
Experimental section
Materials
3-Aminophenylboronic acid hemisulfate (APBA), (3-aminopropyl)triethoxysilane (APTES), poly(acrylic acid) (PAA) and 1-(3-dimethyl aminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) were purchased from Alfa Aesar. Polyvinyl alcohol (PVA) (Mw = 15.9 × 104, Mn = 9.9 × 104), acryloyl chloride, acrylamide (AAm), 5-isothiocyanate fluorescein (FITC), insulin from bovine pancreas (Sigma-Aldrich), 2,2′-azo-bis-isobutyronitrile (AIBN) and other reagents were purchased from local providers. 3-(Acrylamido)phenylboronic acid (AAPBA) was synthesized from acryloyl chloride and APBA according to ref. 37. FITC-labeled insulin (FITC-insulin) was prepared from FITC and insulin according to ref. 38.Polymer synthesis
Poly[acrylamide-co-3-(acrylamido)-phenylboronic acid] (P(AAm–AAPBA)) was prepared according to ref. 24. Briefly, 0.500 g of AAm, 0.071 g of AAPBA and 4.0 mg of AIBN were dissolved in 40 mL of DMF. The mixture was purged with nitrogen for 30 min to remove dissolved oxygen, and then heated to 70 °C to initiate the free radical polymerization under nitrogen bubbling. The reaction was allowed to proceed for 12 h. The product was precipitated in acetone, filtered, washed three times with acetone, and dried under vacuum. The yield was about 70%.Insulin–PVA conjugate was synthesized as follows. 0.250 g of PVA and 0.250 g of EDC were dissolved in 20 mL of 50 mM pH 7.4 phosphate buffer. 7.5 mg of FITC-insulin were dissolved in 5 mL of 50 mM pH 7.4 phosphate buffer. The two solutions were mixed slowly. The pH was adjusted to ∼5.0 using HCl. The reaction was allowed to proceed for 4 h. The resultant product was dialyzed against water for 3 days with frequent water changes, lyophilized, and stored at 4 °C in the dark.
Film fabrication
Quartz slides with a size of 44 mm × 10 mm × 1 mm were used as substrates for the fabrication of LbL films. The substrates were first cleaned in boiling piranha solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 v/v H2O2–H2SO4 mixture) (caution: this solution is extremely corrosive!), rinsed thoroughly with deionized (DI) water and dried. To introduce amino groups, the substrates were immersed in a 1 wt% toluene solution of APTES for 12 h, washed in toluene for two minutes and dried at about 100 °C in an oven. A layer of PAA was assembled by immersing in a 0.1 wt% aqueous solution of PAA (pH 3.0) for 10 min followed by washing with DI water. Phenylboronic acid (PBA) groups were introduced by treating the substrates in an aqueous solution containing 7.5 mM APBA and 12.5 mM EDC for 4 h.
:7 v/v H2O2–H2SO4 mixture) (caution: this solution is extremely corrosive!), rinsed thoroughly with deionized (DI) water and dried. To introduce amino groups, the substrates were immersed in a 1 wt% toluene solution of APTES for 12 h, washed in toluene for two minutes and dried at about 100 °C in an oven. A layer of PAA was assembled by immersing in a 0.1 wt% aqueous solution of PAA (pH 3.0) for 10 min followed by washing with DI water. Phenylboronic acid (PBA) groups were introduced by treating the substrates in an aqueous solution containing 7.5 mM APBA and 12.5 mM EDC for 4 h.
0.1 wt% insulin–PVA and P(AAm–AAPBA) solutions were prepared by dissolving the corresponding polymer in 0.05 M pH 8.5 phosphate buffer. A layer of insulin–PVA was first assembled by immersing the PBA-modified substrates in insulin–PVA for 4 min, then rinsed in DI water or phosphate buffer of the same pH twice, each for 3 min, to remove the loosely bounded insulin–PVA. A layer of P(AAm–AAPBA) was then assembled using the same procedure. Multilayer LbL films were fabricated by repeating this cycle.
