Photoinduced electron transfer in multilayer films composed of conjugated polyelectrolyte and amphiphilic copolymer hosting electron acceptor molecules†
Maciej
Kopeć
a,
Tomasz
Kruk
a,
Szczepan
Zapotoczny
*a,
Andre
Laschewsky
b,
Steven
Holdcroft
c,
Marek
Mac
a and
Maria
Nowakowska
*a
aFaculty of Chemistry, Jagiellonian University, Ingardena 3, 30-060, Kraków, Poland
bFraunhofer-Institut, Angewandte Polymerforschung (FhG-IAP), Geiselbergstrasse 69, D-14476, Potsdam-Golm, Germany
cDepartment of Chemistry, Simon Fraser University, Burnaby, BC, Canada V5A 1S6
First published on 1st November 2011
Abstract
Ultrathin multilayer films composed of conjugated anionic polyelectrolyte, poly(8-(3-thienyl)octanosulfonate) (P3TOSNa), and positively charged poly(N,N-dimethyl-N-4′-vinylbenzyl-N-2-(decanoyl-N′-methylimino)ethylammonium chloride)-co-(N,N,N-trimethyl-N-4′-vinylbenzylammonium chloride) (Ak-St-H) have been fabricated using the layer-by-layer technique. UV-Vis spectroscopy, atomic force microscopy and spectroscopic ellipsometry were applied for thickness and morphology characterization revealing regular growth and smooth surfaces of the films. An electron accepting molecule (butyl viologen) was then successfully entrapped inside the hydrophobic domains formed by Ak-St-Hvia a solubilization procedure. Photoinduced electron transfer from P3TOSNa polymer to butyl viologen molecules has been observed inside the nanostructured multilayers as confirmed by steady-state fluorescence spectroscopy and time-resolved fluorescence decay measurements. The structure of this water-born film helps to protect the hydrophobic acceptor molecules from aggregation and enables efficient charge separation within the film that is one of the crucial steps in organic photovoltaic systems.
Introduction
Nanoscale devices for solar energy conversion have gathered great interest since they are considered to be a possible future generation of clean energy generators.1 Among the wide variety of materials used for such systems, conjugated polyelectrolytes are of particular importance2 as they combine properties typical for Π-conjugated polymers such as fluorescence, electroactivity or charge carriers transport properties with the ability to be easily processed into ultrathin films in a controlled manner provided by the layer-by-layer electrostatic self-assembly technique (LbL).3 Application of the LbL method for fabrication of photovoltaic devices from conjugated polyelectrolytes alternately deposited with electron accepting modified fullerenes,4carbon nanotubes,5nanoparticles,6nanorods7 and organometallic dendrimers8 have been reported.Photoinduced electron transfer, followed by charge separation resulting in the formation of long-lived species, plays an essential role in efficient utilizing of solar energy with the help of organic materials.9 Charge separation occurs at the donor/acceptor interface, thus aggregation of the chromophores is a serious limiting factor as it lowers the area of contact between the phases and facilitates back electron-transfer reaction leading to charge recombination. Some efforts have been undertaken to achieve the photoinduced electron transfer inside the LbL films10–12 and indicated that stratified multilayers, exhibiting a polarity gradient, may facilitate directed electron transfer and efficient charge separation.
