Luminogenic materials constructed from tetraphenylethene building blocks: Synthesis, aggregation-induced emission, two-photon absorption, light refraction, and explosive detection†
Rongrong
Hu
a,
Jose Luis
Maldonado
b,
Mario
Rodriguez
b,
Chunmei
Deng
a,
Cathy K. W.
Jim
a,
Jacky W. Y.
Lam
a,
Matthew M. F.
Yuen
d,
Gabriel
Ramos-Ortiz
*b and
Ben Zhong
Tang
*ac
aDepartment of Chemistry, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
bCentro de Investigaciones en Óptica, A. P. 1-948, 37000, León, Gto, México. E-mail: garamoso@cio.mx
cDepartment of Polymer Science and Engineering, Institute of Biomedical Macromolecules, Key Laboratory of Macromolecular Synthesis and Functionalization of the Ministry of Education, Zhejiang University, Hangzhou, China
dDepartment of Mechanical Engineering, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China
First published on 3rd November 2011
Abstract
Luminogenic molecules [(TPE)3 (1), TPE-C = C-TPE-C = C-TPE (2), and TPE-C≡C-TPE-C≡C-TPE (3)] and their polymers P1–P3 are constructed from tetraphenylethene (TPE) building blocks in high yields by Suzuki, Witting, and Sonogashira coupling reactions. All the compounds are soluble and enjoy high thermal stability, losing little of their weights when they are heated to 290–528 °C under nitrogen or 288–436 °C in air. Analyses by UV spectroscopy and cyclic voltammetry as well as theoretical calculations show that the conjugation of the luminophores is in the order of 2 > 3 > 1, P2 > P3 > P1, and P1–P3 > 1–3. All the molecules and polymers are weakly emissive in solutions. They, however, become strong emitters in the aggregate state with fluorescence quantum yields up to 90%. Both 1–3 and P1–P3 exhibit the feature of aggregation-enhanced two-photon excited fluorescence. Large two-photon absorption cross sections (up to ∼900 GM) are observed in the nanoaggregates of the polymers. Thin solid films of the polymers show high refractive indices (RI = 1.7649 − 1.6873) in a wide wavelength region of 400–1700 nm, high modified Abbé numbers (vD′ up to 3436), and low optical dispersions (D′ down to 2.9 × 10−4). The light emissions of the polymers can be quenched exponentially by picric acid with large quenching constants, suggesting that they can be utilized as efficient chemosensors for explosive detection.
Introduction
Materials with two-photon absorption (TPA) have drawn much attention in recent years given their exceptional utility in three-dimensional micro fabrication and storage,1 optical power limiting,2 three-dimensional imaging,3 photodynamic therapy,4 and two-photon pumped up-converted lasers.5 To realize the full potential of the TPA technology, new TPA materials with large TPA cross sections and good processability, photostability, and durability are needed.6 Thanks to the research efforts of scientists, a number of organic TPA materials with varied molecular structures have been prepared, which provides insight into their structure-property relationship and gives valuable information for further molecular design and synthesis. For example, compounds with higher conjugation have been found to exhibit larger TPA cross sections. Incorporation of electron-donating and accepting units into the molecular structures also helps enhance the absorption. Other factors such as molecular coplanarity, solvent polarity, and hydrogen-bonding are found to play constructive effects in the enhancement of the TPA cross section.7For biophotonic applications, the TPA dyes should possess good water solubility or dispersity and remain highly fluorescent in aqueous media. However, many of them suffer the aggregation-caused emission quenching effect in concentrated solutions or in aqueous media owing to their hydrophobic nature and strong π–π interaction, which has somewhat limited their practical applications.8 To alleviate such problems, scientists utilized polymer nanoparticles as biocompatible encapsulating carriers to achieve a stable dispersion of the hydrophobic dyes. A challenge remaining to be solved in this methodology is the inevitable dilution of the active dye in a nanoparticle carrier, which cannot be offset by increasing the dye loading because of the notorious aggregation-caused quenching (ACQ) effect.9 With such regard, fabrication of silica nanoparticles10 and polyion complexation11 have been proposed as alternative tools to enhance the organo-modified fluorescence and TPA cross section of the luminophors in the aggregate state.
Recently, we and other groups observed a phenomenon of aggregation-induced emission (AIE) that is exactly opposite to the ACQ effect: a series of propeller-shaped molecules are nonemissive in solutions but are induced to emit intensely by aggregate formation.12 Such a novel effect enables the AIE luminogens to find potential high-tech applications as chemosensors, bioprobes, immunoassay markers, stimuli-responsive materials, and solid-state emitters. Although AIE luminogens with various functionalities have been prepared, there are few examples available in the literature that report the synthesis of luminogenic compounds with both AIE features and TPA absorption.13 Collini and coworkers reported the enhancement of TPA cross section of a fluorescent tetrakis(4-sulfonatophenyl)porphyrin diacid in the aggregate state.14 Prasad and coworkers reported the aggregation-enhanced emission and two photon absorption in nanoaggregates of 9,10-bis[4′-(4′′-aminostyryl)styryl]anthracene derivatives.15
To overcome the ACQ limitation of the current TPA luminophors, in this paper, we aim to synthesise AIE luminogens with nonlinear optical properties. We chose tetraphenylethene (TPE), an archetypal AIE luminogen, as a building block because of its facile synthesis, ready functionalization, and high quantum yield. We linked TPE units by different functional bridge groups and generated molecules 1–3 and polymers P1–P3 with different conjugations (Chart 1). We aim to investigate how conjugation affects their properties such as light emission and TPA absorption and meanwhile explore their potential practical applications.
