Carbazole-based coplanar molecule (CmInF) as a universal host for multi-color electrophosphorescent devices
Cheuk-Lam
Ho
ae,
Liang-Chen
Chi
b,
Wen-Yi
Hung
*c,
Wei-Jiun
Chen
c,
Yu-Cheng
Lin
c,
Hao
Wu
ae,
Ejabul
Mondal
b,
Gui-Jiang
Zhou
d,
Ken-Tsung
Wong
*b and
Wai-Yeung
Wong
*ae
aInstitute of Molecular Functional Materials and Department of Chemistry and Centre for Advanced Luminescence Materials, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong, P.R. China. E-mail: rwywong@hkbu.edu.hk
bDepartment of Chemistry, National Taiwan University, Taipei, 106, Taiwan. E-mail: kenwong@ntu.edu.tw
cInstitute of Optoelectronic Sciences, National Taiwan Ocean University, Keelung, 202, Taiwan. E-mail: wenhung@mail.ntou.edu.tw
dMOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter and Department of Chemistry, Xi'an Jiaotong University, Xi'an, 710049, P.R. China
eAreas of Excellence Scheme, University Grants Committee of HKSAR, P.R. China
First published on 26th October 2011
Abstract
The synthesis, isomerism, photophysics and electrophosphorescent characterization of some functional cyclometallated iridium(III) complexes containing 2-[2-(N-phenylcarbazolyl)]pyridine and 2-[3-(N-phenylcarbazolyl)]pyridine molecular frameworks are described. A carbazole-based coplanar molecule (CmInF) obtained through the intramolecular ring closure of aryl substitutions at the C3 and C6 positions exhibits a high triplet energy (ET = 2.77 eV), morphological stability (Tg = 195 °C) and hole mobility in the range of up to 5 × 10−3 cm2 V−1 s−1. Highly efficient multi-color electrophosphorescent devices have been successfully achieved employing CmInF as the universal host material doped with phosphorescent dopants of various colors under the same device configuration of ITO/PEDOT:PSS (300 Å)/TCTA (250 Å)/CmInF: dopant (250 Å)/TAZ (500 Å)/LiF/Al (PEDOT:PSS = poly(ethylene dioxythiophene):polystyrene sulfonate; TCTA = 4,4′,4′′-tri(N-carbazolyl)triphenylamine; TAZ = 3-(4-biphenylyl)-4-phenyl-5-(4-tert-butylphenyl)-1,2,4-triazole). Through the mixing of two phosphorescent dopants of complementary colors, we also fabricated a two-color white organic light-emitting device (WOLED) with the same device structure consisting of 12 wt% FIrpic and 0.3 wt% (Mpg)22Ir(acac) co-doped into CmInF as a single-emitting-layer, which exhibits peak WOLED efficiency of 13.4% (23.4 cd A−1) and 11.2 lm W−1 with the Commission Internationale de L'Eclairage (CIE) coordinates of (0.33, 0.37). In addition, the use of such device structure in full-color OLEDs has the advantages of simplifying manufacturing process and reducing production cost that are the critical issues of commercialization.
Introduction
Organic light-emitting diodes (OLEDs) have drawn tremendous attention due to their potential applications in flat-panel displays and lighting sources.1,2 For full-color OLED display applications, it is essential to deliver a set of primary red-green-blue (RGB) emitters with sufficiently high luminous efficiency. Notably, the use of phosphorescent materials, in which the strong spin–orbit coupling due to the heavy-atom effect leads to singlet–triplet state mixing, can boost the electroluminescence (EL) efficiency.3 In phosphorescent OLEDs (PHOLEDs), triplet emitters of heavy-metal complexes are normally used as the emitting guests in a host material to reduce the quenching effect associated with the relatively long excited-state lifetimes of triplet emitters as well as the triplet–triplet annihilation, etc. Consequently, the selection of a suitable host material dispersed with various phosphorescent guest dopants is of equal importance to achieve high device efficiency. The singlet and triplet levels of the host material should be higher than those of the phosphorescent guest dopants to prevent the reverse energy transfer from guest to host and also confine triplet excitons within the emitting layer.4–7 For blue PHOLEDs, the design of such host material becomes a big challenge, particularly for a blue triplet emitter, in which the conjugation length of the host molecule must be extremely confined to achieve a triplet energy level higher than the photon energies of blue light (≥2.70 eV). In contrast, a host material with triplet energy higher than that of the blue phosphor may not be suitable for red, green, yellow emitters, etc., because of the unbalanced charge carrier transport in the emitting layer.Recently, efficient green and red PhOLEDs with 100% internal quantum efficiency have been disclosed,8–10 but highly efficient and stable blue PHOLEDs still remain a demanding issue. On the other hand, white light emission can be obtained by the mixing of several guest materials that give rise to RGB colors or two complementary colors (blue and red; blue and orange/yellow) through adoption of either the mixed fluorophore/phosphor or all-phosphor system. The design and synthesis of a host material that can work in various PHOLEDs are therefore important not only for blue, green and red phosphors but also for the realization of white OLEDs (WOLEDs) based on their suitable combinations. Up to now, reported literature examples of universal host materials for PHOLEDs are limited. For example, Cheng and Chou recently synthesized a bipolar host material bis-4-(N-carbazolyl)phenyl)phenylphosphine oxide (BCPO) containing a phenylphosphine oxide and two carbazole groups, which exhibits a high triplet energy (ET) of 3.01 eV and a high glass transition temperature (Tg) of 137 °C.11 By using BCPO as the universal host, high external quantum efficiencies (ηext) of 23.5%, 21.6% and 17% were achieved for blue, green and red phosphorescent devices, respectively, using simple device architectures. Cheng et al. also reported three triptycene derivatives 2,6,14-tris-(diphenylamino)triptycene (TATP), 2,6,14-tris-carbazoyltriptycene (TCTP) and 2,6,14-tris-(diphenylphosphoryl)triptycene (TPOTP) with remarkably high ET of 2.94–3.15 eV and Tg of 178–238 °C.12TATP can be employed as a general host for blue, green and red PHOLEDs, providing high EL efficiencies of 7.8%, 11.5% and 9.8%, respectively. Shu et al. reported the fluorene/triarylamine hybrid based host material tris[4-(9-phenylfluoren-9-yl)phenyl]amine (TFTPA) that exhibits a high ET of 2.89 eV and a Tg of 186 °C.13 By employing TFTPA as the host, the devices display ηext as high as 12.3% for blue, 11.8% for green and 9.2% for red. Yang et al. reported a silicon-bridged compound p-BISiTPA, combining p-type triphenylamine and n-type benzimidazole groups, showing a high ET of 2.69 eV and a Tg of 102 °C accompanied by the bipolar charge transporting feature.14 Devices with p-BISiTPA as the host show excellent performance, with ηext as high as 14.2% for blue, 18.9% for orange and 17.4% for white OLEDs at a practical brightness of 1000 cd m−2. Most recently, the same research group has developed three new silicon-bridged compounds (m-BISiTPA, p-OXDSiTPA and m-OXDSiTPA) by integrating electron-donating arylamine and electron-transporting benzimidazole/oxadiazole functionalities into one molecule via a silicon-bridged linkage mode.15 By using these compounds as the universal hosts, they achieved multi-color PHOLEDs for blue, green, orange and all-phosphor white-light electrophosphorescence, exhibiting high ηext.