We have addressed this issue by changing the architecture of the polymers used for the fabrication of LbL films. Recently, we have developed ultrathin multilayers from water-soluble amphiphilic copolymer containing small organic molecule solubilised inside hydrophobic nanodomains.13–15 Using perylene as a fluorescent molecular probe it has been demonstrated that single molecules may be entrapped inside those nanodomains.13 The guest molecules can be excited directly by the absorbed light or as a result of the energy transfer from the host chromophores, e.g.naphthalene in the film formed using poly(styrene sulfonate-stat-vinylnaphthalene). In the current paper we present a similar approach for incorporation of an electron accepting butyl viologen (BV2+) molecule into the multilayer film with sulphonated polythiophene derivative (P3TOSNa) serving as an electron donor. BV2+ is solubilised in the nanodomains formed by amphiphilic polyelectrolyte (Ak-St-H), thus physically separated from the electron donor and the radical cation formed as a result of electron transfer. Even though interpenetration of adjacent layers in the LbL film may occur, the presence of nanocompartments prevents aggregation of the viologen molecules and limits the rate of back electron-transfer reaction also due to the presence of the charged polymeric interface. Photoinduced electron transfer from the excited P3TOSNa polymer to BV2+ is then possible to observe even with a small amount of electron acceptors present in the film. To the best of our knowledge this is the first time that the LbL film of a photoactive conjugated/amphiphilic polyelectrolyte system is presented with efficient charge separation process occurring in there.
Experimental part
Materials
Poly(sodium 8-(3-thienyl)octanosulfonate),16 poly(N,N-dimethyl-N-4′-vinylbenzyl-N-2-(decanoyl-N′-methylimino)ethylammonium chloride)-co-(N,N,N-trimethyl-N-4′-vinylbenzylammonium chloride) (Ak-St-H)17 and 1,1′-n-butyl-bipyridinium bromide (butyl viologen),18 were synthesized as described previously. The chemical structures of those compounds are presented in Scheme 1. Methyl viologen (Aldrich), sodium chloride (POCH Gliwice, Poland, analytical grade), sulfuric acid (96%, puriss, Lach-Ner, Czech Republic), hydrochloric acid (35%, puriss, POCH), hydrogen peroxide (30%, puriss, POCH) and methanol (analytical grade, POCH) were used as received. Deionized water was used in all the experiments.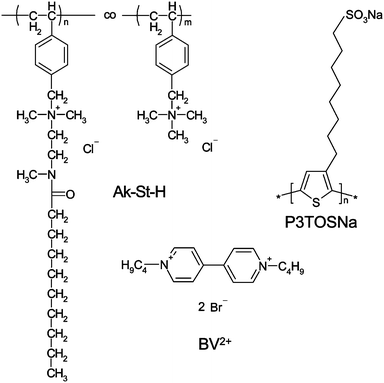 | ||
| Scheme 1 Chemical structure of polymers and molecular probe used in the experiment. | ||
Layer-by-layer deposition
Multilayer films were deposited on quartz slides or silicon wafers which were previously cleaned with “piranha” solution (H2SO4 and H2O2 (30%) mixture in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio) and rinsed with water. Deposition cycles were as follows: the substrate was immersed into the polycation solution (Ak-St-H) (c = 0.1 g L−1 in 0.1 M NaCl) (containing solubilised BV2+ – if specified), sonicated in an ultrasonic bath for 5 min at 40 °C, then rinsed with water and immersed into the polyanion (P3TOSNa) solution for 5 min sonication. All the solutions were at neutral pH. After such a cycle, a single layer pair (bilayer) was deposited on the substrate. The procedure was repeated until the desired number of bilayers were deposited. The schematic structure of the LbL films containing solubilized viologen molecules is presented in Scheme 2.
:1 volume ratio) and rinsed with water. Deposition cycles were as follows: the substrate was immersed into the polycation solution (Ak-St-H) (c = 0.1 g L−1 in 0.1 M NaCl) (containing solubilised BV2+ – if specified), sonicated in an ultrasonic bath for 5 min at 40 °C, then rinsed with water and immersed into the polyanion (P3TOSNa) solution for 5 min sonication. All the solutions were at neutral pH. After such a cycle, a single layer pair (bilayer) was deposited on the substrate. The procedure was repeated until the desired number of bilayers were deposited. The schematic structure of the LbL films containing solubilized viologen molecules is presented in Scheme 2.