 | ||
| Chart 1 Molecular structures of the tetraphenylethylene-containing molecules and polymers. | ||
Experimental section
Materials
Tetrahydrofuran (THF) and toluene were distilled under normal pressure from sodium benzophenone ketyl under argon immediately prior to use. Dimethylformamide (DMF) and triethylamine (Et3N) were distilled and dried over potassium hydroxide. Rhodamine 6G was purchased from Exciton Inc. Dichloromethane (DCM), diphenylmethane (4), 4-bromobenzophenone (5), n-butyllithium (n-BuLi), dichlorobis(triphenylphosphine)palladium(II) [Pd(PPh3)2Cl2], copper(I) iodide (CuI), triphenylphosphine (PPh3), zinc powder, titanium(IV) chloride (TiCl4), tetrabutylammonium fluoride (TBAF), p-toluenesulfonic acid (PTSA), trimethyl borate [B(OCH3)3], sodium carbonate (Na2CO3), tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4], N-bromosuccinimide (NBS), benzoyl peroxide (BPO), N-formylpiperidine, potassium tert-butoxide (t-BuOK), trimethylsilylacetylene, and other chemicals and solvents were all purchased from Aldrich and used as received without further purification. Compound 7, named 1-(4-bromophenyl)-1,2,2-triphenylethene, was synthesized according to the literature method.16Instruments
1H and 13C NMR spectra were measured on Bruker ARX 300 or 400 NMR spectrometers using CDCl3 or CD2Cl2 as solvents and tetramethylsilane (TMS; δ = 0 ppm) as internal standard. IR spectra were recorded on a Perkin-Elmer 16 PC FT-IR spectrophotometer. Thermogravimetric analysis (TGA) was carried out under nitrogen or in air on a Perkin-Elmer TGA 7 analyzer at a heating rate of 10 °C min−1. UV-vis absorption spectra were measured on a Milton Roy Spectronic 3000 array spectrophotometer. Photoluminescence (PL) spectra were recorded on a Perkin-Elmer LS 55 spectrofluorometer. The fluorescence quantum yields of 1–3 and P1–P3 in THF (2.6 μM) and their nanoaggregates dispersed in water were determined by using an integrating sphere calibrated by a fluorescent standard (Rhodamine 6G) with known quantum yield. A HeCd laser (325 nm) was employed as excitation source. The number (Mn) and weight-average (Mw) molecular weights and polydispersity indices (PDI or Mw/Mn) of the polymers were estimated by a Waters Associates gel permeation chromatography (GPC) system equipped with RI and UV detectors. THF was used as eluent at a flow rate of 1.0 mL. A set of monodisperse linear polystyrenes covering the molecular weight range of 103–107 were used as standards for the calibration. High resolution mass spectra were recorded on a GCT premier CAB048 mass spectrometer operating in a MALDI-TOF mode. Cyclic voltammetry was conducted on a CHI600A electrochemical workstation. All the measurements were carried out at room temperature using a conventional three-electrode configuration at a scan rate of 100 mV s−1. The working electrode was a glassy carbon electrode with a diameter of 2 mm. Silver chloride electrode was used as reference and the counter electrode was a platinum wire. The solutions were prepared in distilled DCM. Tetrabutylammonium hexafluorophosphate (0.1 M) was used as supporting electrolyte. The refractive indices of the polymers were determined on a J A Woollam variable angle ellipometry system with a wavelength tunability from 300 to 1700 nm. To fit the acquired Ψ and Δ curves with the data obtained from the 3-layer optical model consisting of a crystalline silicon substrate, 2-nm SiO2 layer and a uniform polymer film, the Levenberg-Marquardt regression algorithm was employed. The Cauchy dispersion law was applied to describe the polymer layer from the visible to IR spectral region. The ground state geometries of 1–3 were optimized via density functional theory (DFT). The DFT calculations were carried out using the B3LYP functional, where Becke's three-parameter hybrid exchange functional was combined with the Lee–Yang–Parr correlation functional,17 and a def2-SV(P) basis set.18 All calculations were performed with the TURBOMOLE 6.0 program.19Two-photon absorption (TPA) of the molecules and polymers in the solution and aggregate states was measured by the two-photon excited fluorescence (TPEF) technique using a Ti:Sapphire laser (Spectra-Physics). This laser provided pulses of 100 fs of duration at a repetition rate of 80 MHz and was tunable in the wavelength range of 740–820 nm. The laser beam was focused into a quartz cell of 1 cm path length by using a 5 cm focal-length lens. A half-wave plate and a polarizer were used to control the excitation intensity. The induced two-photon fluorescence was collimated by a lens at a direction perpendicular to the pump beam. To minimize the attenuation of fluorescence due to linear absorption effects, the excitation beam was focused as close as possible to the lateral wall of the quartz cell. The TPEF was then focused into the input slit of an imaging spectrograph and recorded at the exit with a CCD camera. To calculate the TPEF cross sections, Rhodamine 6G in methanol solution (10 μM) and its nanoaggregates were utilized as references for the calculation. All the samples and standards were tested under the same experimental conditions. The TPEF cross sections were calculated by the equation: σTPEF = σrefcrefnrefF/cnFref, where c and n were the concentration and refractive index of the samples and reference, and F was the integral of the TPEF spectrum. The two photon cross section σTPA was then calculated by the equation σTPA = σTPEF/ΦF, where ΦF was the fluorescence quantum yield of the sample as stated in Table 1.
| Compound | λ ab (nm) | λ em (nm) | Φsoln (%) | Φaggr (%) |
|---|---|---|---|---|
a Abbreviation: λab = absorption maximum in THF solution (10 μM), λem = emission maximum in THF/water mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 v/v), Φsoln and Φaggr = fluorescence quantum yields in THF and aqueous nanoparticle suspension determined by an calibrated integrating sphere. :9 v/v), Φsoln and Φaggr = fluorescence quantum yields in THF and aqueous nanoparticle suspension determined by an calibrated integrating sphere.