Carbazole-based host materials have been used extensively in PHOLEDs due to their large ET and good hole-transporting properties.16–18 Well-known carbazole derivatives are 4,4′-N,N′-dicarbazolebiphenyl (CBP; ET = 2.56 eV)4 and N,N′-dicarbazolyl-3,5-benzene (mCP; ET = 2.9 eV),5,19 but both of them suffer from low thermal and morphological stability (Tg ≈ 65 °C), which are the essential properties for high performance OLED fabrication. To address these issues, many carbazole-based host materials with high ET have been reported for blue PHOLEDs. Therefore, there is still a great demand for new universal host materials which can produce multi-color PHOLEDs including WOLEDs with proper doping concentrations of the emitters.14,15 In this paper, we report an isomeric family of functional carbazole-derived organometallic phosphors and a carbazole-based coplanar host molecule (CmInF, see Scheme 1) obtained through the intramolecular ring closure of aryl substituents at the C3 and C6 positions and its applications in PHOLEDs based on various phosphorescent dopants in appropriate concentrations. The molecular design endows CmInF with several desirable properties: (i) the adoption of a coplanar chromophore, in which carbazole was fused to the neighboring phenylene rings through intramolecular annulation via sp3-hybridised carbon atoms bearing two p-tolyl groups as the peripheral substituents, makes the molecule rigid and bulky, and introduces molecular constraint; (ii) the carbazole unit can simultaneously possess sufficiently large ET and carrier-transport properties, which is often used as the host material; and (iii) the introduction of aryl groups as peripheral substituents of the coplanar backbone also impedes the intermolecular interactions between the molecules leading to their exhibition of amorphous morphologies. We anticipate that the combination of carbazole and fluorene moieties in a coplanar π-conjugated backbone and the presence of two tolyl groups at sp3 C9 position in CmInF could prevent efficiently the intermolecular interactions between molecules, thereby increasing high morphological and thermal stability (Tg = 195 °C). Moreover, a moderate ET value of 2.77 eV makes it a suitable host for PHOLEDs of various emission colors. The application of CmInF as the universal host doped with various phosphorescent emitters in the common device structure shows excellent device performance with ηext of 9.2% for blue, 15.1% for green, 13.7% for red and 17.5% for yellow. A dual complementary emission based all-phosphor WOLED, hosted by CmInF, was achieved with a peak ηext of 13.4%, a current efficiency (ηc) of 23.4 cd A−1 and a power efficiency (ηp) of 11.2 lm W−1.
 | ||
| Scheme 1 Chemical structure of CmInF and various color-tunable phosphorescent iridium(III) complexes used in this study. | ||
Results and discussion
Synthesis and chemical characterization
Scheme 1 lists the structures of CmInF and various color-tunable phosphorescent iridium(III) complexes used in this study. The compound 4-bromo-2-nitrobiphenyl was obtained from the Suzuki–Miyaura coupling of phenylboronic acid with 2,5-dibromonitrobenzene in the presence of catalytic Pd(PPh3)4.20 It was then heated in triethylphosphine under the Cadogan's reductive cyclization conditions to afford 2-bromocarbazole.21Ullmann condensation between 2-bromocarbazole and iodobenzene gave 2-bromo-N-phenylcarbazole in good yield,22 and the synthesis of the cyclometallating ligand was then completed by Stille coupling of 2-bromo-N-phenylcarbazole with 2-(tributylstannyl)pyridine (Scheme 2).23![Synthesis of 2-[2-(N-phenylcarbazolyl)]pyridine.](/image/article/2012/JM/c1jm13794h/c1jm13794h-s2.gif) | ||
| Scheme 2 Synthesis of 2-[2-(N-phenylcarbazolyl)]pyridine. | ||
Following the ligand synthesis, 2-[2-(N-phenylcarbazolyl)]pyridine was easily converted to the new homoleptic iridium(III) complex Ir(2-Cz)33via a one-step cyclometallation with [Ir(acac)3] (Hacac = acetylacetone) in refluxing glycerol at high temperature (>200 °C).24 Here, the reaction was run with an excess of ligand, which can be easily recovered and reused. On the other hand, the iridium-μ-chloro-bridged dimer was synthesized by the reaction of IrCl3·xH2O with 2-[2-(N-phenylcarbazolyl)]pyridine according to a conventional procedure.25 The diiridium(III) complex was then converted to the mononuclear iridium(III) complex (2-Cz)22Ir(acac) by replacing the two bridging chlorides with a bidentate monoanionic acac ligand. The synthetic pathways of the metal complexes are summarized in Scheme 3. Spectroscopic and elemental analyses of both complexes are consistent with the expected formulations of their structures. The facial geometry of the homoleptic complex is confirmed by the simplicity of 1H NMR spectral pattern, which indicates that the number of coupled spins is equal to that of the protons on one ligand because the three ligands are magnetically equivalent due to the 3-fold symmetry of the molecule.26 On the other hand, the preparation of Ir(3-Cz)33 (a geometrical linkage isomer of Ir(2-Cz)33) was reported previously.27 The homoleptic series appears to show higher thermal stability than their heteroleptic analogue as revealed from the onset decomposition temperature (Tdec). Moreover, improvement in the thermostability was noted as we change the cyclometallating ligand from 2-[2-(N-phenylcarbazolyl)]pyridine to 2-[3-(N-phenylcarbazolyl)]pyridine in these complexes.
 | ||
| Scheme 3 Synthesis of new phosphorescent complexes Ir(2-Cz)33 and (2-Cz)22Ir(acac). | ||
The synthesis of the host CmInF was carried out following our previously reported literature.28 From the X-ray crystallographic analysis,28 we observed the coplanar structure of CmInF. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were used to characterize the morphological property and thermal stability of CmInF. The results show a high onset decomposition temperature (Td = 392 °C, corresponding to 5% weight loss) in the TGA curve and a distinctly high glass transition temperature (Tg) at 195 °C and melting point (Tm) at 346 °C from the DSC study. Therefore, this material can form homogeneous and stable amorphous films, which are the two critical factors for its successful use in thin film devices. The amorphous behavior and high Tg value of CmInF reveal that the rigidity of the conjugated backbone and the presence of the peripheral p-tolyl substituents effectively suppress intermolecular interaction.