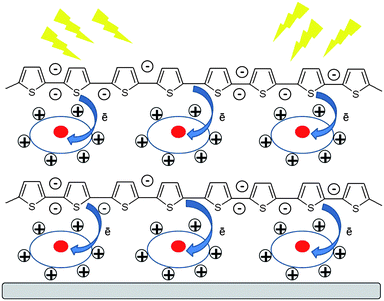 | ||
| Scheme 2 Schematic structure of the LbL films with entrapped viologen molecules (circles) illustrating the photoinduced electron transfer process. | ||
Solubilization
Solubilization was performed by slow addition of microliter quantities of BV2+ solution (c ≈ 10−3 M) in methanol into 10 ml aqueous solution of amphiphilic Ak-St-H polymer (c = 0.1 g L−1 in 0.1 M NaCl) using a syringe under vigorous stirring.Instruments
The UV–Vis absorption spectra were recorded using a Hewlett-Packard HP 8452A diode-array spectrophotometer. Steady-state fluorescence spectra were measured at room temperature in the deoxygenated atmosphere, using an SLM-Aminco 8100 spectrofluorometer in the case of films or Perkin-Elmer LS-55 spectrofluorimeter for solutions. Atomic force microscope (AFM) (Picoforce, Bruker) working in tapping mode was used to characterize the surfaces in air. Standard silicon cantilevers (Bruker) with nominal spring constant equal to 40 N m−1 were used. Dynamic Light Scattering (DLS) measurements were carried out using Zetasizer Nano ZS (Malvern).Time-resolved fluorescence
Fluorescence decay measurements were performed using time-correlated single photon counting (TCSPC) technique. As an excitation source a picosecond diode laser (λ = 400 nm, 70 ps pulse duration) from IBH-UK was used. The experimental fluorescence decays (F′(t)) were fitted to the convolution (F(t)) of the excitation profile (E(t)) with the bi-exponential function (I(t)):| I(t) = A1exp(−t/τ1) + A2exp(−t/τ2) | (1) |
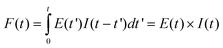 | (2) |
Spectroscopic ellipsometry
Film thickness measurements were performed both in air and in aqueous environment with Woollam M-2000U spectroscopic ellipsometer working in 600–800 nm spectral range to avoid absorption by P3TOSNa. The measurements for each sample were performed at least 7 times followed by fitting procedure. Cauchy function was used as a fitting model. Each film was modelled as a homogeneous organic layer (application of more complex multilayer models have not brought any significantly different results).Results and discussion
LbL film formation and characterization
Multilayer ultrathin films were fabricated using the LbL method with P3TOSNa polymer serving as a polyanion and Ak-St-H polymer as a polycation. We have shown before that due to the presence of a nine carbon atom-long alkyl side chain in the structure of Ak-St-H, the polymer forms micellar systems in aqueous solution19 that are likely to be preserved when the polymer is transferred to the film as shown for a similar copolymer.14The growth of the films was followed using UV-Vis absorption spectroscopy. Fig. 1 shows the absorption spectra of the multilayer films deposited on quartz with the maxima at λ = 420 nm, which is an absorption maximum of P3TOSNa. Linear film growth was observed, indicating that an equal amount of the polymer is adsorbed at each deposition step.
![UV-Vis
absorption spectra of [Ak-St-H/P3TOSNa]x multilayer films. Inset: dependence of the absorption at λ = 420 nm on the number of bilayers.](/image/article/2012/JM/c1jm13785a/c1jm13785a-f1.gif) | ||
| Fig. 1 UV-Vis absorption spectra of [Ak-St-H/P3TOSNa]x multilayer films. Inset: dependence of the absorption at λ = 420 nm on the number of bilayers. | ||
Ellipsometric measurements of the films' thicknesses confirmed the regular depositions of the multilayers (Fig. 2). The measurements were conducted both in air and water. In air, the average thickness per layer was found to be about 3 nm, while the first bilayer was somewhat thicker (4 nm) which reflects the substrate effect. In aqueous environment the film, being composed of hydrophilic polymers, swells and significantly increases its thickness. The growth is not linear since the thicker films swell less due to limited penetration of deeper layers by water molecules. These observations indicate regular growth of the densely packed films. This was further confirmed by atomic force microscopy (Fig. 3). AFM imaging reveals the surface features of size between 15 and 25 nm in diameter (see Fig. 3A and B) that are comparable with that ones measured by dynamic light scattering in the solution (17 nm) thus suggesting that micellar structure of Ak-St-H is preserved in the film. When linear homopolymer, poly(diallyldimethylammonium chloride) was used instead of Ak-St-H as a polycation, smaller features (8–9 nm) have been observed (see ESI†) suggesting that the features mentioned above are very likely to be Ak-St-H micellar structures. Moreover, for the thicker films (Fig. 3C and D), larger aggregates begin to form and dominate the film's surface topography along with a sudden increase of root-mean-square roughness from 0.8 nm and 0.7 nm for 1 and 2 bilayers to 2.5 nm and 2.4 nm for 4 and 6 bilayers, respectively. This effect may partially influence the photophysical properties of the films (see further).