|
||||
| 1 | 341 | 493 | 1.0 | 90 |
| 2 | 360 | 511 | 1.0 | 76 |
| 3 | 352 | 500 | 0.5 | 74 |
| P1 | 356 | 506 | 1.2 | 28 |
| P2 | 382 | 520 | 3.4 | 64 |
| P3 | 360 | 502 | 1.1 | 18 |
Synthesis
Compound 1–3 and polymer P1–P3 were prepared according to the synthetic routes shown in Scheme 1 and 2. Detailed procedures and characterization can be found in the supporting information. | ||
| Scheme 1 Synthetic routes for compounds 1–3. | ||
 | ||
| Scheme 2 Synthesis of polymers P1–P3. | ||
Preparation of aggregates
Stock THF solutions of the molecules and polymers with a concentration of 0.1 mM were prepared. An aliquot (1 mL) of this stock solution was transferred to a 10 mL volumetric flask. After adding an appropriate amount of THF, water was added dropwise under vigorous stirring to furnish 10 μM THF/water mixtures with water fractions (fw) of 0–90 vol %. 10 μM THF/water mixtures with 95% water contents were prepared by 1 mM THF solutions in the same manner. The absorption and emission spectra of the resultant mixtures were measured immediately.The nanoaggregates used in the TPA cross section measurement were prepared by the reprecipitation method. First, solutions of 1–3 and P1–P3 were prepared by dissolving 1 mg of the compounds in 2 mL THF. 0.5 mL of each solution was then injected quickly into 8 mL of poor solvent (aqueous solution of cetyl trimethyl ammonium bromide, 0.08 mM). After sonication, THF was partially removed under vacuum. Through such a procedure, the concentrations of the nanoparticle suspensions were in the range of 2 × 10−7 to 3 × 10−5 M.
Explosive detection
A stock solution of picric acid (PA) with a concentration of 2 mg mL−1 was prepared by dissolving an appropriate amount of PA in THF. Photoluminescence titration was carried out by adding aliquots of PA solution into solutions of P1 in THF and THF/water mixtures with 50 and 90% water contents.Results and discussion
Synthesis
To endow the AIE luminogens with new functionalities and widen their practical applications, we designed the molecular structures of a group of luminogenic molecules (1–3) and polymers (P1–P3) and elaborated multi-step reaction routes for their syntheses (Scheme 1 and 2). Lithiation of 4 followed by reaction with 5 generated 6, which underwent dehydration in the presence of PTSA to form 7 in 95% yield. Treatment of 7 with n-butyllithium followed by reaction with trimethyl borate furnished 8. Meanwhile, McMurry coupling reaction of 5 catalyzed by TiCl4/Zn produced 9 with both cis and trans isomers, whose palladium-catalyzed Suzuki coupling with 8 gave 1 in 62% yield.Similarly, McMurry coupling reaction of 10 afforded 11, which was converted into 12 by bromination with NBS. Reaction of 12 with triphenylphosphine generated phosphonium salt. On the other hand, lithiation of 7 followed by addition of N-formylpiperidine furnished 14. Witting reaction of 13 and 14 in the presence of potassium tert-butoxide afforded 2 in 91% yield. Meanwhile, palladium-catalyzed Sonogashira coupling reaction of 5 with trimethylsilylacetylene produced 15, whose McMurry coupling reaction generated 16. Desilylation of 16 yielded 17 and its coupling reaction afforded the desirable product 3 in 95% yield.
To prepare the polymeric analogs of 1–3, 9 was converted into diboronic acid 18 and dialdehyde 19 by lithiation followed by reaction with trimethyl borate and N-formylpiperidine, respectively. Polymerizations of 9/18 and 13/19 pairs under the conditions for Suzuki and Witting reactions generated P1 and P2 in high yields with moderate molecular weights. On the other hand, Sonogashira coupling of 9 with 17 furnished P3 again in a high isolated yield.
Structural characterization
All the intermediates and products were characterized by standard spectroscopic methods and all gave satisfactory analysis data corresponding to their expected molecular structures (see experimental section for details). Luminogens 1–3, for example, give M+ peaks at m/z 992.4176 (calcd for 1, 992.4382), 1044.6278 (calcd for 2, 1044.4695), and 1040.4373 (calcd for 3, 1040.4382) in their HRMS spectra (Fig. S1–S3†), confirming the formation of the expected products. The IR spectra of 9, 17, and P3 are also given in Fig. 1 as an example. The spectrum of P3 resembles its monomers but exhibits no ≡C–H and terminal C≡C stretching vibrations of 17 at 3281 and 2107 cm−1, respectively, suggesting that all the molecules of 17 have been consumed by the polymerization reaction.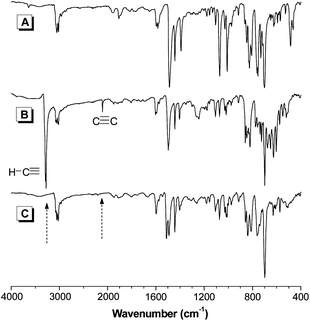 | ||
| Fig. 1 IR spectra of (A) 9, (B) 17, and (C) their polymer P3. | ||
Analysis by 1H NMR spectroscopy gives similar results. The acetylene proton resonance of 17 occurs at δ 3.09, which is absent in the spectrum of P3 (Fig. 2). The absorptions of the aromatic protons in P3 are broad because its TPE units are knitted together by rigid acetylene linkages.
 | ||
| Fig. 2 1H NMR spectra of (A) 9, (B) 17, and (C) their polymer P3 in dichloromethane-d2. | ||
The 13C NMR spectra of 9, 17, and their polymer P3 are shown in Fig. 3. The spectrum of P3 shows no resonance peaks of carbon atoms of 17 at δ 78.1 and 84.2. A new peak associated with the absorptions of the internal triple bond carbons was observed at δ 90.4. No other unexpected signals are found, revealing that the polymeric product is indeed P3, with a molecular structure as shown in Chart 1.