Photophysical and charge transport properties
In this study, we have prepared two new cyclometallated iridium(III) complexes containing 2-[2-(N-phenylcarbazolyl)]pyridine and their photophysical properties were compared to their isomeric congeners derived from 2-[3-(N-phenylcarbazolyl)]pyridine (Table 1). The strongest absorption bands in the ultraviolet region are assigned to the spin-allowed intraligand 1π–π* transitions. The next lower energy in the shoulder region of the 1π–π* transitions and the weaker broad shoulders extending into the visible region with appreciable intensity are associated with an admixture of the typical spin-allowed metal-to-ligand charge transfer 1MLCT, spin–orbit coupling enhanced 3π–π* and spin-forbidden 3MLCT transitions. These assignments are made by analogy with previously reported IrIII complexes.29 The weakest MLCT absorptions are red-shifted from the homoleptic to the corresponding heteroleptic complexes owing to the strong ligand field of acac. By comparing each of the isomeric complexes, different absorption profiles were also observed.| Absorption (293 K) | Emission (293 K) | T d d/°C | |||
|---|---|---|---|---|---|
| λ abs a/nm | λ em a/nm | Φ P b | τ P c/μs | ||
| a Measured in CH2Cl2. b Phosphorescent quantum yield measured in degassed CH2Cl2 relative to fac-[Ir(PPy)3] (ΦP = 0.40), λex = 400 nm. c Triplet emission lifetime measured in degassed CH2Cl2. d Onset decomposition temperature at 5% weight reduction. sh = shoulder peak. | |||||
| Ir(2-Cz)33 | 266, 324, 359sh, 432 | 578 | 0.32 | 0.18 | 458 |
| (2-Cz)22Ir(acac) | 287, 341, 510sh | 591 | 0.29 | 0.21 | 388 |
| Ir(3-Cz)33 27a | 291sh, 316, 398sh | 515 | 0.43 | 0.46 | 477 |
| (3-Cz)22Ir(acac) 27b | 301, 325, 358sh, 435sh | 515 | 0.41 | 0.42 | 404 |
All of the iridium(III) complexes emit strong phosphorescence arising from the lowest energy 3MLCT transition at room temperature in CH2Cl2. The substitution position of the pyridyl ring on N-phenylcarbazole has a great influence on the photoluminescence (PL) spectra of the heavy-metal complexes. Clearly, the PL maximum of the 2-substituted homoleptic derivative is more red-shifted relative to the 3-substituted counterpart (from 515 nm for Ir(3-Cz)33 to 578 nm for Ir(2-Cz)33) due to the more conjugated structure of the former group over the latter one (Fig. 1). This is also the case for the heteroleptic species, (3-Cz)22Ir(acac)versus(2-Cz)22Ir(acac) (from 515 to 591 nm). Here, a prominent color-tuning of phosphorescence emission from green to orange/red is achieved by simply altering the ligation of metal with carbon atom at 2- or 3-position of carbazole unit in these linkage isomers. While the carbon atoms at 2-/7- and 3-/6-position of carbazole have different electronic density, where the 3-/6-position can be activated by the nitrogen atom and hence more electron-rich than the 2-/7-site, the energy level of carbazole-containing complexes can be changed by substitution at the 3-/6- or 2-/7-position. The more electron-donating 3-position on carbon of carbazole pushes up the energy of metal d orbital when it is ligated to the metal ion, giving rise to a higher energy level of the highest occupied molecular orbital (HOMO) than that of the less electron-donating 2-position and this is accompanied by significant bathochromic shifts in λem for these iridium(III) complexes.30 This approach is different from those commonly used for other complexes in which the emission wavelength can be tuned by varying the electronic nature and/or position of the substituents on the cyclometallated ligands.31
![Absorption and PL spectra of the iridium(iii) complexes with 2-[2-(N-phenylcarbazolyl)]pyridine and 2-[3-(N-phenylcarbazolyl)]pyridine groups.](/image/article/2012/JM/c1jm13794h/c1jm13794h-f1.gif) | ||
| Fig. 1 Absorption and PL spectra of the iridium(III) complexes with 2-[2-(N-phenylcarbazolyl)]pyridine and 2-[3-(N-phenylcarbazolyl)]pyridine groups. | ||
The HOMO and the lowest unoccupied molecular orbital (LUMO) energy levels of Ir(2-Cz)33 and (2-Cz)22Ir(acac) were determined electrochemically (HOMO/LUMO: −4.81/–2.31 eV for Ir(2-Cz)33 and −4.83/–2.43 eV for (2-Cz)22Ir(acac)). As compared to [Ir(PPy)3] and (PPy)22Ir(acac) (PPy = 2-phenylpyridyl), the HOMOs of Ir(2-Cz)33 and (2-Cz)22Ir(acac) are elevated when the electron-rich and electron-donating carbazolyl units are attached to the pyridyl rings, signaling that both complexes have a lower ionization potential than their PPy congeners. This leads to a better hole injection/hole transport ability and hence these triplet emitters are potentially dual functional in nature.
Fig. 2 presents the UV-Vis absorption and PL spectra of CmInF in dilute solution (CH2Cl2) and neat film, as well as the corresponding phosphorescence (Phos) spectrum recorded from its EtOH solution at 77 K. The absorption spectra of CmInF in solution and neat film are very similar with peaks appearing at 328, 366 and 385 nm, which imply that no significant intermolecular interactions occur in the ground state. The absorption peaks at 366 and 385 nm are attributed to the π–π* transition of the extended conjugation of coplanar carbazole–fluorene chromophore of CmInF. Its emission spectra in solution and neat film are also similar with peaks appearing at 394 nm and 415 nm. The phosphorescence spectrum of CmInF in EtOH at 77 K indicates the high-energy phosphorescence emission at 448 nm, corresponding to the ET value of 2.77 eV. The decrease in triplet energy agrees with the extended π-conjugation at C3 and C6 positions on the central carbazole core and reveals that CmInF has a lower ET value as compared to CzSi (ET = 3.02 eV),32 but a higher value than those in the earlier reported host materials DFC (ET = 2.53 eV)33 and CPhBzIm (ET = 2.48 eV).34 This ET value is sufficiently high to prevent the reverse triplet energy transfer from the dopant back to CmInF. Thus, CmInF should be a suitable host for various phosphorescent devices from blue to red.
 | ||
| Fig. 2 Absorption and PL spectra of CmInF in solution and neat film, as well as the corresponding phosphorescence (Phos) spectrum recorded from its EtOH solution at 77 K. | ||
We also studied the electrochemical properties of CmInF using cyclic voltammetry (CV) and its cyclic voltammogram is shown in Fig. 3. CmInF exhibits one reversible oxidation process at 1.09 eV (vs. Ag/AgCl). No reduction peak was observed. It is worth mentioning that no electropolymerization occurred during multiple cycles of CV scanning. It was previously reported that without blocking the active sites C3 and C6 of carbazole, the oxidation process is not reversible.35 The observed reversible oxidation potential of CmInF indicates that carbazole is π-conjugated with fluorene at 3 and 6 positions. Thus, the rigid peripheral fluorene moieties conjugated to the carbazole core not only emerge with a significant impact on the oxidation potential of the whole molecule, but their addition also efficiently blocks the electrochemically active sites (C3 and C6) of carbazole and may improve its electrochemical stability in the EL devices. Moreover, the existence of stable radical cationic species suggests that it can be useful for potential hole-transporting material. The HOMO energy level (−5.4 eV) of CmInF was estimated by atmospheric photoelectron spectroscopy. From the equation LUMO = HOMO + ΔEg, where ΔEg (3.1 eV) is the optical band gap determined from the absorption threshold, we estimated the LUMO energy level to be −2.3 eV.