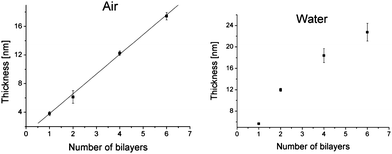 | ||
| Fig. 2 Ellipsometrically measured film thickness in air (left) and water (right) as a function of number of bilayers. | ||
 | ||
| Fig. 3 AFM images in air of one (A), two (B), four (C) and six (D) bilayers-thick films composed of Ak-St-H and P3TOSNa. Image size is 2 μm × 2 μm and z-scale is 10 nm for all the images. | ||
Electron transfer in nanostructured self-assembled films
[Ak-St-H/P3TOSNa]x films were used as platforms to perform photoinduced electron transfer (PET) in a confined environment. For that purpose, hydrophobic electron acceptor molecule, butyl viologen (BV2+), has been incorporated by the solubilization procedure into the aqueous Ak-St-H solution prior to its deposition. Methyl viologen (MV2+) is known to quench fluorescence of anionic conjugated polyelectrolytes with extreme efficiency – the effect often referred to as the amplified fluorescence quenching.20 This phenomenon, first described by Whitten et al., occurs due to ion-pair formation between the polymer and quencher molecules and a “molecular wire” effect, described as ultrafast exciton diffusing along a polymer chain that visits multiple (up to 1000) monomeric units before it is quenched. Such an effect was also evidenced for P3TOSNa in solution (see ESI†). More recently, fluorescence quenching of an electropolymerized poly(pyrrole-N-propanoic acid) film with MV2+ in aqueous solution has been also demonstrated using confocal laser scanning microscopy.21We report here on fabrication of ultrathin films with already embedded viologen molecules that are able to quench the conjugated polyelectrolyte's fluorescence. Since BV2+ contains butyl side chains it is more hydrophobic than MV2+ and it can be solubilised in the alkyl nanodomains formed in aqueous solution of Ak-St-H. It is expected that when Ak-St-H with incorporated BV2+ is used to form LbL films, the guest molecules are still present inside the hydrophobic (alkyl) domains in the film. That expectation is based on the results of our previous studies in which we have demonstrated that the perfluorinated probe molecules solubilised in aqueous solution of a perfluorinated derivative of Ak-St-H are present in the LbL films formed from this polymer.14 One could also consider the possibility that BV2+ is coadsorbed along with Ak-St-H during the film formation. Such a procedure for preparation of ordered nanocompartmentalized polyelectrolyte multilayers, in which molecules of dyes were coadsorbed with one of the polyelectrolyte has been demonstrated by Jonas and coworkers.22 However, taking into account the repulsion forces functioning between BV2+ and cationic Ak-St-H polymer and the fact that the films formed are carefully washed with water, the coadsorption of the low-molecular-weight compound is unlikely.