 | ||
| Fig. 3 13C NMR spectra of (A) 9, (B) 17, and (C) their polymer P3 in dichloromethane-d2. | ||
Solubility and stability
Though 1–3 and P1–P3 are constructed from aromatic rings, they dissolve readily in common organic solvents, such as toluene, dichloromethane, chloroform, and THF. Polymers P1–P3 can also form tough films by spin-coating their solutions. The good solubility of the molecules and polymers may be stemmed from the twisted conformation of their TPE units, which reduces intermolecular interactions and generates large free volume to accommodate more solvent molecules.Since 1–3 and P1–P3 possess many aromatic rings, they are anticipated to show high resistance to thermolysis.20 This is indeed the case, as revealed by their TGA thermograms shown in Fig. 4. Molecules 1–3 show 5% of their weight loss (Td) at 290–308 °C under nitrogen atmosphere. Similar Td values are recorded when the measurements are performed in air. Compared to their low molecular weight counterparts, P1–P3 enjoy higher thermal stability. They start to decompose at higher temperatures with Td values of 463–528 °C under nitrogen. Although they exhibit lower thermal stability in air, their Td values are all beyond 430 °C and still much higher than those of 1–3. It is noteworthy that more than 70% of their weights are retained when they are heated under nitrogen at 600 °C, making them promising as precursors for ceramic materials.
 | ||
| Fig. 4 TGA thermograms of (A) 1–3 and (B) P1–P3 recorded under nitrogen and in air at a heating rate of 10 °C min−1. | ||
Photophysical properties
The absorption spectra of 1–3 and P1–P3 in their dilute THF solutions are depicted in Fig. 5. The absorption spectra of 1–3 peaked at 341, 360, and 352 nm, respectively, revealing that the functional bridge group has influenced strongly the conjugation of the molecules. The absorption maxima of P1–P3 are located at 356, 382, and 360 nm, which are 15, 22, and 8 nm red-shifted from those of 1–3, indicative of a higher conjugation in the polymeric system.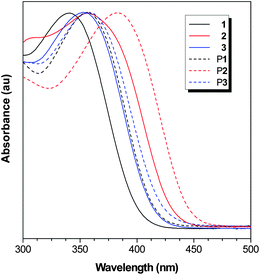 | ||
| Fig. 5 Normalized absorption spectra of 1–3 and P1–P3 in THF solutions. Concentration: 10 μM. | ||
All the molecules and polymers, similar to TPE, are AIE-active, as suggested by the fluorescent images of their solutions in THF and THF/water mixtures. The photographs of solutions of 1 and P1 are given in Fig. 6 and 7 as examples, while those of 2, 3, P2, and P3 are provided in Fig. S4–S7†. The THF solution of 1 emits no light under UV illumination. Addition of a poor solvent such as water into the THF solution has, however, aggregated its molecules and enhanced its emission intensity. The emission is still weak in aqueous mixtures at low water fractions (≤50%) but becomes stronger afterwards. A similar phenomenon was also observed in P1 but its solution is already somewhat emissive in the presence of a small amount of water (30%), presumably due to its comparatively lower solubility in the aqueous mixture.
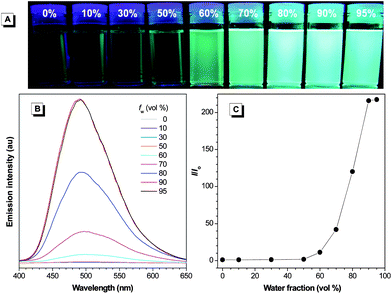 | ||
| Fig. 6 (A) Photographs of 1 in THF/water mixtures with different fractions of water (fw) taken under UV illumination. (B) Emission spectra of 1 in THF/water mixtures. (C) Plot of (I/Io) values versus the compositions of the aqueous mixtures. Io = emission intensity in pure THF solution. Solution concentration: 10 μM; excitation wavelength: 341 nm. | ||
 | ||
| Fig. 7 (A) Photographs of P1 in THF/water mixtures with different fractions of water (fw) taken under UV illumination. (B) Emission spectra of P1 in THF/water mixtures. (C) Plot of (I/Io) values versus the compositions of the aqueous mixtures. Io = emission intensity in pure THF solution. Solution concentration: 10 μM; excitation wavelength: 356 nm. | ||
In addition to the visual observations, we also studied the emission behaviors of 1–3 and P1–P3 in the solution and aggregate states by using a spectrofluorometer. As shown in Fig. 6B, 1 is practically non-emissive in THF. When 60% of water is added to its THF solution, an emission peak, however, emerges at 493 nm, whose intensity is enhanced by further increasing the water content in the aqueous mixture. From the isolated species in THF solution to nanoaggregates in 90% aqueous mixture, the emission intensity rises by 216-fold (Fig. 6C). Unlike 1, P1 emits at 506 nm in pure THF solution albeit in a low intensity (Fig. 7B). We have previously proposed that the AIE phenomenon is caused by the restriction of intramolecular rotation in the aggregate state, which blocks the nonradiative relaxation channel and populates the radiative decay. The TPE units in P1 are knitted together by covalent bonds, which partially restricts their intramolecular rotation and hence makes the polymer somewhat emissive in the solution state. The emission intensity starts to rise when a small amount of water is added to the THF solution and reaches its maximum value at 90% water content, which is 68-fold higher than that in pure THF solution (Fig. 7C). Similar observations are also found in 2, 3, P2, and P3 (Fig. S4–S7†).