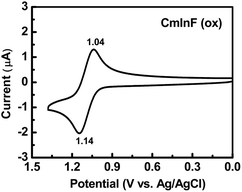 | ||
| Fig. 3 Cyclic voltammogram of CmInF. | ||
To evaluate the carrier mobility of CmInF, we carried out time-of-flight (TOF) measurements using the device structure of ITO/CmInF (3 μm)/Ag (100 nm). Fig. 4(a) shows a representative TOF transient for holes which reveals the nondispersive hole-transport characteristics and this implies that the carriers move with constant velocity across the organic film. To determine the carrier mobilities, the transit time, tT, can be clearly evaluated from the intersection point of two asymptotes in the double logarithmic representation of the TOF transient as shown in the inset of Fig. 4(a). The hole mobilities of CmInF thus determined as a function of the electric field are shown in Fig. 4(b) and the hole mobilities are in the range from 2 × 10−3 to 5 × 10−3 cm2 V−1 s−1 for fields varying from 105 to 3.3 × 105 V cm−1. It is worth noting that a hole mobility of up to 5 × 10−3 cm2 V−1 s−1 is very high among the carbazole-based oligomers reported in the literature.
 | ||
| Fig. 4 (a) Representative TOF transient for CmInF (hole, 3 μm thickness, E = 2.3 × 105 V cm−1). Inset: double logarithmic plot. (b) Hole mobility vs. E1/2. | ||
Electrophosphorescent properties
To evaluate the performance of CmInF as a universal host material for multi-color emission, several PHOLED devices have been fabricated with the same device configuration of ITO/PEDOT:PSS (300 Å)/TCTA (250 Å)/CmInF doped with various phosphor dyes (250 Å)/TAZ (500 Å)/LiF (5 Å)/Al. The conducting polymer poly(ethylene dioxythiophene):polystyrene sulfonate (PEDOT:PSS) was used as the hole-injection layer,364,4′,4′′-tri(N-carbazolyl)triphenylamine (TCTA) as the hole-transport layer as well as an exciton blocker (ET: 2.76 eV, HOMO/LUMO: −5.7/–2.4 eV) to confine triplet excitons within the emitting layer (EML).37,38 The 1,2,4-triazole-based material, 3-(4-biphenylyl)-4-phenyl-5-(4-tert-butylphenyl)-1,2,4-triazole (TAZ) (ET: 2.7 eV, HOMO/LUMO: −6.3/–2.7 eV), was used as the electron-transport and hole-blocking layer to provide exciton and carrier confinement.39 The EML consists of the large-gap CmInFhost doped with the blue iridium(III) bis[(4,6-di-fluorophenyl)-pyridinato-N,C2′]picolinate FIrpic,4 green bis(2-phenylpyridine)iridium(III) acetylacetonate (PPy)22Ir(acac),40 red (Mpq)22Ir(acac),41 and a series of our tailor-made dopants (Ir(3-Cz)33, Ir(TPA)33,42Ir(2-Cz)33 and (2-Cz)22Ir(acac), see Scheme 1) emitting from green to red. The green dopant (3-Cz)22Ir(acac) was not tested for PHOLED here since it has very similar emission features as Ir(3-Cz)33. The ET estimated from the highest energy peaks of the phosphorescence spectra for light from blue to red colors are 2.65–2.00 eV, which are lower than CmInF (ET = 2.77 eV) and can be confined in EML. There are no residual emissions from the host and/or adjacent layers, even at high drive currents, indicating that the EL emission originates from the dopant and there is complete energy and/or charge transfer from the host exciton to the phosphor upon electrical excitation (see Fig. 5). | ||
| Fig. 5 EL spectra of devices incorporating the host CmInF doped with various dopants. | ||
Fig. 6 shows the current density–voltage–luminance (J–V–L) characteristics, and external quantum and power efficiencies as a function of brightness for each of the PHOLED devices. The pertinent EL properties analyzed from these graphs are summarized in Table 2. All of the devices in this study exhibited low turn-on voltages (3–3.5 V) and the difference in the J–V performance is presumably because of the different emitting dopants that influence the charge carrier transport of EML. We initially examined the performance of the FIrpic-based blue device with Commission Internationale de L'Eclairage (CIE) coordinates of (0.17, 0.37). The ηext can reach 9.2% photons/electron with the ηc and ηP values of 20.2 cd A−1 and 11.8 lm W−1. These values are higher than those obtained using the same triplet emitter in CBP (6.1%, 7.7 lm W−1) and mCP (7.5%, 8.9 lm W−1) hosts.5 The values are moderate among FIrpic-based PHOLEDs, presumably because the ET of CmInF is not high enough relative to FIrpic. However, this is a worthy sacrifice to acquire a compromise among the ET of RGB phosphors.