Driving force for PET from polythiophene derivative to butyl viologen can be calculated using Rehm–Weller equation:23
| ΔG0 = EoxD − EredA − E* − C | (3) |
PET in the system was experimentally studied by steady-state fluorescence and life-time measurements of the P3TOSNa polymer. Even though viologens are known to form an intensely blue-colored reduced state, it was impossible to follow its absorption nor emission changes because of too low concentration in the film. Steady-state fluorescence spectra of LbL films deposited on quartz substrate were measured at excitation wavelength, λ = 420 nm, absorbed only by P3TOSNa. The samples were constantly illuminated and the spectra were taken in 120 s intervals for total time of 30 min. Fig. 4A shows changes in the emission maximum intensity for 2 bilayers-thick films with and without BV2+ solubilized. In the absence of viologen only a slight decrease (of about 10%) of fluorescence intensity is visible, caused by the photobleaching of the polythiophene derivative. When the viologen molecule was present in the film, a significant drop in the P3TOSNa emission intensity can be observed upon irradiation. As the exposure time was equal for both samples, quenching efficiency can be estimated. The plot obtained by subtracting fluorescence intensity in the presence of acceptor molecule from the intensity without BV2+ reaches asymptotically a value of 0.4 over time for the 2 bilayer film. (Fig. 4B). This observation is most likely the effect of a very low concentration of viologen in the film that is too low to accept at once all the photogenerated electrons from P3TOSNa. Thus, after BV2+ cations are reduced via PET they need some time to “regenerate” viaoxidation by the surrounding molecules. Eventually, the system reaches an equilibrium as the plot reaches its plateau, so it can be assumed that the maximal efficiency of PET reaction is achieved in the system with given viologen content in the film. Similar characteristics have been observed for thicker films although efficiency of the process was significantly lower – for 4 bilayer-thick film it drops to c.a. 0.15 and undergoes little changes for 6, and 10 bilayers (Table 1). This is likely because the thinner multilayers display a more stratified structure which then becomes disrupted after adsorption of consecutive layers as has been shown before. Interpenetrated polymer chains may perturb the acceptor entrapment by compressing polythiophene chains and the nanodomains leading to aggregation and thus increasing probability of back electron transfer and charge recombination. These observations correlate well with the AFM data showing significant growth in the polymer aggregates' size between 2 and 4 bilayers-thick films (Fig. 3B and C). Curiously, we have found that the thinnest films exhibit a slight blue shift in their emission maxima that decreases from 13 nm for 2 bilayers to zero for 8 bilayers and thicker films (see ESI†). It is known that the conjugated polyelectrolytes are in equilibrium between their neutral and self-doped, more conductive form,26,27 exhibiting blue-shifted emission. The observed blue shift in the films may suggest that the equilibrium, usually governed by changes in pH value (see ESI†), may be slightly disturbed by other means (e.g.polymer–surface interactions) that consequently influence also the electron transfer efficiency.
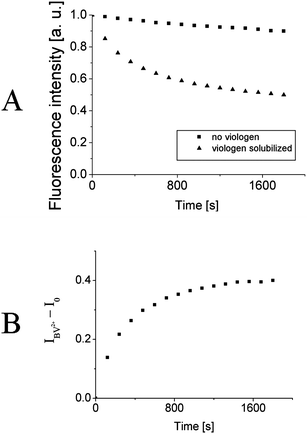 | ||
| Fig. 4 A) Normalized fluorescence intensity at emission maximum as a function of time for 2 bilayers-thick film with (triangles) and without (squares) butyl viologen solubilized. B) The difference in fluorescence intensities without and with viologen solubilized as a function of time. | ||
| Number of bilayers | Maximum PET efficiency (±10%) |
|---|---|
| 2 | 0.40 |
| 4 | 0.15 |
| 6 | 0.12 |
| 10 | 0.10 |
Precise quantitative calculations of the electron transfer efficiency are hardly possible since the exact concentration of the viologen in the film is difficult to determine. What is more, the effect of light scattering (more pronounced for thicker and rougher films) influences the measured fluorescence intensities. Interestingly, no quenching effect was observed when the films without solubilized acceptor molecules were immersed into the aqueous solution of butyl viologen, suggesting that the molecular probe can be embedded inside the film exclusively by being introduced into the amphiphilic copolymer solution prior to deposition.
Importantly, if the irradiation was carried out under aerobic conditions, the fluorescence decay of P3TOSNa was not dependent on the presence of the viologen molecules and was very likely the result of the irreversible photooxidation of the conjugated polyelectrolyte.
To confirm the occurrence of PET in the system, time correlated-single photon counting (TCSPC) technique was employed and fluorescence intensity decays have been measured. Thick, 10 bilayers films were used for these experiments as the fluorescence intensity of the thinner ones was below the instrument's sensitivity threshold. The emission decay at 564 nm which is around emission maximum of P3TOSNa for the films without and with viologen were measured (Fig. 5). The emission decay profiles were clearly not monoexponential and could be fitted with two exponential components. By integration of the decay profiles average fluorescence lifetimes were calculated to be τ0 = 1.85 ns and τ = 1.48 ns for the films without and with the viologen, respectively. Thus, shortening of the P3TOSNa fluorescence lifetime in the presence of viologen indicates quenching of the fluorescence due to electron transfer. The measured reduction of the fluorescence lifetime by 20% may be correlated with the efficiency of the electron transfer. This value is twice as high compared to the efficiency for the same 10 bilayers film estimated from the steady-state measurements. However, the difference may be caused by high sensitivity of the steady-state apparatus to small deviations in sample positioning and light scattering. It has to be noted that a significant quenching effect is observed with TCSPC technique for the film exhibiting the lowest efficiency in the steady-state measurements. That supports the idea that BV2+ acceptor molecules are localized within defined nanodomains formed by the polymer chains and can take part in PET reaction. The spectacular effect of nanocompartmentalization of dye on the photophysical properties of the polyelectrolyte multilayers were observed by Jonas et al.22 They have obtained a very elegant system for unidirectional Förster resonance energy transfer (FRET) in which the dye-loaded compartments were separated by dye-impermeable barriers introduced to the films. In our system separation is realized by solubilization of BV2+ in hydrophobic polymeric nanodomains.
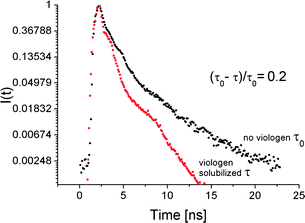 | ||
| Fig. 5 Fluorescence decays of Ak-St-H/P3TOSNa 10 bilayers-thick film without (right) and with (left) viologen solubilized. | ||
Conclusions
In this report we have presented a simple and versatile method for introducing electron accepting molecules (viologen derivatives) into the polyelectrolyte multilayer film in order to perform efficient photoinduced electron transfer within such ultrathin film. The films consisting of the conjugated polyelectrolyte serving as an electron donor and nanostructured amphiphilic polyelectrolyte hosting the electron acceptor molecules were formed using LbL method and characterized by spectroscopic and microscopic techniques. The PET process within such films was evidenced by observation of quenching of fluorescence of the conjugated polyelectrolyte, P3TOSNa, by steady-state and time-resolved techniques. The nanocompartmentalization of the acceptor molecules limits their aggregation and increases the efficiency of the PET process. Such nanostructured LbL systems seem to be very promising for construction of ultrathin devices such as photovoltaic cells or photoelectrochemical nanosensors.Acknowledgements
The research was carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08). The authors would like to thank Polish Ministry of Science and Higher Education for the financial support. The authors are also grateful to MSc Agnieszka Puciul (Jagiellonian Univeristy) for ellipsometric measurements.References
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834 CAS.
- (a) H. Jiang, P. Taranekar, J. R. Reynolds and K. S. Schanze, Angew. Chem., Int. Ed., 2009, 48, 4300 CrossRef CAS; (b) C. V. Hoven, A. Garcia, G. C. Bazan and T.-Q. Nguyen, Adv. Mater., 2008, 20, 3793 CrossRef CAS; (c) K. S. Schanze and A. H. Shelton, Langmuir, 2009, 25, 13698 CrossRef CAS; (d) A. Duarte, K.-Y. Pu, B. Liu and G. C. Bazan, Chem. Mater., 2011, 23, 501 CrossRef CAS.