To have a quantitative comparison, we measured the fluorescence quantum yields (ΦF) of the molecules and polymers in the solution and aggregate states. Whereas the ΦF values of 1–3 in THF are generally below 1%, those in the aggregate state are much higher (74–90%), manifesting their AIE feature (Table 1). The ΦF values of P1–P3 in the solution state are also low (1.1–3.4%) but in relative terms, they are higher than those of 1–3, in agreement with their stronger emissions in the THF solutions. The polymers show higher fluorescence quantum yields in the aggregated state. Compared with those of 1–3, their ΦF values, however, are much lower, probably due to the emission quenching caused by the defects in the polymer chain.
Electronic structure
To better understand the photophysical properties of 1–3, theoretical calculations on their energy levels were performed. The molecular orbital amplitude plots of the HOMO and LUMO energy levels of trans1–3 are shown in Fig. 8, while those of the cis counterparts are provided in Fig. S8†. All the trans isomers show similar orbital distribution and their HOMO and LUMO energy levels are dominated by the central TPE core and the vinyl cores of the two terminal TPE units. The six peripheral phenyl rings are twisted and contribute little to the energy levels of the molecules. This orbital distribution results in a linear and high conjugation in 1–3. The two peripheral TPE units are also conjugated with the central TPE core in cis1–3. The orbitals of their HOMO and LUMO, however, are distributed in a V-shaped manner. The calculated HOMO and LUMO values of the cis and trans isomers are almost the same (Table S1†), indicating that their mixing would not affect the overall photophysical properties of the molecules. The calculated energy band gaps for trans1, 2 and 3 are 3.53, 3.05, and 3.20 eV, respectively, which nicely explain the bathochromic shifts in the absorption and emission of 2 from those of 3 and 1. | ||
| Fig. 8 Molecular orbital amplitude plots of HOMO and LUMO energy levels of trans1–3 calculated using B3LYP/6-31G(d) basis set. | ||
Electrochemical properties
We also investigated the electrochemical properties of 1–3 by cyclic voltammetry. Such a method is widely used for estimating the energy levels of organic semiconductors. All the molecules exhibit similar voltammograms. Their HOMO energy levels can be estimated from their onset oxidation potentials based on the value of −4.8 eV for ferrocene as internal standard with respect to the zero vacuum level. The onset oxidation potentials of 1–3 are found at 1.14, 1.14, and 1.21 eV from which HOMO values of −5.45, −5.45, and −5.52, are derived, respectively (Table 2). The band gaps of 1–3 can be determined from their onset absorption wavelengths and are calculated to be 3.11, 2.89, and 3.00 eV, respectively. Although the experimental values are somewhat lower than the calculated ones, the conjugation of the molecules is still in the order of 2 > 3 > 1.| Compound | E onset-ox (V) | HOMO (eV) | λ onset(nm) | E g(eV) | LUMO (eV) |
|---|---|---|---|---|---|
| a Abbreviation: Eonset-ox = onset oxidation potential measured by cyclic voltammetry, HOMO = highest occupied molecular orbital derived by the equation: HOMO = −(Eonset-ox + 4.8 − Eferrocene) eV, where the value of Eferrocene measured in our experiment was 0.49 eV, λonset = onset absorption wavelength, Eg = energy band gap determined from λonset, LUMO = lowest unoccupied molecular orbital = Eg + HOMO. | |||||
| 1 | 1.14 | −5.45 | 399 | 3.11 | −2.34 |
| 2 | 1.14 | −5.45 | 429 | 2.89 | −2.56 |
| 3 | 1.21 | −5.52 | 414 | 3.00 | −2.52 |
Nonlinear optical properties
Since 1–3 and P1–P3 are conjugated, we are intrigued to know whether they show two-photon excited fluorescence and absorption. With this in mind, we thus investigated the nonlinear optical properties of the molecules and polymers in the solution and aggregate states by a two-photon excited technique. Delightfully, all the molecules and polymers exhibit two-photon excited fluorescence (TPEF). An example of the TPEF spectra of P2 in THF and aqueous nanoparticle suspension are given in Fig. 9. Both the spectra peaked at 520 nm. The TPEF intensity of the latter, however, is 8.6-fold higher than the former, demonstrating of a phenomenon of aggregation enhanced two-photon excited fluorescence. | ||
| Fig. 9 TPEF spectra of THF solution and nanoaggregates of P2. Solution concentration: 2.6 mM; excitation wavelength: 800 nm. | ||
The two-photon absorption cross sections (σTPA) of the molecules and polymers are given in Table 3. The σTPA values of 1–3 are low in THF (0.5–4.7 GM) when photoexcited at 800 nm. On the contrary, P1–P3 exhibit much higher absorptions under the same experimental conditions, presumably due to their higher conjugation. All the compounds show higher σTPA values in the aggregate state, particularly for P2, which absorbs intensely with a σTPA value of 275.8 GM. Their AIE effect can be compared by using the γAIE value, which is defined by the equation: γAIE = σTPA, aggr/σTPA, soln. The γAIE values of 1–3 range from 1.4 to 2.8, while those of P1–P3 reach 8.7, 8.7, and 7.5, respectively, which are larger than the values of most compounds reported previously.13,15,21
| Compound | λ ex, 800 nm | λ ex, 750 nm | ||||
|---|---|---|---|---|---|---|
| σ TPA, soln(GM) | σ TPA, aggr(GM) | γ AIE | σ TPA, soln (GM) | σ TPA, aggr (GM) | γ AIE | |
| a In THF solution (2.6 μM). b In aqueous nanoparticle suspension (31.25 μg mL−1). c Abbreviation: λex = excitation wavelength, σTPA = two-photon absorption cross section, γAIE =σTPA, aggr/σTPA, soln, nd = not determined because of the weak excitation intensity at 750 nm. | ||||||
| 1 | 0.5 | 1.4 | 2.8 | nd | 2.9 | nd |
| 2 | 4.7 | 13.2 | 2.8 | 49.1 | 63.9 | 1.3 |