 | ||
| Fig. 6 (a) J–V–L characteristics, (b) external quantum efficiency and (c) power efficiency versus brightness for PHOLED devices with different dopants. | ||
| Dopant (%) | V on a/V | Voltage and ηext at 1000 nitb/V, % | L max/cd m−2 | I max/mA cm−2 | η ext (%) | η c/cd A−1 | η p/lm W−1 | CIE (x, y) |
|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage at which emission became detectable. b The values of driving voltage and ηext of device at 1000 cd m−2. | ||||||||
| (PPy)22Ir(acac), 9% | 3 | 5.5, 14.5 | 122![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (13 V) 000 (13 V) |
700 | 15.1 | 55.6 | 42.2 | 0.34, 0.62 |
| FIrpic, 12% | 3 | 8.3, 6 | 19800 (14.5 V) |
704 | 9.2 | 20.2 | 11.8 | 0.17, 0.37 |
| (Mpq)22Ir(acac), 9% | 3 | 8.3, 10 | 21300 (14.5 V) |
820 | 13.3 | 14.1 | 9.7 | 0.65, 0.35 |
| Ir(3-Cz)33, 9% | 3.5 | 7, 11.8 | 130000 (15 V) |
910 | 12.4 | 43.6 | 27 | 0.30, 0.62 |
| Ir(2-Cz)33, 9% | 3 | 7.5, 16.2 | 80200 (14.5 V) |
880 | 17.5 | 44 | 29 | 0.56, 0.44 |
| Ir(TPA)33, 8% | 3.5 | 7.45, 11.1 | 95000 (16 V) |
860 | 12 | 45 | 24 | 0.38, 0.60 |
| (2-Cz)22Ir(acac), 8% | 3 | 6.5, 10.8 | 51000 (15 V) |
1240 | 13.7 | 25.6 | 19 | 0.60, 0.39 |
| FIrpic, 12% + (Mpq)22Ir(acac) 0.3% | 3 | 9, 7.7 | 15900 (14.5 V) |
570 | 13.4 | 23.4 | 11.2 | 0.33, 0.37 |
Next, we employed CmInF as a green host for (PPy)22Ir(acac), Ir(3-Cz)33 and Ir(TPA)33 dopants with the same device configuration. The (PPy)22Ir(acac)-based device exhibited a maximum brightness (Lmax) of 122000 cd m−2 at 13 V and attractive EL efficiencies (15.1%, 55.6 cd A−1, 42.2 lm W−1) that are superior to those of the Ir(3-Cz)33-based device (12.4%, 43.6 cd A−1, 27 lm W−1) and Ir(TPA)33-based device (12%, 45 cd A−1, 24 lm W−1). It is worth noting that these green-emitting devices show a low roll-off in ηext (Fig. 6(b)). When the brightness reaches 1000 cd m−2, ηext still remains as high as 14.5% with a low efficiency roll-off value of 4% for the (PPy)22Ir(acac)-based device. The Ir(3-Cz)33- and Ir(TPA)33-based devices also exhibited low efficiency roll-off values of only 4.8% and 7.5%, respectively, at a practical brightness of 1000 cd m−2. To further evaluate the suitability of CmInF as the host material for low-energy triplet emitters, we fabricated PHOLED devices by using Ir(2-Cz)33 (orange), (2-Cz)22Ir(acac) (red) and (Mpq)22Ir(acac) (red) as phosphorescent dopants. The Ir(2-Cz)33-based device showed an impressive performance with a low turn-on voltage of 3 V, Lmax of 80200 cd m−2 at 14.5 V, and peak EL efficiencies of 17.5%, 44 cd A−1, and 29 lm W−1 with the CIE coordinates at (0.56, 0.44). It is also worth mentioning that the Ir(2-Cz)33-based device gave better performance than other recently reported PHOLEDs in the orange chromaticity region.43–45 For the red emitters, the (2-Cz)22Ir(acac)-based device exhibited a Lmax of 51000 cd m−2 at 15 V and respectable EL efficiencies (13.7%, 25.6 cd A−1, 19 lm W−1) with the CIE coordinates of (0.60, 0.39) while (Mpq)2Ir(acac)-based device exhibited a Lmax of 21300 cd m−2 at 14.5 V and very good EL efficiencies (13.3%, 14.1 cd A−1, 9.7 lm W−1) with the CIE coordinates of (0.65, 0.35).
Through the mixing of two dopants showing complementary colors,46 we also fabricated a two-color white OLED (WOLED) under the same device structure consisting of 12 wt% FIrpic and 0.3 wt% (Mpg)22Ir(acac) co-doped into CmInF as a single-emitting-layer. The WOLED exhibited a Lmax of 15900 cd m−2 at 14.5 V, with maximum efficiencies (ηext, ηc, and ηp) of 13.4%, 23.4 cd A−1, and 11.2 lm W−1 (Fig. 7). Although a gradual decrease in efficiency with an increase of current density was observed, the EL efficiency of WOLED remained at 7.7% at the practical brightness of 1000 cd m−2. The inset of Fig. 7 displayed the EL spectra of the WOLED device recorded at various brightness levels. The relative intensity of the red emission decreased slightly when the brightness increased, leading to a slight shift in the CIE coordinates from (0.36, 0.37) at a value of L = 1000 cd m−2 to (0.33, 0.37) at 11300 cd m−2. This color shift originated from the shifting of the recombination zone upon increasing the applied voltage and from the easier formation of high-energy excitons at higher voltage.47,48 Although the performance of these devices has yet to be optimized, it is our intention to highlight the potential merits of CmInF as a universal host towards color-tunable OLED applications.
 | ||
| Fig. 7 EL efficiencies for two-color WOLED. Inset: EL spectra of WOLED device at various brightness levels. | ||
Conclusions
In summary, we have investigated the effect of the position of pyridyl ring substituent at C2 and C3 sites of the carbazole ring on the emission color, photophysics and electrophosphorescent properties of some iridium(III) cyclometallates bearing N-phenylcarbazole moieties. An interesting color-tuning strategy was developed by a change of the chelating carbon atom by a different binding site of the carbazolyl group. The origin of the wavelength shift arises from the striking difference of electron density on the carbon atom bound to the iridium centre. We also reported nicely a prominent multi-color spectrum employing CmInF as a universal host material doped with various phosphor dyes with the same device configuration to reach the ultimate target for highly efficient, color-switchable OLEDs. The present work furnished OLED colors spanning from sky-blue to red (475–620 nm) with high EL efficiencies (9.2–17.5%) which have great potential for application in multi-color displays and possibly in white light illumination sources through the mixing of two phosphorescent dopants of complementary colors.Experimental
Photophysical and thermal measurements
All reactions were carried out under nitrogen atmosphere with the use of standard Schlenk techniques. Glassware was oven-dried at about 120 °C. Analytical grade solvents were purified by distillation over appropriate drying agents under an inert nitrogen atmosphere prior to use. All reagents and chemicals, unless otherwise stated, were purchased from commercial sources and used without further purification. The positive-ion fast atom bombardment (FAB) mass spectra were recorded in m-nitrobenzyl alcohol matrices on a Finngin-MAT SSQ710 mass spectrometer. NMR spectra were measured in deuterated solvents as the lock and reference on a JEOL JNM-EX270 FT NMR system or a Varian INOVA 400 MHz FT-NMR spectrometer, with 1H and 13C NMR chemical shifts quoted relative to Me4Si standard. Steady-state spectroscopic measurements were conducted both in solution and solid films prepared by vacuum (2 × 10−6 Torr) deposition on a quartz plate. Absorption spectra were recorded with a U2800A spectrophotometer (Hitachi). Fluorescence spectra at 300 K and phosphorescent spectra at 77 K were measured on a Hitachi F-4500 spectrophotometer upon excitation at the absorption maxima. The differential scanning calorimetry (DSC) analyses were performed on a Universal V2,6D TA Instrument at a heating rate of 10 °C min−1 from 50 to 400 °C under a nitrogen atmosphere. Thermogravimetric analysis (TGA) was undertaken with a Universal V2,6D TA Instrument or a Perkin-Elmer TGA6 thermal analyzer. The thermal stability of the samples under a nitrogen atmosphere was determined by measuring their 5% weight loss while heating proceeded at a rate of 10 °C min−1 up to 700 °C.Cyclic voltammetry