Insulin release
Test tubes with a diameter of 14 mm were used in the release experiments, to which 6 mL of release media were added. 10 bilayer LbL films were then immersed in the media. Temperature was controlled at 37 °C with a refrigerated circulator. At appropriate intervals, the release media were removed and the same volume of fresh media was added. The concentration of insulin–PVA in the release media was measured by fluorescence spectrometry. To measure the total amount of insulin–PVA in the films, the films were completely disintegrated by immersion in 50 mM fructose. Before use, the media containing glucose were left overnight to ensure that they had reached mutarotation equilibrium.39 The experiments were all carried out in triplicate. The results were reported as mean ± standard deviation.Characterizations
UV-vis absorption spectra were measured on a TU 1810PC UV-Vis spectrophotometer (Purkinje General, China). Fluorescence spectra were measured on a Shimadzu RF-5301PC spectrofluorometer using an excitation wavelength of 490 nm. 1H NMR spectra of P(AAm–AAPBA) were recorded on a Varian UNITY-plus 400 NMR spectrometer using D2O as solvent. The molecular weight of the polymers was determined on a Viscotek chromatograph (GPCmax, Houston, TX) using a Triple Detector Array 302 (Viscotek, Houston, TX) with refractive index, viscosity, and static light scattering at 30 °C. Column Set: TSK GMPWXL. Eluent: 0.2 M NaNO3/0.1 M NaH2PO4. Flow rate: 1.0 mL min−1.Results and discussion
Synthesis of the polymers
The LbL films were fabricated from poly[acrylamide-co-3-(acrylamido)-phenylboronic acid] (P(AAm–AAPBA)) and FITC-labeled insulin–PVA conjugates. P(AAm–AAPBA) was synthesized by free radical copolymerization of AAm and AAPBA. The apparent Mw of the copolymer was measured to be 8.5 × 104 by GPC. The content of the PBA groups in the copolymer was determined by NMR to be 5.3 mol% from the peak integration ratio between the benzene protons and the methylene protons of the main chain.Insulin was labeled with FITC to facilitate its detection by fluorescence spectroscopy. It was then conjugated with PVA so it could be integrated into the film during film fabrication. An insulin molecule bears two carboxylic acid groups and three amino groups, providing reaction sites for modification. Previously insulin conjugates with various small molecules38,40,41 and macromolecules42–44 have been synthesized to improve their physicochemical, immunological, and pharmacological properties. It has been shown that conjugation with PEG can increase its physical stability, reduce immunogenicity and antigenicity, and extend its circulation half-life.45 In this work, FITC–insulin was synthesized according to ref. 38. Then a PVA conjugate was synthesized by EDC coupling as shown in Scheme 1. GPC traces of the parent PVA and the conjugate are shown in Fig. 2S and 3S,† respectively. Mn and Mw of the parent PVA were measured to be 9.9 × 103 and 15.9 × 103, respectively, while Mn and Mw of the conjugate are 14.4 × 103, and 19.4 × 103, respectively. From the difference in Mn upon conjugation, one can estimate that about 72.9% PVA chain was conjugated with one FITC–insulin molecule assuming a homogeneous insulin distribution.
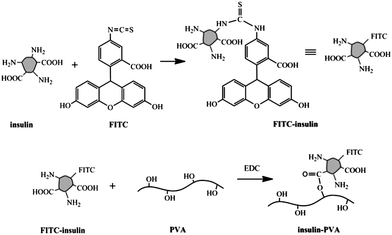 | ||
| Scheme 1 Synthesis of FITC-labeled insulin–PVA conjugate. | ||
Fabrication of insulin–PVA/P(AAm–AAPBA) multilayer films
The films were fabricated by alternately soaking the substrates in solutions of insulin–PVA and P(AAm–AAPBA). Before film fabrication, the substrates were modified sequentially with (3-aminopropyl)triethoxysilane, poly(acrylic acid) and 3-aminophenylboronic acid to introduce PBA groups onto the substrate surface. Then insulin–PVA and P(AAm–AAPBA) were deposited sequentially as a result of the in situ formation of phenylboronate ester bonds between the 1,3-diol groups on insulin–PVA and PBA groups on P(AAm–AAPBA) as shown in Scheme 2. | ||
| Scheme 2 In situ formation of phenylboronate ester bonds between PVA and P(AAm–AAPBA). | ||
The fabrication process was followed by UV-vis spectroscopy. As shown in Fig. 1, the absorption of the film increases with increasing number of dipping cycles. The peak at ∼247 nm and the shoulder at ∼212 nm are the absorption of phenyl groups, mainly from P(AAm–AAPBA). From the inset of Fig. 1, one can see the absorbance of the film, either at 212 nm or at 247 nm, increases linearly with increasing number of dipping cycles. The result indicates the amount of P(AAm–AAPBA) deposited in each dipping cycle is almost the same.