| 3 | 1.9 | 2.7 | 1.4 | nd | 8.7 | nd |
| P1 | 2.1 | 18.2 | 8.7 | nd | 56.5 | nd |
| P2 | 31.7 | 275.8 | 8.7 | 106.9 | 895.9 | 8.4 |
| P3 | 20.8 | 155.7 | 7.5 | 88.7 | 714.2 | 8.1 |
Since 1–3 and P1–P3 show no absorption or weakly absorb at 800 nm, one would expect they show stronger TPEF and higher TPA values at shorter excitation wavelengths. Indeed, the σTPA values of the molecules and polymers are much higher (enhancement up to 10-fold) when determined at 750 nm excitation wavelength. Again, the nanoaggregates show larger TPA cross sections than their corresponding isolated species in THF solutions. The σTPA values of P2 and P3 are impressively high (895.9 GM for P2 and 714.2 GM for P3) with γAIE values larger than 8, taking into the account that the absorptions of most TPA materials in the aggregate state lie in the range of 90–330 GM.10,11,13–15,21 This is quite unusual because P2 and P3 possess no electron-donating or accepting unit in their molecular structures and compounds with similar structures normally have low σTPA values. For example, the σTPA value of stilbene is merely 12 GM.7b
Preliminary results show that the σTPA values of the molecules and polymers are further enhanced if lower excitation wavelengths are used. For example, the σTPA value for the nanoaggregates of P2 determined at 740 nm excitation wavelength is as high as 1360.9 GM. A more detailed investigation on the TPA properties of the compounds is under investigation. It is noteworthy that the light emissions of the nanoaggregates are very stable, suffering no spectral change when they are dispersed in the aqueous nanoparticle suspension for several months. This enables them to find potential biophotonic applications, for example, as markers for multiphoton microscopy or biosensors.
Light refraction
P1–P3 are conjugated and possess many aromatic rings and thus may show high refractive indices (RI). Indeed, as can be seen from Fig. 10, the thin film of P2 prepared by spin-coating of its solution displays high RI values (n = 1.7649 − 1.6873) in a wide wavelength region (400–1700 nm). Similarly high RI values are also observed in P3 (n = 1.7795 − 1.6430). The RI values of P2 and P3 at 632.8 nm are ≥1.69, which is much higher than those of the commercially important optical plastics (e.g., n ∼ 1.49 for PMMA).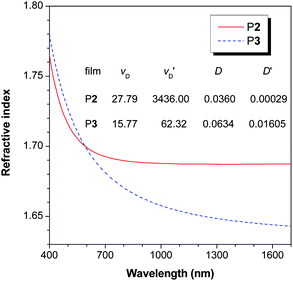 | ||
| Fig. 10 Wavelength dependence of refractive index of thin films of P2 and P3. | ||
For a material to be useful for practical applications, its optical aberrations should be small. The Abbé number (νD) of a material is a measure of the variation or dispersion in its RI value with wavelength and is defined as: νD = (nD − 1)/(nF − nC), where nD, nF and nC are the RI values at wavelengths of Fraunhofer D, F and C spectral lines of 589.2, 486.1 and 656.3 nm, respectively. A modified Abbé number (νD′) has also been proposed to evaluate the potential application of an optical material, using its RI values at the non-absorbing wavelengths of 1064, 1319 and 1550 nm.22 The modified Abbé number is defined as: νD′ = (n1319 − 1)/(n1064 − n1550), where n1319, n1064 and n1550 are the RI values at 1319, 1064 and 1550 nm, respectively. The chromatic dispersion (D′) is the constringence of the Abbé number: (D′ = 1/νD′). Both P2 and P3 show low optical dispersions. The D value of P2 is 36.0 × 10−3, which is comparable to those of the commercially important “organic glasses” such as PC (D = 29.7 × 10−3) and PMMA (D = 17.5 × 10−3).
Explosive detection
Sensors based on fluorescent conjugated polymers have attracted much attention due to their amplified response and superior sensitivity to analytes, in comparison to their low molar mass congeners. Generally, intrinsic autoaggregation of chains of conjugated polymers and/or their analyte-induced aggregation causes self-quenching problems that greatly reduce the sensing performance. However, aggregation is beneficial to the emission of P1–P3. With such regard, we explored their utility as chemosensors for explosives because their sensitive detection has antiterrorism implications.23 The nanoaggregates of P1 in THF/water mixtures with 50 and 90% water contents are utilized as probes. For comparison, the detection was also carried out in pure THF solution. Picric acid (PA) was employed as a model compound due to its commercial availability.As shown in Fig. 11A, the emission of P1 is weakened when PA is gradually added into its nanoaggregates in the aqueous mixture. The fluorescence quenching can be clearly discerned at a PA concentration as low as 1 ppm. At a PA concentration of 0.29 mM, virtually no light can be detected by the spectrofluorometer. The Lewis acid–base interactions between the TPE units of P1 with the electron-deficient PA molecules may play a key role in the quenching process. The Stern–Volmer plot of relative PL intensity (Io/I − 1) versus PA concentration in THF solution as well as the nanoaggregates suspended in the aqueous mixtures give curves bending upward instead of linear lines (Fig. 11B), indicating that the PL quenching becomes more efficient with increasing quencher concentration. The emission of the aggregates decreases in a much faster rate than their isolated chains in THF solution (Fig. S9†), indicative of a higher sensitivity. The nanoaggregates have more cavities to bind with more quencher molecules and provide additional interchain diffusion pathways for excitions to migrate,24 thus making the quenching a highly efficient process. The aggregate-based sensor in the aqueous mixture with fw = 50% shows a higher sensing performance than that with fw = 90%, probably because the polymer packing in the former is looser than in the latter and the looser packing allows voids to interact with more PA molecules.