- (a) Y. Lvov, G. Decher and H. Möhwald, Langmuir, 1993, 9, 481 CrossRef CAS; (b) G. Decher, Science, 1997, 277, 1232 CrossRef CAS; (c) P. Bertrand, A. Jonas, A. Laschewsky and R. Legras, Macromol. Rapid Commun., 2000, 21, 319 CrossRef CAS; (d) X. Zhang, H. Chen and H. Zhang, Chem. Commun., 2007, 1395 RSC; (e) K. Ariga, J. P. Hill and Q. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319 RSC; (f) X. Chi, M. Shen and H. Möhwald, Prog. Polym. Sci., 2004, 29, 987 CrossRef; (g) L. L. del Mercato, P. Rivera-Gil, A. Z. Abbasi, M. Ochs, C. Ganas, I. Zins, C. Sonnichsen and W. J. Parak, Nanoscale, 2010, 2, 458 RSC; (h) Y. Wang, L. Hosta-Rigau, H. Lomas and F. Caruso, Phys. Chem. Chem. Phys., 2011, 13, 4782 RSC.
- (a) H. Li, Y. Li, J. Zhai, G. Cui, H. Liu, S. Xiao, Y. Liu, F. Lu, L. Jiang and D. Zhu, Chem.–Eur. J., 2003, 9, 6031 CrossRef CAS; (b) J. K. Mwaura, M. R. Pinto, N. Ananthakrishnan, K. S. Schanze and J. R. Reynolds, Langmuir, 2005, 21, 10119 CrossRef CAS.
- V. Sgobba, A. Troeger, R. Cagnoli, A. Mateo-Alonso, F. Parenti, A. Mucci, L. Schenetti and D. M. Guldi, J. Mater. Chem., 2009, 19, 4319 RSC.
- (a) H. Ding, M. K. Ram and C. Nicolini, J. Mater. Chem., 2002, 12, 3585 RSC; (b) Z. Liang, K. L. Dzienis, J. Xu and Q. Wang, Adv. Funct. Mater., 2006, 16, 542 CrossRef CAS; (c) S. Wang, C. Li, F. Chen and G. Shi, Nanotechnology, 2007, 18, 185707 CrossRef; (d) R. Kniprath, J. T. McLeskey, J. P. Rabe and S. Kirstein, J. Appl. Phys., 2009, 105, 124313 CrossRef.
- S. A. McClure, B. J. Worfolk, D. A. Rider, R. T. Tucker, J. A. M. Fordyce, M. D. Fleischauer, K. D. Harris, M. J. Brett and J. M. Buriak, ACS Appl. Mater. Interfaces, 2010, 2, 219 CAS.
- C. W. Tse, K. Y. Kitty Man, K. W. Cheng, C. S. K. Mak, W. K. Chan, C. T. Yip, Z. T. Liu and A. B. Djurisic, Chem.–Eur. J., 2007, 13, 328 CrossRef.
- (a) J.-L. Bredas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691 CrossRef CAS; (b) M. M. Alam and S. A. Jenekhe, J. Phys. Chem. B, 2001, 105, 2479 CrossRef CAS; (c) J. Peet, A. J. Heeger and G. C. Bazan, Acc. Chem. Res., 2009, 42, 1700 CrossRef CAS.
- D. M. Kaschak, J. T. Lean, C. C. Waraksa, G. B. Saupe, H. Usami and T. E. Mallouk, J. Am. Chem. Soc., 1999, 121, 3435 CrossRef CAS.
- C. Tedeschi, H. Möhwald and S. Kirstein, J. Am. Chem. Soc., 2001, 123, 954 CrossRef CAS.
- C. Tedeschi, L. Li, H. Möhwald, C. Spitz, D. von Seggern, R. Menzel and S. Kirstein, J. Am. Chem. Soc., 2004, 126, 3218 CrossRef CAS.