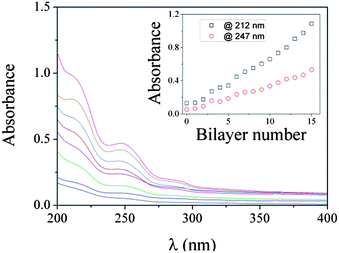 | ||
| Fig. 1 UV-vis absorption spectra of insulin–PVA/P(AAm–AAPBA) films with various bilayer numbers, which are 0, 2, 4, 6, 8, 10, 12, and 14, respectively, from bottom to top. The films were fabricated in pH 8.5 50 mM phosphate buffer. Inset: absorbance at 212 nm and 247 nm as a function of bilayer number. | ||
The effects of pH and ionic strength on the film buildup were studied. As shown in Fig. 2, film fabrications were all successful, and linear film growth was observed in all cases. Fig. 2A indicates that the film growth rate increases with decreasing pH. The faster film growth at a lower pH can be explained by the fewer binding sites between the film surface and the depositing polymer chains, because the binding constant between a PBA group and a diol group decreases with decreasing pH.46 Therefore there will be a high density of chain segments extending from the surface, providing a larger effective surface area for the deposition of the next layer. Lower pH also results in a lower dissociation degree of the PBA groups (pKa of PBA was reported to be 8.6, ref. 47), and, in turn, less negatively charged P(AAm–AAPBA) polymer. Therefore the polymer will adopt a less extended conformation, which is also favourable for producing thick films.14 Since both P(AAm–AAPBA) polymer and the film are negatively charged, a lower dissociation degree of the PBA groups also results in weaker electrostatic repulsion between them, which is favourable for film buildup too. Because of all these reasons, film growth is faster at a lower pH. Fig. 2B shows film growth is faster when assembled in a higher concentration buffer. The faster growth can be attributed to the higher ionic strength which screens the electrostatic repulsion among the phenylboronate ions. Therefore the P(AAm–AAPBA) chains will adopt a less extended conformation, which is favourable for producing thick films. Higher ionic strength also screens the repulsion between the surface and the P(AAm–AAPBA) chain, thus facilitating the film buildup. As a result, more film materials will be deposited in each cycle in a higher concentration buffer.
 | ||
| Fig. 2 Film growth as monitored by absorption increase at 212 nm in 50 mM phosphate buffer of various pHs (A) and various concentrations of pH 8.5 phosphate buffer (B). | ||
Since insulin was fluorescently labeled, film growth was also monitored using fluorescence spectroscopy. As shown in Fig. 3, the self-assembled film exhibits an emission band peaked at 525 nm. The fluorescence intensity increases as the film grows (Fig. 4), which is in agreement with the results shown in Fig. 1 and 2. Also a linear relationship between the fluorescence intensity and bilayer number was found, suggesting again that the amount of film material deposited in each cycle is almost the same.
 | ||
| Fig. 3 Fluorescence excitation spectra of insulin–PVA/P(AAm–AAPBA) films with various dipping cycles as marked in the figure. The excitation wavelength is 490 nm. | ||
 | ||
| Fig. 4 Fluorescence intensity of LbL films fabricated from P(AAm–AAPBA) and FITC-labeled insulin–PVA conjugate (□), and P(AAm–AAPBA) and a mixed solution of FITC-insulin and PVA (○). | ||
The above results indicate LbL films containing insulin were successfully fabricated from P(AAm–AAPBA) and insulin–PVA conjugate. As a control, LbL films were also fabricated by alternate dipping of the substrates in a P(AAm–AAPBA) solution and a mixed solution of PVA and FITC–insulin. As shown in Fig. 4, fluorescence intensity of the film does not increase regularly as in the case of the insulin–PVA conjugate. The weak fluorescence of the film can be attributed to the physical adsorption of a small amount of FITC–insulin during the film fabrication. This result suggests that insulin cannot be effectively integrated into the film by simple blending with PVA. In contrast, when covalently conjugated with PVA, insulin can be integrated successfully (Fig. 4).
Insulin release from insulin–PVA/P(AAm–AAPBA) LbL films
As insulin–PVA and P(AAm–AAPBA) are linked with reversible phenylboronate ester bonds, the resulting films are dynamic in nature. When the films are soaked in an aqueous solution, the films disintegrate gradually, thus releasing insulin into the solution (Scheme 3). Fig. 5S† shows a typical result. The fluorescence intensity of the solution increases with time, indicating the successful release of insulin. | ||
| Scheme 3 Disintegration of insulin–PVA/P(AAm–AAPBA) film when immersed in aqueous solution as a result of the breakage of phenylboronate ester bonds. | ||
Fig. 5 shows the release profile of insulin in phosphate buffer with various pHs. In all cases the release rate is relatively fast at the beginning and slows down gradually. The gradually decreased release rate may be attributed to the polydispersity of the polymers. It was known that the intermolecular interaction among the polymer chains increases with increasing molecular weight. Therefore shorter chains, with a relatively weaker interaction with other chains, will be released at a quicker rate. Previously we have shown that the erosion rate of the hydrogen-bonded poly(acrylic acid)/poly(vinyl pyrrolidone) films in water decreases with increasing molecular weight.21 The initial quick release of the short chains leaves a film with a relatively higher molecular weight. Therefore the disintegration rate decreases gradually, and so does the insulin release rate.
 | ||
| Fig. 5 Release profiles of insulin from insulin–PVA/P(AAm–AAPBA) films soaked in 50 mM phosphate buffer with various pHs. T = 37 °C. | ||
Fig. 5 also shows that the release rate increases with increasing pH. Increasing pH has two effects on the film disintegration. On one hand, more phenylboronate ester bonds may form between insulin–PVA and P(AAm–AAPBA) in the film because high pH is favourable for their formation.46 In this context, the disintegration of the film should be slower at a higher pH. On the other hand, as pH increases, more PBA groups in the film will disassociate. As the P(AAm–AAPBA) chains bear more negative charges, the electrostatic repulsion among them will increase. Therefore, the film disintegration should be quicker at higher pH. The final results should be a combination of the two opposite effects. It seems that the second effect of pH dominates the first one. Therefore the film disintegration becomes faster at higher pH, resulting in a faster insulin release.
To study the effect of ionic strength, insulin release was measured in pH 8.5 phosphate buffers with various concentrations. As shown in Fig. 6, the release rate decreases with increasing buffer concentration. The reduced release rate can be explained by the screening of the electrostatic repulsion among the negatively charged groups in the film as a result of the increased ionic strength of the release media. The solubility of the polymers also decreases as buffer concentration increases, which can be regarded as another reason for the slower insulin release.
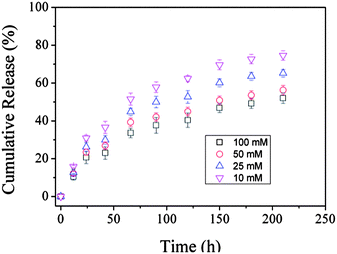 | ||
| Fig. 6 Release profiles of insulin from insulin–PVA/P(AAm–AAPBA) films soaked in various concentrations of pH 8.5 phosphate buffers. T = 37 °C. | ||
Glucose-regulated insulin release
The above results show that insulin can be released from the insulin–PVA/P(AAm–AAPBA) film as a result of the film disintegration in aqueous solutions, and the release rate can be tuned by pH and ionic strength. For insulin release, it is more desirable if the release rate can be controlled by glucose. To test the effect of glucose, release kinetics of insulin in pH 8.5 release media containing various amount of glucose were measured. As expected, insulin release rate increases with increasing glucose concentration, as shown in Fig. 7. | ||
| Fig. 7 Release profiles of insulin from insulin–PVA/P(AAm–AAPBA) films soaked in 50 mM pH 8.5 phosphate buffer containing various concentrations of glucose. T = 37 °C. | ||
The acceleration of insulin release in the presence of glucose can be attributed to the reaction between glucose and PBA groups in the film.24,48 Glucose can react with PBA groups in two ways (Scheme 4). It may bind with the free PBA groups and convert them from a neutral, hydrophobic form to a negatively charged, hydrophilic form.49,50 In this way, the P(AAm–AAPBA) polymer will bear more negative charges, resulting in increased electrostatic repulsion among the polymer chains. Therefore the film disintegrates at a higher speed and releases insulin at a higher speed. Glucose may also compete with PVA for the PBA binding sites.51,52 As a result, the crosslink density of the film will decrease, which is also favourable for film disintegration.
 | ||
| Scheme 4 Two ways glucose reacts with PBA groups in the films: binding with the free PBA groups (upper) and competing with PVA for the PBA binding sites (lower). | ||
As we mentioned above, the binding constant between PBA and diol compounds usually increases with increasing pH, because only the charged borates can form a stable complex with diol.46,53 The affinity of glucose to PBA also increases with increasing pH, as already revealed in the literature.50,54 Therefore PBA-based glucose-sensitive materials, which have a pKa of ∼8.6, usually show glucose sensitivity at relatively high pHs, but not at physiological pH. Glucose-regulated insulin release from the insulin–PVA/P(AAm–AAPBA) films was also observed at pH 9.0 (data not shown), but at pH 7.4, the influence of glucose on insulin release is indistinctive (Fig. 8).
 | ||
| Fig. 8 Release profiles of insulin from insulin–PVA/P(AAm–AAPBA) films soaked in 50 mM pH 7.4 phosphate buffer containing various concentrations of glucose. T = 37 °C. | ||
As mentioned above, glucose may react with PBA groups in two ways, i.e., binding with free PBA groups or competing with PVA for PBA binding sites. Both reactions will result in the acceleration of the film disintegration. The loss of glucose sensitivity of the film at pH 7.4 may suggest that the response of the film to glucose should be mainly attributed to the binding of glucose with free PBA groups, not its competition with PVA for PBA binding sites. The equilibrium constant of the competition reaction is the quotient of the association constant of the PBA–glucose complex and the association constant of the PBA–PVA complex. Both constants will decrease when the pH drops from 8.5 to 7.4, therefore pH changes may not significantly influence this reaction. If this reaction is the main reason for the glucose responsivity of the film, the glucose responsivity of the film should be relatively pH-independent. In contrast, the reaction between glucose and free PBA groups depends only on the association constant of the PBA–glucose complex, which decreases significantly when the pH drops from 8.5 to 7.4.46 Therefore, the pH-dependent glucose-sensitive behaviour of the film may suggest the glucose sensitivity of the film should be mainly attributed to the binding of glucose with free PBA groups.
Glucose-regulated release was also observed when the LbL films were immersed alternately in a glucose-free medium and a glucose-containing medium. As shown in Fig. 9, except for the first cycle, the amount of insulin released in glucose-containing medium is always larger than that in glucose-free medium. An opposite result was observed in the first cycle, i.e., the amount of insulin released in glucose-free medium is larger than that in glucose-containing medium, which can be explained by the fast initial release of this system, as already shown in Fig. 5–8. Either in glucose-containing medium or glucose-free medium, the amount of insulin released reduces gradually. This trend is also in agreement with the trends observed in Fig. 5–8.
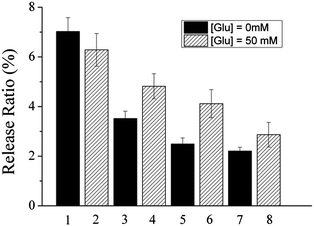 | ||
| Fig. 9 Release of insulin from insulin–PVA/P(AAm–AAPBA) films immersed alternately in 50 mL of fresh 50 mM pH 9.0 phosphate buffer and 50 mL of fresh 50 mM pH 9.0 phosphate buffer containing 50.0 mM glucose, each for 3 h. Release ratio is the ratio of the amount of insulin released to the total amount of insulin loaded. T = 37 °C. | ||
Conclusions
In conclusion, we demonstrated that a dynamically linked LbL film from insulin–PVA and P(AAm–AAPBA) can be used for self-regulated insulin release. The films were fabricated using the phenylboronate ester bond between insulin–PVA and P(AAm–AAPBA) as the driving force. As the bond can break under conditions of equilibrium control, the films gradually disintegrate and release insulin when they are soaked in aqueous solutions. The release rate of insulin can be tuned by pH and ionic strength. More importantly, it increases with increasing glucose concentration. The present system provides a lot of advantages for insulin release, as it allows for a long time release of insulin and tunable release rate according to glucose level. The amount of drug loaded can also be facilely adjusted by the number of assembly cycles. However, the present system lacks glucose responsivity at physiological pH. Further improvement of the system is currently under way in our laboratory.Acknowledgements
We are grateful for financial support for this work from the National Natural Science Foundation of China (Grant nos.20974049, 20974050 and 2117407), Tianjin Committee of Science and Technology (10JCYBJC02000), Program for New Century Excellent Talents in University (NCET-11-0264), and the Ministry of Science and Technology of China (Grant no. 2007DFA50760).Notes and references
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS.
- J. Lehn, Prog. Polym. Sci., 2005, 30, 814–831 CrossRef CAS.
- T. Maeda, H. Otsuka and A. Takahara, Prog. Polym. Sci., 2009, 34, 581–604 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- W. J. Tong and C. Y. Gao, J. Mater. Chem., 2008, 18, 3799–3812 RSC.
- A. P. R. Johnston, C. Cortez, A. S. Angelatos and F. Caruso, Curr. Opin. Colloid Interface Sci., 2006, 11, 203–209 CrossRef CAS.
- Z. Y. Tang, Y. Wang, P. Podsiadlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203–3224 CrossRef CAS.
- Y. J. Zhang, Y. Guan and S. Q. Zhou, Biomacromolecules, 2005, 6, 2365–2369 CrossRef CAS.
- C. Y. Wang, S. Q. Ye, L. Dai, X. X. Liu and Z. Tong, Biomacromolecules, 2007, 8, 1739–1744 CrossRef CAS.
- Y. J. Zhang, S. G. Yang, Y. Guan, W. X. Cao and J. Xu, Macromolecules, 2003, 36, 4238–4240 CrossRef CAS.
- P. Kohli and G. J. Blanchard, Langmuir, 2000, 16, 4655–4661 CrossRef CAS.
- Y. J. Zhang, Y. Guan, S. G. Yang, J. Xu and C. C. Han, Adv. Mater., 2003, 15, 832–835 CrossRef CAS.
- H. Lee, R. Mensire, R. E. Cohen and M. F. Rubner, Macromolecules, 2011 Search PubMed.
- W. B. Stockton and M. F. Rubner, Macromolecules, 1997, 30, 2717–2725 CrossRef CAS.
- L. Wang, Z. Wang, X. Zhang, J. Shen, L. Chi and H. Fuchs, Macromol. Rapid Commun., 1997, 18, 509–514 CrossRef CAS.
- Y. J. Zhang and W. X. Cao, Langmuir, 2001, 17, 5021–5024 CrossRef CAS.
- Y. Shimazaki, M. Mitsuishi, S. Ito and M. Yamamoto, Langmuir, 1997, 13, 1385–1387 CrossRef CAS.
- X. Zhang, H. Chen and H. Y. Zhang, Chem. Commun., 2007, 1395–1405 RSC.
- S. A. Sukhishvili and S. Granick, J. Am. Chem. Soc., 2000, 122, 9550–9551 CrossRef CAS.
- H. Y. Zhang, Y. Fu, D. Wang, L. Y. Wang, Z. Q. Wang and X. Zhang, Langmuir, 2003, 19, 8497–8502 CrossRef CAS.
- Y. Guan, S. G. Yang, Y. J. Zhang, J. Xu, C. C. Han and N. A. Kotov, J. Phys. Chem. B, 2006, 110, 13484–13490 CrossRef CAS.
- W. Lin, Y. Guan, Y. J. Zhang, J. Xu and X. X. Zhu, Soft Matter, 2009, 5, 860–867 RSC.
- Y. Guan, Y. J. Zhang, T. Zhou and S. Q. Zhou, Soft Matter, 2009, 5, 842–849 RSC.
- Z. B. Ding, Y. Guan, Y. Zhang and X. X. Zhu, Soft Matter, 2009, 5, 2302–2309 RSC.
- A. N. Zelikin, ACS Nano, 2010, 4, 2494–2509 CrossRef CAS.
- A. A. Antipov and G. B. Sukhorukov, Adv. Colloid Interface Sci., 2004, 111, 49–61 CrossRef CAS.
- S. Ye, C. Wang, X. Liu, Z. Tong, B. Ren and F. Zeng, J. Controlled Release, 2006, 112, 79–87 CrossRef CAS.
- Z. F. Dai, A. Heilig, H. Zastrow, E. Donath and H. Mohwald, Chem.–Eur. J., 2004, 10, 6369–6374 CrossRef CAS.
- G. Ibarz, L. Dahne, E. Donath and H. Mohwald, Adv. Mater., 2001, 13, 1324–1327 CrossRef CAS.
- C. S. Peyratout and L. Dahne, Angew. Chem., Int. Ed., 2004, 43, 3762–3783 CrossRef CAS.
- S. De Koker, L. J. De Cock, P. Rivera-Gil, W. J. Parak, R. Auzély Velty, C. Vervaet, J. P. Remon, J. Grooten and B. G. De Geest, Adv. Drug Delivery Rev., 2011, 63, 748–761 CrossRef CAS.
- X. Chen, W. Wu, Z. Guo, J. Xin and J. Li, Biomaterials, 2011, 32, 1759–1766 CrossRef CAS.
- Z. B. Ding, Y. Guan, Y. J. Zhang and X. X. Zhu, Polymer, 2009, 50, 4205–4211 CrossRef CAS.
- J. Blacklock, H. Handa, D. S. Manickam, G. Mao, A. Mukhopadhyay and D. Oupicky, Biomaterials, 2007, 28, 117–124 CrossRef CAS.
- R. M. Flessner, C. M. Jewell, D. G. Anderson and D. M. Lynn, Langmuir, 2011, 27, 7868–7876 CrossRef CAS.
- R. C. Smith, M. Riollano, A. Leung and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 8974–8977 CrossRef CAS.
- K. Shiomori, A. E. Ivanov, I. Y. Galaev, Y. Kawano and B. Mattiasson, Macromol. Chem. Phys., 2004, 205, 27–34 CrossRef CAS.
- W. W. Bromer, S. K. Sheehan, A. W. Berns and E. R. Arquilla, Biochemistry, 1967, 6, 2378–2388 CrossRef CAS.
- M. Lee, S. Kabilan, A. Hussain, X. Yang, J. Blyth and C. R. Lowe, Anal. Chem., 2004, 76, 5748–5755 CrossRef CAS.
- D. Shiino, Y. Murata, K. Kataoka, Y. Koyama, M. Yokoyama, T. Okano and Y. Sakurai, Biomaterials, 1994, 15, 121–128 CrossRef CAS.
- D. Shiino, Y. Murata, A. Kubo, Y. J. Kim, K. Kataoka, Y. Koyama, A. Kikuchi, M. Yokoyama, Y. Sakurai and T. Okano, J. Controlled Release, 1995, 37, 269–276 CrossRef CAS.
- F. Liu, S. C. Song, D. Mix, M. Baudys and S. W. Kim, Bioconjugate Chem., 1997, 8, 664–672 CrossRef CAS.
- K. Hinds, J. J. Koh, L. Joss, F. Liu, M. Baudys and S. W. Kim, Bioconjugate Chem., 2000, 11, 195–201 CrossRef CAS.
- K. D. Hinds, K. M. Campbell, K. M. Holland, D. H. Lewis, C. A. Piche and P. G. Schmidt, J. Controlled Release, 2005, 104, 447–460 CrossRef CAS.
- K. D. Hinds and S. W. Kim, Adv. Drug Delivery Rev., 2002, 54, 505–530 CrossRef CAS.
- G. Springsteen and B. H. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- K. Kataoka, H. Miyazaki, T. Okano and Y. Sakurai, Macromolecules, 1994, 27, 1061–1062 CrossRef CAS.
- X. Zhang, Y. Guan and Y. Zhang, Biomacromolecules, 2012, 13, 92–97 CrossRef CAS.
- S. A. Asher, V. L. Alexeev, A. V. Goponenko, A. C. Sharma, I. K. Lednev, C. S. Wilcox and D. N. Finegold, J. Am. Chem. Soc., 2003, 125, 3322–3329 CrossRef CAS.
- A. Matsumoto, R. Yoshida and K. Kataoka, Biomacromolecules, 2004, 5, 1038–1045 CrossRef CAS.
- T. Konno and K. Ishihara, Biomaterials, 2007, 28, 1770–1777 CrossRef CAS.
- S. Kitano, K. Kataoka, Y. Koyama, T. Okano and Y. Sakurai, Makromol. Chem. Rapid Commun., 1991, 12, 227–233 CrossRef CAS.
- K. Kataoka, H. Miyazaki, M. Bunya, T. Okano and Y. Sakurai, J. Am. Chem. Soc., 1998, 120, 12694–12695 CrossRef CAS.
- S. Xing, Y. Guan and Y. Zhang, Macromolecules, 2011, 44, 4479–4486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GPC traces of the polymers, assembly of P(AAm–AAPBA) and EDC-treated FITC-insulin, and the fluorescence spectra of the release media. See DOI: 10.1039/c2jm33413e |
| This journal is © The Royal Society of Chemistry 2012 |