![(A) Emission spectra of P1 in THF/water mixture with 90% water fraction (fw) containing different amounts of picric acid (PA). (B) Plots of (Io/I − 1) values versus PA concentrations in THF/water mixtures with fws of 0, 50, and 90 vol%. Io = intensity at [PA] = 0 mM. Concentration: 10 mM; excitation wavelength: 356 nm.](/image/article/2012/JM/c1jm13556b/c1jm13556b-f11.gif) | ||
| Fig. 11 (A) Emission spectra of P1 in THF/water mixture with 90% water fraction (fw) containing different amounts of picric acid (PA). (B) Plots of (Io/I − 1) values versus PA concentrations in THF/water mixtures with fws of 0, 50, and 90 vol%. Io = intensity at [PA] = 0 mM. Concentration: 10 mM; excitation wavelength: 356 nm. | ||
Conclusions
In this work, luminogenic molecules (1–3) and polymers (P1–P3) are synthesized in high yields from tetraphenylethene building blocks by Suzuki, Witting, and Sonogashira coupling reactions. All the compounds are soluble and show high thermal stability, losing little of their weights when they are heated to 290–528 °C under nitrogen or 288–436 °C in air. UV and CV analyses as well as theoretical calculations demonstrate that the conjugation of the molecules and polymers is in the order of 2 > 3 > 1, P2 > P3> P1, and P1–P3 > 1–3. Whereas both 1–3 and P1–P3 are weakly emissive in solutions, they become strong emitters when aggregated in poor solvent with fluorescence quantum yields up to 90%, demonstrative of a phenomenon of aggregation-induced emission. Both molecules and polymers exhibit the feature of aggregation-enhanced two-photon excited fluorescence. Large two-photon absorption cross sections (up to ∼900 GM) are observed in the nanoaggregates of P1–P3, whose values can be further enhanced by using an excitation source of shorter wavelengths. Thin solid films of P2 and P3 show high refractive indices (RI = 1.7649 − 1.6873) in a wide wavelength region of 400–1700 nm, high modified Abbé numbers, and low optical dispersions. The light emission of P1 can be quenched efficiently by picric acid with a large quenching constant. Materials with such attributes are anticipated to find an array of high-tech applications.Acknowledgements
This work was partially supported by the Research Project Competition of HKUST (RPC11SC09 and RPC10SC13), the Research Grants Council of Hong Kong (604711, 603509, HKUST2/CRF/10), the University Grants Committee of Hong Kong (AoE/P-03/08), and the National Science Foundation of China (20634020 and 20974028). B. Z. T. is thankful for the support from Cao Guangbiao Foundation of Zhejiang University. Gabriel Ramos-Ortiz is thankful for the CONACYT grant (grant J49512F).References
- (a) G. S. He, T. S. Tan, Q. D. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS; (b) S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777 CrossRef CAS; (c) D. A. Parthenopoulos and P. E. Rentzepis, Science, 1989, 245, 843 CAS; (d) S. Kawata, H. B. Sun, T. Tanaka and K. Takada, Nature, 2001, 412, 697 CrossRef CAS; (e) B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I. Y. S. Lee, D. McCord-Maughon, J. Qin, H. Rockel, M. Rumi, X. L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51 CrossRef CAS; (f) H. B. Sun, T. Kawakami, Y. Xu, J. Y. Ye, S. Matuso, H. Misawa, M. Miwa and R. Kaneko, Opt. Lett., 2000, 25, 1110 CrossRef CAS; (g) W. H. Teh, U. During, G. Salis, R. Harbers, U. Drechsler, R. F. Mahrt, C. G. Smith and H. J. Guntherodt, Appl. Phys. Lett., 2004, 84, 4095 CrossRef CAS; (h) A. S. Dvornikov, E. P. Walker and P. M. Rentzepis, J. Phys. Chem. A, 2009, 113, 13633 CrossRef CAS.
- (a) A. C. Walker, A. K. Kar, W. Ji, U. Keller and S. D. Smith, Appl. Phys. Lett., 1986, 48, 683 CrossRef CAS; (b) E. W. Van Stryland, Y. Y. Wu, D. J. Hagan, M. J. Soileau and K. Mansour, J. Opt. Soc. Am. B, 1988, 5, 1980 CrossRef CAS; (c) P. A. Bouit, G. Wetzel, G. Berginc, B. Loiseaux, L. Toupet, P. Feneyrou, Y. Bretonniére, K. Kamada, O. Maury and C. Andraud, Chem. Mater., 2007, 19, 5325 CrossRef CAS; (d) Y. Morel, A. Irimia, P. Najechalski, Y. Kervella, O. Stephan, P. L. Baldeck and C. Andraud, J. Chem. Phys., 2001, 114, 5391 CrossRef CAS; (e) N. Venkatram, D. N. Rao and M. A. Akundi, Opt. Express, 2005, 13, 867 CrossRef CAS.
- (a) W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73 CAS; (b) D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434 CrossRef CAS; (c) D. J. Bharali, D. W. Lucey, H. Jayakumar, H. E. Pudavar and P. N. Prasad, J. Am. Chem. Soc., 2005, 127, 11364 CrossRef CAS.
- (a) J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795 CrossRef CAS; (b) H. Stiel, K. Teuchner, A. Paul, W. Freyer and D. Leupold, J. Photochem. Photobiol., A, 1994, 80, 289 CrossRef CAS.
- A. Mukherjee, Appl. Phys. Lett., 1993, 62, 3423 CrossRef CAS.
- B. A. Reinhardt, L. L. Brott, S. J. Clarson, A. G. Dillard, J. C. Bhatt, R. Kannan, L. X. Yuan, G. S. He and P. N. Prasad, Chem. Mater., 1998, 10, 1863 CrossRef CAS.
- (a) H. M. Kim and B. R. Cho, Chem. Commun., 2009, 153 Search PubMed; (b) M. Albota, D. Beljonne, J. L. Bredas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Rockel, M. Rumi, G. Subramaniam, W. W. Webb, X. L. Wu and C. Xu, Science, 1998, 281, 1653 CrossRef CAS.
- (a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, Chichester, UK 1970 Search PubMed; (b) W. S. Li and T. Aida, Chem. Rev., 2009, 109, 6047 CrossRef CAS; (c) B. Gu, W. Ji, X. Q. Huang, P. S. Patil and S. M. Dharmaprakash, J. Appl. Phys., 2009, 106, 33511 CrossRef.
- (a) A. Mishra, C. Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141 CrossRef CAS; (b) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS.
- (a) L. Li, Y. Tian, J. Yang, P. Sun, L. Kong, J. Wu, H. Zhou, S. Zhang, B. Jin, X. Tao and M. Jiang, Chem. Commun., 2010, 46, 1673 RSC; (b) S. Kim, Q. D. Zheng, G. S. He, D. J. Bharali, H. E. Pudavar, A. Baev and P. N. Prasad, Adv. Funct. Mater., 2006, 16, 2317 CrossRef CAS.
- N. Kato, Y. Katayama, H. Semba and K. Limura, Jpn. J. Appl. Phys., 2010, 49, 031601 CrossRef.
- (a) J. Luo, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (c) D. Oelkrug, A. Tompert, J. Gierschner, H. J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902 CrossRef CAS; (d) D. Oelkrug, A. Tompert, H. J. Egelhaaf, M. Hanack, E. Steinhuber, m. Hohloch, H. Meier and U. Stalmach, Synth. Met., 1996, 83, 231 CrossRef CAS; (e) M. K. Chaudhuri and S. C. Ganguly, J. Phys. C: Solid State Phys., 1969, 2, 1560 CAS; (f) T. Ikeyama and T. Azumi, J. Phys. Chem., 1985, 89, 5332 CrossRef CAS.
- (a) H. H. Fang, Q. D. Chen, J. Yang, H. Xia, B. R. Gao, J. Feng, Y. G. Ma and H. B. Sun, J. Phys. Chem. C, 2010, 114, 11958 CrossRef CAS; (b) Y. H. Jiang, Y. C. Wang, J. L. Hua, J. Tang, B. Li, S. X. Qian and H. Tian, Chem. Commun., 2010, 46, 4689 RSC.
- E. Collini, C. Ferrante and R. Bozio, J. Phys. Chem. B, 2005, 109, 2 CrossRef CAS.
- (a) S. Kim, T. Y. Ohulchanskyy, H. E. Pudavar, R. K. Pandey and P. N. Prasad, J. Am. Chem. Soc., 2007, 129, 2669 CrossRef CAS; (b) S. Kim, H. E. Pudavar, A. Bonoiu and P. N. Prasad, Adv. Mater., 2007, 19, 3791 CrossRef CAS; (c) S. B. Noh, R. H. Kim, W. J. Kim, S. Kim, K. S. Lee, N. S. Cho, H. K. Shim, H. E. Pudavar and P. N. Prasad, J. Mater. Chem., 2010, 20, 7422 RSC.
- M. Banerjee, S. J. Emond, S. V. Lindeman and R. Rathore, J. Org. Chem., 2007, 72, 8054 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- (a) A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef; (b) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- (a) R. Ahlrichs, M. Baer, M. Haeser, H. Horn and C. Koelmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS; (b) O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346 CrossRef CAS; (c) F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433 CrossRef CAS.
- (a) P. Kundu, K. R. J. Thomas, J. T. Lin, Y. T. Tao and C. H. Chien, Adv. Funct. Mater., 2003, 13, 445 CrossRef CAS; (b) J. Y. Shen, C. Y. Lee, T. H. Huang, J. T. Lin, Y. T. Tao, C. H. Chien and C. Tsai, J. Mater. Chem., 2005, 15, 2455 RSC.
- (a) J. S. Park, R. H. Kim, N. S. Cho, H. K. Shim and K. S. Lee, J. Nanosci. Nanotechnol., 2008, 8, 4793 CrossRef CAS; (b) Z. J. Liu, P. Shao, Z. L. Huang, B. Liu, T. Chen and J. G. Qin, Chem. Commun., 2008, 2260 RSC.
- (a) C. J. Yang and S. A. Jenekhe, Chem. Mater., 1995, 7, 1276 CrossRef CAS; (b) C. J. Yang and S. A. Jenekhe, Chem. Mater., 1994, 6, 196 CrossRef CAS.
- (a) J. C. Sanchez, A. G. Dipasquale, A. L. Rheingold and W. C. Trogler, Chem. Mater., 2007, 19, 6459 CrossRef CAS; (b) H. Sohn, M. J. Sailor, D. Magde and W. C. Trogler, J. Am. Chem. Soc., 2003, 125, 3821 CrossRef CAS.
- (a) S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef; (b) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS; (c) J. Liu, Y. Zhong, P. Lu, Y. Hong, J. W. Y. Lam, F. Mahtab, Y. Yu, K. S. Wong and B. Z. Tang, Polym. Chem., 2010, 1, 426 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterizations; high resolution mass spectra of 1–3; photographs of 2, 3, P2, and P3 in THF and THF/water mixtures taken under UV illumination and their associated emission spectra, molecular orbital plots of HOMO and LUMO energy levels of cis1–3; emission spectra of P1 in THF and THF/water mixtures (1/1 v/v) containing different amounts of picric acid.. See DOI: 10.1039/c1jm13556b |
| This journal is © The Royal Society of Chemistry 2012 |