The oxidation potential was determined by cyclic voltammetry (CV) in CH2Cl2 solution (1.0 mM) containing 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte at a scan rate of 100 mV s−1. A glassy carbon electrode and a platinum wire were used as the working and counter electrodes, respectively. The ferrocene/ferrocenium (Fc/Fc+) redox couple in THF/TBAP occurs at E1/2 = +0.51 V. The potential value was recorded relative to the oxidation potential of ferrocene, which was added to the electrolyte as an internal standard. All potentials were recorded versusAg/AgCl (saturated) as a reference electrode.Time-of-flight (TOF) mobility measurements
The samples for the TOF measurements were prepared through vacuum deposition in the configuration of ITO/CmInF (3 μm)/Ag (100 nm) and then placed inside a cryostat and kept under vacuum. The thickness of the organic film was monitored in situ using a quartz crystal sensor and calibrated using a thin film thickness measurement system (K-MAC ST2000). A nitrogen laser was used as the excitation source (λ = 337 nm) and was incident on the sample through the ITO. Under an applied dc bias, the transient photocurrent was swept across the bulk of the organic film towards the collection electrode (Al); it was then recorded using a digital storage oscilloscope. Depending on the polarity of the applied bias, the selected carriers (holes or electrons) were swept across the sample with a transit time of tT. For an applied bias V and a sample thickness D, the applied electric field E is equal to V/D; the carrier mobility is then given by the equation μ = D/(tTE) = D2/(VtT) from which the carrier transit times, tT, can be extracted from the intersection points of the two asymptotes to the plateau and tail sections in the double logarithmic plot.OLED device fabrication
All chemicals were purified through vacuum sublimation prior to use. The OLEDs were fabricated through vacuum deposition of the materials at 10−6 Torr onto ITO-coated glass substrates having a sheet resistance of 15 Ω sq−1. The ITO surface was cleaned ultrasonically—sequentially with acetone, methanol, and deionized water—and then it was treated with UV-ozone. The deposition rate of each organic material was ca. 1–2 Å s−1. Subsequently, LiF was deposited at 0.1 Å s−1 and then capped with Al (ca. 5 Å s−1) through shadow masking without breaking the vacuum. The J–V–L characteristics of the devices were measured simultaneously in a glove-box using a Keithley 6430 source meter and a Keithley 6487 picoammeter equipped with a calibration Si-photodiode. EL spectra were measured using a photodiode array (OTO SD1200).Synthesis of 4-bromo-2-nitrobiphenyl
Phenylboronic acid (1.50 g, 12.3 mmol), 2,5-dibromonitrobenzene (3.45 g, 12.3 mmol) and Pd(PPh3)4 (1.42 g, 1.42 mmol) were dissolved in a mixture of toluene (250 mL) and 2 M aq. Na2CO3 (12 mL). The degassed mixture was heated at 90 °C for 5.5 h, cooled to 20 °C and then diluted with distilled H2O. The organic phase was separated and the organic products were extracted into diethyl ether. The combined extracts were dried with MgSO4 and concentrated under reduced pressure to give a dark brown liquid. The main product was separated by chromatography eluting with a mixture of hexane and CH2Cl2 and characterized by fast-atom bombardment (FAB) mass spectrometry. The mixture was used in the next step without further purification (yield: 70%). MS (FAB): m/z 278 (M+). 1H NMR (CDCl3): δ [ppm] 8.00 (d, J = 2.0 Hz, 1H, Ar), 7.75 (dd, J = 8.2 and 2.0 Hz, 1H, Ar), 7.45–7.41 (m, 3H, Ar), 7.33 (d, J = 8.2 Hz, 1H, Ar), 7.31–7.27 (m, 2H, Ar). 13C NMR (CDCl3): δ [ppm] 136.5, 135.6, 135.5, 133.5, 131.8, 129.5, 129.1, 128.8, 128.0, 127.3, 124.2, 121.6 (Ar).2-Bromocarbazole
4-Bromo-2-nitrobiphenyl (1.50 g, 5.42 mmol) and P(OEt)3 (4.80 g, 37.9 mmol) were heated under reflux for 10 h. The excess P(OEt)3 was distilled off under vacuum. The remaining residue was diluted with a 1:1 mixture of MeOH and distilled H2O to give a precipitate which was filtered off and washed several times with a 1:1 mixture of MeOH/H2O, followed by hexane. Column chromatography using hexane as eluent gave the title compound as a white solid (500 mg, 38%). MS (FAB): m/z 246 (M+). 1H NMR (CDCl3): δ [ppm] 11.40 (s, 1H, NH), 8.15–8.11 (dd, J = 8.0 Hz, 2H, Ar), 7.68 (d, J = 1.7 Hz, 1H, Ar), 7.54–7.19 (m, 4H, Ar). 13C NMR (CDCl3): δ [ppm] 140.6, 139.9, 126.1, 121.9, 121.8, 121.6, 121.3, 120.3, 119.1, 118.2, 113.5, 111.2 (Ar).
2-Bromo-N-phenylcarbazole
The N-arylation of 2-bromocarbazole (2.50 g, 10.2 mmol) was carried out by the Ullmann condensation of 2-bromocarbazole with iodobenzene (3.12 g, 15.3 mmol) in the presence of CuI (1.35 g, 7.14 mmol), KOH (4.00 g, 71.4 mmol) and 1,10-phenanthroline (1.42 g, 7.14 mmol) in p-xylene (50 mL) and the mixture was stirred at 140 °C for two days. The cooled mixture was extracted with CH2Cl2. The organic layer obtained was washed with water and dried over anhydrous MgSO4 and evaporated. The product was purified by flash chromatography on silica gel using hexane as eluent to give a pure white solid (90%, 2.95 g). MS (FAB): m/z 322 (M+). 1H NMR (CDCl3): δ [ppm] 8.10–8.08 (d, 1H, J = 7.6 Hz, Ar), 7.98–7.96 (d, 1H, J = 8.4 Hz, Ar), 7.63–7.59 (m, 2H, Ar), 7.53–7.48 (m, 4H, Ar), 7.42−7.35 (m, 3H, Ar), 7.30–7.27 (m, 1H, Ar). 13C NMR (CDCl3): δ [ppm] 141.68, 141.08, 137.03, 130.05, 127.91, 127.11, 126.34, 123.05, 122.71, 122.25, 121.44, 120.36, 120.26, 119.50, 112.78, 109.95 (Ar).Synthesis of 2-[2-(N-phenylcarbazolyl)]pyridine
2-Bromo-N-phenylcarbazole (1.43 g, 4.45 mmol) and 2-(tributylstannyl)pyridine (2.15 g, 6.68 mmol) were mixed in dry toluene (50 mL) and Pd(PPh3)4 (0.51 g, 0.45 mmol) was then added to the solution. The resulting mixture was stirred at 110 °C for 2 days. After cooling to room temperature, the reaction mixture was poured into a separating funnel and CH2Cl2 was added followed by washing with water. The organic phase was dried over MgSO4. Solvent was then removed and the residue was purified by column chromatography eluting with CH2Cl2/hexane (1:1, v/v). The title product was obtained as a white solid (1.36 g, 95%). MS (FAB): m/z 320 (M+). 1H NMR (CDCl3): δ [ppm] 8.66–8.65 (d, J = 4.4 Hz, 1H, Ar), 8.22–8.14 (m, 2H, Ar), 8.04 (s, 1H, Ar), 7.87 (m, 1H, Ar), 7.76 (m, 2H, Ar), 7.72 (m, 4H, Ar), 7.47 (m, 3H, Ar), 7.30 (m, 1H, Ar), 7.19 (m, 1H, Ar). 13C NMR (CDCl3): δ [ppm] 158.09, 149.60, 141.74, 141.42, 137.50, 136.68, 129.97, 127.59, 127.34, 126.24, 123.96, 122.97, 121.83, 120.99, 120.52, 120.48, 120.06, 119.09, 109.87, 108.34 (Ar).