- S. Zapotoczny, M. Golonka and M. Nowakowska, Langmuir, 2008, 24, 5868 CrossRef CAS.
- W. Niemiec, S. Zapotoczny, K. Szczubiałka, A. Laschewsky and M. Nowakowska, Langmuir, 2010, 26, 11915 CrossRef CAS.
- (a) N. Ma, H. Zhang, B. Song, Z. Wang and X. Zhang, Chem. Mater., 2005, 17, 5065 CrossRef CAS; (b) A. Guyomard, G. Muller and K. Glinel, Macromolecules, 2005, 38, 5737 CrossRef CAS; (c) A. Guyomard, B. Nysten, G. Muller and K. Glinel, Langmuir, 2006, 22, 2281 CrossRef CAS.
- M. I. Arroyo-Villan, G. A. Diaz-Quijada, M. S. A. Abdou and S. Holdcroft, Macromolecules, 1995, 28, 975 CrossRef CAS.
- A. Kotzev, A. Laschewsky and R. H. Rakotoaly, Macromol. Chem. Phys., 2001, 202, 3257 CrossRef CAS.
- P. M. S. Monk, The Viologens: Physicochemical Properties, Synthesis and Applications of the Salts of 4,4′-Bipyridine, John Wiley and Sons, New York, 1998 Search PubMed.
- K. Szczubialka, Ł. Moczek, A. Goliszek, M. Nowakowska, A. Kotzev and A. Laschewsky, J. Fluorine Chem., 2005, 126, 1409 CrossRef CAS.
- (a) L. Chen, D. W. McBranch, H.-L. Wang, R. Helgeson, F. Wudl and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12287 CrossRef CAS; (b) B. S. Harrison, M. B. Ramey, J. R. Reynolds and K. S. Schanze, J. Am. Chem. Soc., 2000, 122, 8561 CrossRef CAS; (c) M. B. Ramey, J. A. Hiller, M. F. Rubner, C. Tan, K. S. Schanze and J. R. Reynolds, Macromolecules, 2005, 38, 234 CrossRef CAS; (d) K. E. Achyuthan, T. S. Bergstedt, L. Chen, R. M. Jones, S. Kumaraswamy, S. A. Kushon, K. D. Ley, L. Lu, D. McBranch, H. Mukundan, F. Rininsland, X. Shi, W. Xia and D. G. Whitten, J. Mater. Chem., 2005, 15, 2648 RSC; (e) Y. Liu, K. Katsu Ogawa and K. S. Schanze, J. Photochem. Photobiol., C, 2009, 10, 173 CrossRef CAS.
- H. Dong, X. Cao and C. M. Li, ACS Appl. Mater. Interfaces, 2009, 1, 1599 CAS.
- S. Peralta, J.-L. Habib-Jiwan and A. M. Jonas, ChemPhysChem, 2009, 10, 137 CrossRef CAS.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CAS.
- M. Koch, R. Nicolaescu and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 11507 CAS.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1637 CrossRef CAS.
- A. O. Patil, Y. Ikenoue, F. Wudl and A. J. Heeger, J. Am. Chem. Soc., 1987, 109, 1858 CrossRef CAS.
- (a) J. Lukkari, A. Viinikanoja, J. Paukkunen, M. Salomäki, M. Janhonen, T. Ääritalo and J. Kankare, Chem. Commun., 2000, 571 RSC; (b) J. Lukkari, M. Salomäki, A. Viinikanoja, T. Ääritalo, J. Paukkunen, N. Kocharova and J. Kankare, J. Am. Chem. Soc., 2001, 123, 6083 CrossRef CAS; (c) A. Viinikanoja, S. Areva, N. Kocharova, T. Aäritalo, M. Vuorinen, A. Savunen, J. Kankare and J. Lukkari, Langmuir, 2006, 22, 6078 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1jm13785a |
| This journal is © The Royal Society of Chemistry 2012 |