Synthetic procedures of new iridium(III) compounds
:2, v/v). The mixture was refluxed for 24 h and then cooled to room temperature. A precipitate gradually formed during the reaction. The precipitate was collected by filtration and washed with ethanol followed by hexane. The solid was then pumped dry completely to give the crude iridium(III) dimer for subsequent reaction. Without further purification, this crude dimer was mixed with 10 equivalents of Na2CO3 (196 mg, 0.185 mmol) in 2-ethoxyethanol (8 mL) and 3 equivalents of acetylacetone (0.056 mL, 0.554 mmol) were added. The reaction mixture was then refluxed for 16 h. After the mixture was cooled to room temperature, water was added and extracted with CH2Cl2. The organic layer was collected and the solvent was subsequently removed. The residue was then purified using preparative TLC plates eluting with a mixture of CH2Cl2/hexane to give a reddish orange solid in 41% yield. MS (FAB): m/z 930 (M+). 1H NMR (CDCl3): δ [ppm] 8.67 (d, J = 4.0 Hz, 2H, Ar), 7.93–7.91 (m, 2H, Ar), 7.79–7.54 (m, 15H, Ar), 7.25 (m, 8H, Ar), 7.02–6.93 (m, 3H, Ar), 5.26 (s, 1H, acac), 1.83 (s, 6H, CH3). 13C NMR (CDCl3): δ [ppm] 184.55 (CO), 168.96, 148.36, 142.67, 141.33, 138.21, 137.68, 136.50, 134.94, 129.70, 127.05, 126.78, 125.63, 123.05, 122.57, 121.41, 120.45, 118.92, 118.62, 109.73, 109.02, 105.88 (Ar), 100.49 (CH), 28.85 (CH3). Anal. Calcd for C51H37N4O2Ir: C, 65.86; H, 4.01; N, 6.02; found: C, 66.02; H, 3.92; N, 6.20%.
Acknowledgements
W.-Y. Wong acknowledges the financial support from Hong Kong Baptist University (FRG2/09-10/091), Hong Kong Research Grants Council (HKBU202709 and HKUST2/CRF/10) and Areas of Excellence Scheme, University Grants Committee of HKSAR, P.R. China (Project No. [AoE/P-03/08]). W.-Y. Hung and K.-T. Wong are grateful for the financial support from the National Science Council (NSC 98-2119-M-002-007-MY3, 100-2112-M-019 -002 -MY3) of Taiwan.References
- C. W. Tang and S. A. Van Slyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082 CrossRef CAS.
- R. J. Holmes, S. R. Forrest, Y.-J. Tung, R. C. Kwong, J. J. Brown, S. Garon and M. E. Thompson, Appl. Phys. Lett., 2003, 82, 2422 CrossRef CAS.
- S. Tokito, T. Iijima, Y. Suzuri, H. Kita, T. Tsuzuki and F. Sato, Appl. Phys. Lett., 2003, 83, 569 CrossRef CAS.
- K. Goushi, R. Kwong, J. J. Brown, H. Sasabe and C. Adachi, J. Appl. Phys., 2004, 95, 7798 CrossRef CAS.
- S.-J. Su, C. Cai and J. Kido, Chem. Mater., 2011, 23, 274 CrossRef CAS.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304 CrossRef CAS.
- Y. Tao, Q. Wang, L. Ao, C. Zhong, J. Qin, C. Yang and D. Ma, J. Mater. Chem., 2010, 20, 1759 RSC.
- H.-H. Chou and C.-H. Cheng, Adv. Mater., 2010, 22, 2468 CrossRef CAS.
- H.-H. Chou, H.-H. Shih and C.-H. Cheng, J. Mater. Chem., 2010, 20, 798 RSC.
- P.-I. Shih, C.-H. Chien, F.-I. Wu and C.-F. Shu, Adv. Funct. Mater., 2007, 17, 3514 CrossRef CAS.
- S. Gong, Y. Chen, C. Yang, C. Zhong, J. Qin and D. Ma, Adv. Mater., 2010, 22, 5370 CrossRef CAS.
- S. Gong, Y. Chen, J. Luo, C. Yang, C. Zhong, J. Qin and D. Ma, Adv. Funct. Mater., 2011, 21, 1168 CrossRef CAS.
- S. J. Lee, J. S. Park, K. J. Yoon, Y. I. Kim, S. H. Jin, S. K. Kang, Y. S. Gal, S. Kang, J. Y. Lee, J. W. Kang, S. H. Lee, H. D. Park and J. J. Kim, Adv. Funct. Mater., 2008, 18, 3922 CrossRef CAS.
- F. M. Hsu, C. H. Chen, P. I. Shih and C. F. Shu, Chem. Mater., 2009, 21, 1017 CrossRef CAS.
- T. J. Park, W. S. Jeon, J. J. Park, S. Y. Kim, Y. K. Lee, J. Jang and J. H. Kwon, Thin Solid Films, 2008, 517, 896 CrossRef CAS.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D'Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171 RSC.
- M. Tavasli, S. Bettington, M. R. Bryce, A. S. Batsanov and A. P. Monkman, Synthesis, 2005, 10, 1619 Search PubMed.
- (a) J. I. G. Cadogan and M. Cameron-Wood, J. Chem. Soc., 1962, 361 CAS; (b) J. I. G. Cadogan, M. Cameron-Wood, R. K. Mackie and R. J. G. Searle, J. Chem. Soc., 1965, 4831 RSC; (c) J.-F. Morin and M. Leclerc, Macromolecules, 2001, 34, 4680 CrossRef CAS; (d) F. Dierschke, A. C. Grimsdale and K. Müllen, Synthesis, 2003, 2470 CAS.
- (a) M. Sonntag and P. Strohriegl, Chem. Mater., 2004, 16, 4736 CrossRef CAS; (b) F. Ullmann and M. Zlokasoff, Chem. Ber., 1905, 38, 2211 CrossRef CAS.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef.
- K. Dedeian, P. I. Djurovich, F. O. Garces, G. Carlson and R. J. Watts, Inorg. Chem., 1991, 30, 1685 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. F. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS.
- M. G. Colombo, T. C. Brunold, T. Riedener, H. U. Güdel, M. Förtsch and H. Bürgi, Inorg. Chem., 1994, 33, 545 CrossRef CAS.
- (a) W.-Y. Wong, C.-L. Ho, Z.-Q. Gao, B.-X. Mi, C.-H. Chen, K.-W. Cheah and Z. Lin, Angew. Chem., Int. Ed., 2006, 45, 7800 CrossRef CAS; (b) C.-L. Ho, Q. Wang, C.-S. Lam, W.-Y. Wong, D. Ma, L. Wang, Z.-Q. Gao, C.-H. Chen, K.-W. Cheah and Z. Lin, Chem.–Asian J., 2009, 4, 89 CrossRef CAS.
- K.-T. Wong, T.-C. Chao, L.-C. Chi, Y.-Y. Chu, A. Balaiah, S.-F. Chiu, Y.-H. Liu and Y. Wang, Org. Lett., 2006, 8, 5033 CrossRef CAS.
- (a) H. Z. Xie, M. W. Liu, O. Y. Wang, X. H. Zhang, C. S. Lee, L. S. Hung, S. T. Lee, P. F. Teng, H. L. Kwong, H. Zhen and C. M. Che, Adv. Mater., 2001, 13, 1245 CrossRef CAS; (b) Y. H. Song, S.-J. Yeh, C.-T. Chen, Y. Chi, C.-S. Liu, J.-K. Yu, Y.-H. Hu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Adv. Funct. Mater., 2004, 14, 1221 CrossRef CAS; (c) W.-Y. Wong and C.-L. Ho, J. Mater. Chem., 2009, 19, 4457 RSC; (d) H. Yersin, Top. Curr. Chem., 2004, 241, 1 CAS; (e) P.-T. Chou and Y. Chi, Chem.–Eur. J., 2007, 13, 380 CrossRef CAS; (f) B. M. W. Langeveld and U. S. Schubert, Adv. Mater., 2005, 17, 1109 CrossRef; (g) M. A. Baldo, M. E. Thompson and S. R. Forrest, Pure Appl. Chem., 1999, 71, 2095 CrossRef CAS; (h) R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093 CrossRef CAS; (i) P. L. Burn, S.-C. Lo and I. D. W. Samuel, Adv. Mater., 2007, 19, 1675 CrossRef CAS; (j) Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2007, 36, 1421 RSC; (k) J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596 CrossRef; (l) H. Wu, L. Ying, W. Yang and Y. Cao, Chem. Soc. Rev., 2009, 38, 3391 RSC; (m) Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC.
- (a) G. Zhou, W.-Y. Wong and X. Yang, Chem.–Asian J., 2011, 6, 1706 CrossRef CAS; (b) C. Yang, X. Zhang, H. You, L. Zhu, L. Chen, L. Zhu, Y. Tao, D. Ma, Z. Shuai and J. Qin, Adv. Funct. Mater., 2007, 17, 651 CrossRef CAS; (c) K. Zhang, Z. Chen, C. Yang, X. Zhang, Y. Tao, L. Duan, L. Chen, L. Zhu, J. Qin and Y. Cao, J. Mater. Chem., 2007, 17, 3451 RSC; (d) S. Bettington, M. Tavasli, M. R. Bryce, A. Beeby, H. Al-Attar and A. P. Monkman, Chem.–Eur. J., 2006, 13, 1423 CrossRef.
- W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2009, 253, 1709 CrossRef CAS.
- M.-H. Tsai, H.-W. Lin, H.-C. Su, T.-H. Ke, C. Wu, F.-C. Fang, Y.-L. Liao, K.-T. Wong and C.-I. Wu, Adv. Mater., 2006, 18, 1216 CrossRef CAS.
- K.-T. Wong, Y.-M. Chen, Y.-T. Lin, H.-C. Su and C.-C. Wu, Org. Lett., 2005, 7, 5361 CrossRef CAS.
- W.-Y. Hung, L.-C. Chi, W.-J. Chen, Y.-M. Chen, S.-H. Chou and K.-T. Wong, J. Mater. Chem., 2010, 20, 10113 RSC.
- J. He, H. Liu, Y. Dai, X. Ou, J. Wang, S. Tao, X. Zhang, P. Wang and D. Ma, J. Phys. Chem. C, 2009, 113, 6761 CAS.
- A. Elscher, F. Bruder, H.-W. Heuer, F. Jonas, A. Karbach, S. Kirchmeyer, S. Thurm and R. Wehrmann, Synth. Met., 2000, 111, 139 CrossRef.
- S.-J. Su, E. Gonmori, H. Sasabe and J. Kido, Adv. Mater., 2008, 20, 4189 CAS.
- Y.-Y. Lyu, J. Kwak, W. S. Jeon, Y. Byun, H. S. Lee, D. Kim, C. Lee and K. Char, Adv. Funct. Mater., 2009, 19, 420 CrossRef CAS.
- J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332 CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS.
- W.-Y. Hung, T.-C. Tsai, S.-Y. Su, K.-T. Wong and L.-C. Chi, Phys. Chem. Chem. Phys., 2008, 10, 5822 RSC.
- G.-J. Zhou, Q. Wang, C.-L. Ho, W.-Y. Wong, D. Ma, L. Wang and Z. Lin, Chem.–Asian J., 2008, 3, 1830 CrossRef CAS.
- C.-H. Lin, Y. Chi, M.-W. Chung, Y.-J. Chen, K.-W. Wang, G.-H. Lee, P.-T. Chou, W.-Y. Hung and H.-C. Chiu, Dalton Trans., 2011, 40, 1132 RSC.
- C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang and Z. Lin, Adv. Funct. Mater., 2008, 18, 928 CrossRef CAS.
- W.-Y. Wong, G.-J. Zhou, X.-M. Yu, H.-S. Kwok and B. Tang, Adv. Funct. Mater., 2006, 16, 838 CrossRef CAS.
- G.-J. Zhou, W.-Y. Wong and S. Suo, J. Photochem. Photobiol., C, 2010, 11, 133 CrossRef CAS.
- Y. Shao and Y. Yang, Appl. Phys. Lett., 2005, 86, 073510 CrossRef.
- W.-Y. Hung, Z.-W. Chen, H.-W. You, F.-C. Fan, H.-F. Chen and K.-T. Wong, Org. Electron., 2011, 12, 575 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |