Green chemistry in analytical atomic spectrometry: a review
C.
Bendicho
*a,
I.
Lavilla
a,
F.
Pena-Pereira
ab and
V.
Romero
a
aDepartamento de Química Analítica y Alimentaria, Área de Química Analítica, Facultad de Química, Universidad de Vigo, Campus As Lagoas-Marcosende s/n, 36310 Vigo, Spain. E-mail: bendicho@uvigo.es; Fax: +34-986-812556; Tel: +34-986-812281
bCESAM & Department of Chemistry, University of Aveiro, 3810-193 Aveiro, Portugal
First published on 4th September 2012
Abstract
As a result of the greater consciousness within the analytical community on the impact of chemicals on human health and environment, green issues are increasingly taken into account when choosing an established analytical method or developing a new one. Apart from typical analytical characteristics (e.g., sensitivity, limit of detection, repeatability, etc.), other features such as the amount of sample/reagents, operation time, use of energy-effective apparatus, waste production, etc. should be highlighted in order to meet the principles of Green Chemistry. Although conventional approaches for trace element analysis by atomic spectrometry usually involve well-established sample pre-treatments based on ‘wet chemistry’, and high consumption of gases, reagents, etc. is inherent to many techniques in this group, there are still many avenues where green issues can be implemented. For greening atomic spectrometry, green chemistry principles should be applied to every step of the analytical process, i.e., from sampling and sample pre-treatment to data processing. In this review, main pathways for greening atomic spectrometry such as downsizing of instrumentation, use of portable instruments, solid sampling, application of clean energies (ultrasound, microwaves, etc.) for sample pre-treatment, development of on-site, on-line and at-line approaches vs. typical off-line methods, application of modern extraction techniques (e.g., solid- and liquid-phase microextraction), green solvents and derivatization agents and use of chemometric tools for method optimization, signal processing, etc. are discussed in a critical way.
1. Introduction
In recent years, an increased interest has arisen in the analytical community for the implementation of the principles of Green Chemistry. Several of the twelve principles established by Anastas and Warner1 more than 10 years ago are directly connected with Analytical Chemistry, such as prevention of wastes, safer solvents and reagents, energy efficiency, renewability, reducing derivatives, real-time analysis and accident prevention through implementation of safer chemistry. In the last two years, the subject has deserved attention in several books2,3 and reviews,4–15 and the concept ‘Green Analytical Chemistry (GAC)’ has been increasingly employed.Several trends have driven for long the research on new detection methods such as miniaturization, automation, simplification and acceleration, which in turn are related to many of those principles. Implementation of those trends in the analytical methods has usually provided not only enhanced analytical characteristics, but also significantly improved greenness profile.
Although some workers have addressed the greening of analytical techniques such as chromatography8,14 or molecular spectroscopy12 and mainly oriented toward the determination of organic analytes,13 to the best of our knowledge, no review or monograph has been specifically focused on the implementation of GAC principles in analytical atomic spectrometry.
At present, there is a growing interest in developing green methods using atomic spectrometric techniques. Fig. 1 shows the evolution of the publications devoted to green analytical chemistry in atomic spectrometry since 2000 following the subjects: ‘green’, ‘greener’, ‘clean’, ‘cleaner’ and ‘environmentally friendly’ atomic spectrometry. Fig. 2 shows the corresponding literature sources.
 | ||
| Fig. 1 Evolution of the publications devoted to green analytical chemistry in atomic spectrometry since 2000 using the subjects ‘green’, ‘greener’, ‘clean’, ‘cleaner’ and ‘environmentally friendly’ atomic spectrometry (source: ISI Web of knowledge (Web of Science) – Thomson Reuters). | ||
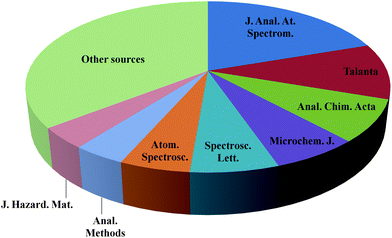 | ||
| Fig. 2 Evaluation of the publications devoted to green analytical chemistry in atomic spectrometry as a function of the corresponding literature sources since 2000 using the subjects ‘green’, ‘greener’, ‘clean’, ‘cleaner’ and ‘environmentally friendly’ atomic spectrometry (source: ISI Web of knowledge (Web of Science) – Thomson Reuters). | ||
The implementation of GAC principles in the analytical atomic spectrometry field demands for a close knowledge of the ways for greening every step of the analytical procedure, including sampling, preservation, sample pre-treatment, measurement and data processing. In order to establish a metric of the greenness related to any analytical protocol, issues such as the type of solvent, apparatus and method should be focused. Some attempts have already been made to assign a greening profile, e.g., National Environment Methods Index (NEMI) database,16 according to the properties of reagents and wastes generated. Recently, Namieśnik's group has proposed a novel metric approach so that new or modified analytical methods can be compared in respect to greenness.17 Ideally, a green method in atomic spectrometry should involve small reagent/sample consumption, energy-efficient apparatus, safe operations, short total times for analysis and avoid the use of toxic/hazardous reagents.
An assessment of different strategies carried out in every stage of the analytical process according to their ‘greenness profile’ for trace element analysis and speciation by atomic spectrometry is provided in Table 1.
| Greenness profile | Preservation | Sample dissolution | Derivatization | Separation | Measurement | Data processing |
|---|---|---|---|---|---|---|
| Low | Addition of chemicals for preservation | Wet digestion with mineral acids | Harmful, unstable, toxic derivatization agents | Solvent extraction using large volumes of toxic organic solvents (e.g., benzene, CHCl3, etc.) | Conventional application of atomic spectrometric techniques such as FAAS, ICP-OES, etc. | Univariate approach for method optimization |
| Open vessel digestion | Organic solvents and multistage procedures for derivatization (e.g., Grignard method for organometals, Westöö method for Hg, etc.) | |||||
| Conductive heating (e.g. hotplate) | ||||||
| Intermediate | — | Energy-efficient treatments (MW, US, UV radiation, etc.) | Derivatization in aqueous phase (e.g., ethylation for speciation of organometals by hyphenated techniques) | Miniaturized extraction techniques (e.g., SPE, LPME, SBSE, SPME) | Application of sensitive techniques instead of using preconcentration with classical methods (e.g. use ETAAS instead of FAAS combined with solvent extraction) | — |
| Small volume digestion | Use of energy-efficient methods (focused MWs, ultrasound, etc.) for extraction of species | Membrane separations | ||||
| Soft extractions (e.g. UAE instead of complete dissolution) | Surfactants (e.g., cloud point extraction) | |||||
| Advanced oxidation (e.g., UV/H2O2, UV–US, photocatalysis, etc. instead of chemical oxidants) | New sorbents based on nanomaterials | Efficient sample introduction systems instead of conventional nebulizers | ||||
| Modern solid–liquid extraction techniques (SFE, ASE, etc.) | Automated on-line systems allowing sample pre-treatment, separations, etc. (FIA, SIA, multicommutation) | Non-chromatographic approaches for speciation analysis of samples with a few species | ||||
| High | On site analysis without preservation | Solid sampling: direct solid sampling and slurry sampling (SS-ETAAS, ETV-ICP-OES, ETV-ICP-MS, GD-ICP-OES, TXRF, LIBS, etc.) | Photochemical vapour generation | Solventless separation techniques (e.g., SPME with thermal desorption) | Miniatomizers with low consumption of gases | Multivariate approaches for method optimization (e.g., factorial design) |
| Electrochemical vapour generation | Miniaturized separations (e.g., microchips) | ‘In-atomizer’ trapping techniques instead of preconcentration by chemical procedures | Chemometric tools for treatment of large amount of data in order to extract hidden information | |||
| Ultrasound-promoted cold vapour generation | Green solvents (e.g., ionic liquids) | Miniaturized flow systems (lab-on-valve, lab-on-a-chip) coupled to atomic detectors | ||||
| Green interfaces between HPLC and ICP-MS, AFS, etc. for speciation based on nanomaterials (e.g. UV/nano-TiO2) | Solventless methods for speciation (purge-and-trap, cryotrapping, etc.) | Screening analysis using portable instruments (on-site) | Multivariate calibration approaches | |||
| Nanoflow HPLC for hyphenated techniques in speciation analysis |
Undoubtedly, the maximum greening profile is achieved with methods carried out on-site using portable instrumentation.2,3 In this case, neither preservation nor pre-treatment procedures are required and transport to the lab is also circumvented. Unfortunately, unlike other analytical techniques (e.g., photometry, electroanalysis), the instrumentation available for atomic spectrometry is difficult to adapt for field analysis without any sample pre-treatment, although some efforts have been made in this direction (e.g., portable X-ray fluorescence spectrometers, portable laser-induced breakdown spectrometers, miniaturized atomizers for atomic absorption spectrometry, etc.). In the present state of the art, portable instruments based on atomic spectrometry principles are, in general, far from fulfilling the outstanding analytical performance of conventional instruments available in labs.
The solid state is perhaps the one that requires the most stringent sample pre-treatment for determination of trace elements by the wide-spread atomic spectrometric techniques. Therefore, one of the most realistic options to meet GAC requirements is direct analysis with little or no sample pre-treatment. In this way, solid analysis including direct solid introduction and slurry sampling into the atomization system has been known for long as an efficient way to overcome wet chemistry that includes non-green operations, i.e., use of corrosive mineral acids, potential risks inherent to the application of high pressures and heating, high energy consumption, etc. When no direct analysis is feasible, an enhanced greening profile can be reached using activation of sample pre-treatment by efficient energies such as microwaves, ultrasound, UV radiation, etc.11,15 These processes generally involve less concentrated reagents and safer operation conditions.
As an intermediate situation, on-line and at-line analyses represent a jump toward GAC concepts in atomic spectrometry, involving automated or semi-automated operation and generally lower consumption of reagents. On the opposite side, off-line methods, which are typically recommended in many official methods of analysis, do not fulfil most GAC requirements.
Examples where pre-treatment is maintained but a drastic decrease in the amount of reagents occurs are the group of modern miniaturized separation techniques. Preconcentration, matrix removal, derivatization and other typical processes required for trace element analysis and speciation can be easily simplified and integrated by resorting to solid and liquid-phase microextraction approaches (e.g., Ref. 9).
Fig. 3 shows a variety of green strategies resulting from the passage of atomic spectrometry through the optics of green chemistry. According to this, pathways toward the achievement of greener atomic spectrometry methods would include the following.
 | ||
| Fig. 3 Relevant areas of improvement for increasing the greenness of atomic spectrometry after filtering through the standpoint of green chemistry. | ||
(a) Development of on-site, on-line and at-line methods in contrast to traditional off-line methods.
(b) Decreasing the use of reagents, sample consumption (use of microsamples) and derivatizing agents, or even better, to apply direct analysis without reagents.
(c) Replacing traditional separation methods (e.g., liquid–liquid extraction, LLE) by other methods involving miniaturized approaches (e.g., solid- and liquid-phase microextraction), with the subsequent reduction of solvents.
(d) Use of green solvents and derivatization reagents (e.g., ionic liquids, ILs) in the analytical methodology.
(e) Removal or simplification of the sample pre-treatment stages, using energy-efficient procedures (e.g., application of ultrasound, microwaves, UV light in pre-treatment operations).
(f) Extraction of the maximum information from analytical data using chemometric tools that facilitate calibration, method optimization, signal acquisition, etc.
(g) Development of automated analytical methods using flow-injection, sequential injection or multicommutation approaches.
(h) Development of automated and miniaturized methods based on lab-on-valve (LOV) approaches and micro-total analytical systems (μ-TASs).
(i) Design of new instrumentation for greening atomic spectrometry.
The aim of this review is to show these possibilities so that users of atomic spectroscopy techniques can acquire criteria in order to implement green chemistry concepts in labs devoted to trace element analysis and speciation.
2. Green sample preparation in atomic spectrometry
Sample preparation is undoubtedly considered as an essential stage in the analytical process. The isolation and preconcentration of target analytes, as well as the performance of a clean-up step when dealing with complex matrices, are the main objectives pursued at this step of the analytical process. In the last few years, many efforts have been taken towards the development of environmentally friendly sample preparation approaches in analytical chemistry. In this section we provide an overview of those sample treatment approaches and related green issues that are directly relevant to the development of sustainable analytical procedures for total element analysis and speciation.2.1. Green extraction techniques: different variants of solid and liquid phase extraction
Sample preparation techniques such as solid-phase extraction (SPE), solid-phase microextraction (SPME) and liquid-phase microextraction (LPME) have represented a step forward in the development of versatile treatment techniques and, in general, they also fulfil the requirements so as to be considered as green sample preparation techniques.The development of the above mentioned sample preparation approaches has allowed the downscaling of the sample and organic solvent volumes needed to perform a single analysis, thus giving rise to a significant reduction in the amounts of residues typically generated.
The SPE technique allows the preconcentration of analytes in short extraction times for total element and speciation analysis.18 It should be highlighted that in spite of high reduction in solvent consumption when compared with LLE, the amounts of solvent needed to perform a single SPE are generally in the range of 5–15 mL.19 In the last few years, the performance of SPE systems has been improved with the introduction of novel materials that show higher adsorption capability, selectivity and/or lower cost of preparation, including ion-imprinted polymers, biosorbents and nano-sized particles.20 The application of on-line SPE procedures with green sorbents has also been reported. Chen et al.21 proposed the employment of a hydrophilic ionic liquid (1-chlorovinyl-3-methylimidazolium chloride, NmimCl) immobilized onto a polyvinyl chloride (PVC) substrate as a green SPE sorbent to carry out speciation analysis. In this work, Cr(VI) was determined by retention via anion exchange and electrostatic interaction with a mini-column containing PVC–NmimCl particles. Cr(III) was pre-eliminated by using a strong acidic styrene type cation exchange resin mini-column, thus allowing the speciation analysis of Cr by electrothermal atomic absorption spectrometry (ETAAS) and inductively coupled plasma-mass spectrometry (ICP-MS). The on-line SPE system improved the sample throughput and provided an enhancement factor of 23.4 when using 2 mL sample volumes. Tian et al.22 employed mungbean-coat as a biodegradable adsorbent for on line-SPE. Cadmium was retained and enriched in the mini-column, presumably via coordinative interactions with the carboxylic acid groups of the bean-coat. The retained cadmium is then eluted with 70 μL of 1 mol L−1 HNO3 and determined by ETAAS.
The inception of miniaturized sample preparation techniques such as SPME23–25 and LPME9,26,27 has represented a breakthrough in GAC. In fact, both SPME and LPME are (virtually) solventless techniques that allow the achievement of large enrichment factors mainly as a result of their highly reduced extractant-to-sample volume ratio. Hence, the SPME process can be completely carried out without the employment of organic solvents when thermal desorption is performed after the microextraction process, while LPME techniques generally use an almost negligible volume of extractant phase (1–100 μL). Several microextraction modes are nowadays feasible for the extraction of a given analyte as a function of its physicochemical properties. However, the different microextraction modes can differ significantly in terms of greenness depending on the volume and properties of the extractant phase, additional reagents needed to perform the extraction process, the number of steps involved and, in the case of SPME, the desorption conditions. A derivatization step is generally required to efficiently extract and preconcentrate target analytes by SPME or LPME when dealing with total element and speciation analysis. Thus, both neutral complex formation and in situ chemical vapour generation (CVG) are mainly employed with this aim.28
Another sample preparation technique that has been employed with the aim of total element determination and speciation analysis is stir bar sorptive extraction (SBSE).29–31 Like SPME, this technique is based on the extraction of analytes onto a polymer-based coating. As a larger mass of polymer is involved as compared to SPME, enhanced extraction efficiency is achieved. Like SPME, SBSE is characterized as a fully solventless sample preparation technique when thermal desorption is selected. A special thermal desorption unit is generally used when SBSE is combined with gas chromatography (GC) due to the non-fitting geometry of the coated stir bar and the injection port, respectively.32
Fig. 4 shows a schematic representation of the main miniaturized extraction techniques discussed above.
 | ||
| Fig. 4 Schematic representation of relevant miniaturized sample preparation techniques. | ||
Even though the miniaturization of a conventional method generally involves positive aspects in terms of solvent consumption and waste reduction, we should stress that the employment of a miniaturized sample preparation technique is not enough to classify an analytical method as green. In fact, several examples can be found in the literature where reagents and/or organic solvents displaying certain toxicity are used with microextraction approaches, as can be shown in Section 2.4.
2.2. Green methods for treatment of solid samples
Conventional sample preparation methods for decomposition of solid samples for analysis by atomic spectrometric techniques involve the addition of high amounts of additional reagents and oxidizing acids, as well as high temperatures for matrix decomposition. These conventional pre-treatment methods are not free from drawbacks, including risk of contamination, analyte losses, extended decomposition times and large energy consumption.The use of microwave (MW) and ultrasound (US) energies, as well as the use of organic solvents, carbon dioxide and water at both subcritical and supercritical conditions has led to the development of a plethora of sample preparation approaches for solid samples that meet the greenness criteria to a lesser or greater extent.
MW energy is commonly used for both digestion and extraction procedures. Unlike conductive heating systems typically used, MW-assisted digestion (MAD) involves homogeneous heating of the sample by dipole rotation and ionic conduction, thus improving the matrix decomposition process in terms of time and energy consumption, lower volume of acids needed, and lower blanks obtained.19
The application of small volume polytetrafluoroethylene (PTFE) closed vials for greener MAD of breast biopsies has been recently proposed by Millos et al.33 The use of three vials of low capacity (6 mL) inserted into a commonly used MW digestion vessel allowed the simultaneous matrix decomposition of small biological sample sizes (20–30 mg) prior to multielemental determination by ICP-MS using reduced volumes (0.3 mL per sample) of HNO3.
MW-assisted extraction (MAE) exploits the increased solvent diffusion of an extractant heated by MW energy to extract and solubilize target analytes present in a sample matrix. Both pressurized (PMAE) and focused (FMAE) MAE can be performed, FMAE being recognized as highly efficient for the extraction of organometallic compounds.19 As a proof of concept, FMAE has been exploited for the solid–liquid extraction of organometallic species of Hg and Sn in solid environmental samples.34 The use of disposable MW glass vessels avoided possible contamination risks and allowed the extraction of a batch of 10 samples in less than 1 h using acetic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (3:1) or diluted tetramethylammonium hydroxide as the extractant.
:methanol (3:1) or diluted tetramethylammonium hydroxide as the extractant.
As pointed out above, US energy is also employed for digestion and extraction purposes. Acoustic cavitation produced by US irradiation provides unique conditions that are exploited for improving the greenness profile of analytical methodologies, including reduced operation times, lower energy requirements, reduced amounts of solvents and lower risk of contamination and/or analyte losses.15 A variety of US-based systems, namely, US bath, US probe and cup horns/sonoreactors, are nowadays commercially available.15,35 When applied to solid matrix decomposition, US energy avoids the use of drastic temperature and pressure conditions, even though the use of concentrated acids is still mandatory. On-line MAD can be used to speed up matrix decomposition of liquid extracts and slurries, hence avoiding the high cooling times required before the digestion vessels can be opened. For instance, Gómez-Ariza et al.36 performed chiral speciation of selenomethionine in pre-treated breast and formula milk samples by coupling high performance liquid chromatography (HPLC)–hydride generation (HG)–atomic fluorescence spectrometry (AFS) with on-line MAD.
The use of US energy in combination with enzymes (hydrolases) has been recently proposed for the rapid decomposition of solid samples under mild conditions. US-assisted enzymatic hydrolysis has been applied for total trace element analysis and speciation, even though further work is needed to carefully control the effect of US irradiation on the stability and activity of hydrolases. As an example, Moreda-Piñeiro et al.37 have applied ultrasound assisted-enzymatic hydrolysis of seafood samples for As speciation. The use of pepsin as a proteolytic enzyme together with ultrasound irradiation allowed achieving the enzymatic process in short times.
US irradiation can also contribute to accelerate the solid–liquid extraction of elements from solid matrices. When compared with the related sample preparation techniques mentioned above, the extraction time is commonly reduced with US-assisted extraction (UAE), and moreover treatments are performed at atmospheric pressure and room temperature. Diluted acids and soft extractants under mild conditions are employed in UAE for total trace element analysis and speciation, respectively. Costas et al.38 employed UAE as a sample pre-treatment for extraction of rare earth elements in seafood tissues prior to ICP-MS analysis. UAE involved the use of diluted acids and reduced extraction times, yielding much lower volumes of acidic waste than MAD. A clean-up of the extracts with a C18 cartridge was mandatory to remove organic matter prior to ICP-MS analysis.
The use of fluids in the supercritical state has also been considered as an efficient sample preparation technique for the extraction of target compounds from complex matrices. The unique properties of supercritical fluids, namely, much lower viscosity and diffusion coefficients than in the liquid state, cause an enhancement of mass transfer and solubilisation processes. Thus, supercritical fluid extraction (SFE) has been used in several areas, CO2 being by far the most employed supercritical fluid due to its non-toxicity, non-flammability, economy and availability. Even though SFE involves short extraction times and minimal organic solvent consumption, as well as suitability for automation, it has been superseded by alternative sample preparation techniques, mainly due to the poor robustness of early commercial SFE systems, the lack of standard extraction procedures and the requirement of clean-up procedures after SFE of certain samples.39 As for the extraction of metal species and organometallic compounds by SFE, the use of an organic modifier of supercritical CO2 is generally mandatory due to its limited capability of leaching polar or ionic analytes. In addition, ion-pair formation and complexation are usually performed in SFE to extract charged species.40
The use of fluids at high temperature and pressure in such a way that they are kept at subcritical conditions has also been reported for extraction of analytes from solid samples. Pressurized liquid extraction (PLE) makes use of both organic solvents and water as extractants. The physicochemical properties of water are highly modified when the temperature is increased between its boiling point and critical temperature, the relative permeability being decreased on increasing the temperature. Extraction of metals by pressurized hot water extraction is commonly performed by using diluted acids as modifiers.41 In general, PLE involves the use of relatively low volumes of extractants (10–40 mL), causing the extraction process to occur in reduced times. However, the obtained extracts using PLE usually require a clean-up to remove co-extracted compounds, which extends the sample pre-treatment time before the analysis.42
2.3. Membrane and surfactant-based sample preparation
Membranes act as selective barriers that allow the contact of the sample solution with an acceptor phase. They allow the enrichment of target analytes and sample clean-up using lower volumes of organic solvents than conventional sample preparation methods, such as LLE or SPE, being also suitable for miniaturization. Thus, membrane-based extraction techniques are being considered as environmentally friendly sample preparation approaches.6,43 Accordingly, membranes can contribute to greening analytical procedures. However, the green profile of membrane-based methods will depend to a large extent on the reagents and solvents used.Membranes have been used in a variety of configurations for sample pre-treatment, including hollow fibers, flat-sheet membranes and membrane bags, and employed for trace element analysis and speciation.44,45
A flat sheet supported liquid membrane (SLM) was reported for Hg separation using polyvinylidenefluoride as the supporting material, trioctylamine as the carrier, coconut oil as the diluent, and a diluted NaOH aqueous solution as the stripping phase.46 Peng et al.47 employed a hollow fiber (HF) SLM extraction system in combination with ETAAS for determining Cd in diluted seawater samples. A liquid membrane was prepared by filling the pores of a polypropylene HF with a 1-octanol solution containing a mixture of dithizone (used as a carrier) and oleic acid, using a 0.05 mol L−1 HNO3 solution (20 μL) as the stripping solution.
Miniaturized sample preparation techniques have been developed with the use of polymeric HFs. Thus, HF-LPME has been employed for preconcentration of metals and organometallic compounds. For instance, HF-LPME has been used in combination with electrothermal vaporization (ETV)-ICP-MS for the determination of ultratrace levels of Cu, Zn, Pd, Cd, Hg, Pb and Bi in environmental and biological samples.48 HFs caused an enhancement of the extraction efficiency, providing an appropriate sample clean-up and improved stability of the extractant phase in comparison with related miniaturized LPME approaches such as single-drop microextraction.
Surfactants are characterized as being non-volatile, non-flammable and, in general, as showing negligible toxicity. Several analytical separation processes have been improved in terms of greenness by using surfactants as extraction media, cloud point extraction (CPE) being the most relevant and popular.49 CPE is based on the formation of a turbid solution when a sample containing a surfactant is heated over the cloud-point temperature. Above this temperature, which depends on the type of surfactant used and its concentration, two immiscible phases are formed, the surfactant-rich phase being the one capable of extracting a variety of hydrophobic compounds. Thus, surfactants can be used as green extractant phases in CPE for enriching target analytes prior to their determination by atomic spectrometric techniques, thereby avoiding the use of volatile organic solvents commonly used in LLE. Several methods for total element analysis and speciation involving CPE can be found in the literature.49
On line-CPE avoids the equilibration, cooling and centrifugation procedures typically needed in batch mode, hence providing an increase in sample throughput. Furthermore, the elution step can be easily performed. Ortega et al.50 employed a flow injection (FI)-CPE method in combination with inductively coupled plasma-optical emission spectrometry (ICP-OES) for the determination of total Gd in previously digested urine samples. Gd(III) was complexed with 2-(5-bromo-2-pyridylazo)-5-diethylaminophenol and extracted with the non-ionic surfactant poly(ethylene glycol) mono-nonylphenyl ether. A homemade collection column was used to retain the surfactant rich phase containing the analyte, which was finally eluted with 4 mol L−1 HNO3. Multielemental analysis has also been performed by combination of on-line CPE with ICP-OES. Thus, Yamini et al.51 performed the on-line preconcentration of Cd(II), Co(II), Cr(III), Cu(II), Fe(III) and Mn(II) by complexation with 1-(2-theonyl)-3,3,3-trifluoroacetone and extraction by using a non-ionic surfactant (Triton X-114).
The use of MW irradiation in combination with surfactant-based procedures has demonstrated to be highly efficient, thus allowing an important reduction in the extraction time, incubation temperature and energy consumption as compared to conventional hot plate CPE. For instance, Simitchiev et al.52 reported a MW-assisted CPE-ICP-MS method for the determination of Rh, Pd and Pt in pharmaceuticals. In addition, Meeravali and Jiang53 determined Au and Tl in soils and water samples by MW-assisted mixed micelle CPE-ICP-MS.
Recently, surfactants have been applied for extraction of metals by means of admicelles and hemimicelles adsorbed onto the surface of metallic nanoparticles. For instance, Faraji et al.54 determined Hg(II) by FI-ICP-OES after SPE of mercury–Michler’s thioketone complex by means of sodium dodecyl sulphate (SDS)-coated magnetic nanoparticles. Furthermore, Karatapanis et al.55 employed cetylpyridinium bromide-coated Fe3O4@SiO2 nanoparticles to extract Cu(II), Ni(II), Co(II), Cd(II), Pb(II) and Mn(II) as their complexes with 8-hydroxyquinoline, prior to their determination by ETAAS. Specific features to be emphasized are the low consumption of surfactant per analysis (∼30 mg), the high enrichment factors achieved and the renewability of magnetite nanoparticles.
Emulsification has been exploited for the extraction of metals and metalloids in complex samples such as lubricating oils56 or cosmetics.57 Emulsification of oily samples with surfactants and water can be considered as a green sample preparation technique since it avoids the use of organic solvents and the destruction of the organic phase, mainly performed in the literature by acid digestion. The formation of water-in-oil emulsions has been exploited for the development of greener methodologies compatible with atomic spectrometry. Thus, Aranda et al.58 presented a methodology based on the combination of emulsion formation and cold vapour (CV)-AFS for total and inorganic Hg determination in biodiesel samples. In addition, Cassella et al.59 determined Cu, Fe, Ni and Pb by ETAAS in diesel oil samples by the formation of a water-in-oil emulsion with Triton X-114 in acidic media and subsequent breaking of the emulsion by heating.
2.4. Green solvents and reagents
Green chemistry also deals with risk reduction and pollution prevention. Risk is defined as the product of hazard and exposure.60 Exposure to a given hazardous substance may be reduced, for instance, by using miniaturized sample preparation techniques. However, exposure controls are capable of failing, then maximizing the risk when hazardous reagents and solvents are used. The removal of toxic solvents and reagents from the analytical procedures is thus a challenging task that has been faced by researchers, especially in the last two decades. In spite of being highly desirable, the full removal of certain solvents and reagents is not always achievable without significant worsening of the analytical characteristics. In these cases, the replacement of harmful solvents and reagents by greener alternatives is advisable. For instance, volatile organic solvents have been replaced by water,41 ILs,61 supramolecular solvents62 and carbon dioxide63 in a variety of sample preparation techniques for total element analysis and speciation, thus giving rise to greener analytical methodologies. However, toxic organic solvents, such as benzene64 or chlorinated solvents65 are being systematically employed in certain sample preparation techniques. In general, the selection of a given solvent for its application in atomic spectrometry is performed by comparison of certain physicochemical properties, such as solubility, polarity, density, viscosity or vapour pressure. Given the environmental, health and safety impact of organic solvents, their toxicological features should be seriously taken into consideration. Therefore, we should also consider aspects such as toxicity, flammability, explosivity, stratospheric ozone depletion and/or atmospheric ozone production, in order to also fulfil the criterion of reduced hazards.66 In addition to the impact on health and environment, the energy required to manufacture the solvent and the cumulative energy demand should also be evaluated for potentially feasible solvents.67 The use of a solvent selection guide is highly recommended for the development of green analytical methods, such as those recently proposed by GlaxoSmithKline (GSK)68 and Pfizer,69 respectively. A quick view of green and less green solvents commonly used in the industry and analytical labs is shown in Table 2. Detailed information of the relevant aspects considered to establish the greenness of organic solvents can also be found in the literature.70| Solvent | Solvent selection guide | |
|---|---|---|
| GSKa | Pfizerb | |
| a Ref. 68. b Ref. 69. c The GSK solvent selection guide establishes three types of solvents ranked from the greenest to the least green: few issues > some issues > major issues. In the same way, the Pfizer solvent selection guide establishes three types of solvents ranked from the greenest to the least green: preferred > usable > undesirable. | ||
| Water | Few issues (greenest option) | Preferred |
| Hydrocarbons | ||
| Pentane | Undesirable | |
| Hexane | Major issues | Undesirable |
| Heptane | Some issues | Usable |
| 2-Methylpentane | Major issues | |
| Isooctane | Some issues | Usable |
| Cyclohexane | Some issues | Usable |
| Methylcyclohexane | Usable | |
| Benzene | Major issues | Undesirable |
| Toluene | Some issues | Usable |
| Xylenes | Usable | |
| p-Xylene | Some issues | |
| Alcohols | ||
| Methanol | Some issues | Preferred |
| Ethanol | Some issues | Preferred |
| 1-Propanol | Some issues | Preferred |
| 2-Propanol | Some issues | Preferred |
| 1-Butanol | Few issues | Preferred |
| 2-Butanol | Few issues | |
| tert-Butanol | Some issues | Preferred |
| Ethylene glycol | Usable | |
| 2-Methoxyethanol | Major issues | |
| Halogenated solvents | ||
| Dichloromethane | Major issues | Undesirable |
| Chloroform | Major issues | Undesirable |
| Carbon tetrachloride | Major issues | Undesirable |
| 1,2-Dichloroethane | Major issues | Undesirable |
| Ketones | ||
| Acetone | Some issues | Preferred |
| Methyl ethyl ketone | Major issues | Preferred |
| Methyl isobutyl ketone | Some issues | |
| Carboxylic acids | ||
| Acetic acid | Usable | |
| Ethers | ||
| Diethyl ether | Major issues | Undesirable |
| Diisopropyl ether | Major issues | Undesirable |
| tert-Butyl methyl ether | Some issues | Usable |
| Cyclopentyl methyl ether | Some issues | |
| 1,2-Dimethoxyethane | Major issues | Undesirable |
| 1,4-Dioxane | Major issues | Undesirable |
| Tetrahydrofuran | Major issues | Usable |
| 2-Methyl tetrahydrofuran | Some issues | Usable |
| Esters | ||
| Methyl acetate | Some issues | |
| Ethyl acetate | Some issues | Preferred |
| Propyl acetate | Few issues | |
| Dimethyl acetate | Undesirable | |
| Isopropyl acetate | Few issues | Preferred |
| tert-Butyl acetate | Few issues | |
| Nitrogen-containing solvents | ||
| Acetonitrile | Major issues | Usable |
| Pyridine | Undesirable | |
| N-Methyl formamide | Major issues | |
| N-Methyl pyrrolidone | Major issues | Undesirable |
| Dimethyl formamide | Major issues | Undesirable |
| Dimethyl acetamide | Major issues | |
| Sulfur-containing solvents | ||
| Dimethyl sulfoxide | Some issues | Usable |
When reagents cannot be replaced by environmentally friendly alternatives, their minimization should be considered as a viable option. Thus, the employment of multicommutation,71,72 sequential injection,73 and lab-on-valve systems,74,75 the immobilization of reagents onto a solid substrate74 or the introduction of the necessary reagents as part of the extractant phase in microextraction techniques76 allows an important reduction of the mass of reagents needed to perform a single analysis and, therefore, a drastic reduction of costs and wastes produced.
A ‘reagent free’ photo-induced CVG method was reported by Li et al.77 for the determination of mercury in alcoholic beverages by exploiting the reducing capacity of the ethanol present in wine and liquor samples when exposed to UV irradiation.
The development of ligandless analytical procedures combined with atomic spectrometry has also been reported in the literature. Thus, formation of metal hydroxides78,79 or insoluble chlorides80 has been employed for the development of ligandless analytical methodologies by pH adjustment and addition of NaCl, respectively.
The application of unrefined natural reagents as greener and cheaper reagents has also been referenced in the literature.81 For instance, Tuzen et al.82,83 employed SPE resins containing Bacillus sphaericus and Streptococcus pyogenes for Cr and Hg speciation, respectively.
Green reagents have also been employed to increase the efficiency of certain analytical processes. Thus, ILs have been used in combination with complexing agents84,85 to achieve the enhancement in the CVG efficiency of transition and noble metals.
2.5. Green derivatization methods for total element analysis and speciation
In accordance with the 8th principle of green chemistry, unnecessary derivatization should be avoided whenever possible in order to limit the use of additional reagents that, in turn, can generate wastes.43 The improvement of analytical methods for total element analysis and speciation has also been derived from the application of greener derivatization methods. Thus, several strategies have been proposed in the literature in order to avoid conventional derivatization methodologies that involve the use of non-green reagents and/or the formation of hazardous by-products.Conventional methods, such as HG or CV generation, involve the use of toxic and expensive reagents, namely, tetrahydroborate(III), tin chloride or potassium permanganate. The replacement of CVG by powerful and greener derivatization alternatives has been a hot topic in analytical chemistry in recent years.86
Photo-CVG, based on the direct conversion of non-volatile precursors into volatile species by means of photochemical reactions, has been studied and employed to the development of greener alternatives to classical CVG for sample introduction in analytical atomic spectrometry.11,87–92 The use of low molecular weight organic acids, alcohols and aldehydes as organic precursors to assist UV reduction allows the formation of volatiles of analytical interest, being employed for the determination of a group of elements, including Ba, Fe, Co, Rh, Ni, Pd, Cu, Ag, Au, Cd, Hg, In, Sn, Pb, As, Sb, Bi, S, Se, Te and I.86
Ultrasound-promoted cold vapour generation has also been successfully employed for the conversion of Hg(II) into Hg0 in the presence of formic acid, thus avoiding the use of chemical reducing agents.93 The mechanism is based on the decomposition of formic acid by means of US irradiation and subsequent reduction of Hg(II) to Hg0 by the reducing volatiles generated. It is interesting to note that reagentless formation of Hg0 is achieved by sonication of the sample in the absence of any chemical reagent even though the conversion efficiency is reduced in comparison with the use of formic acid. This method has been applied so far to the determination of Hg in waters and ophthalmic solutions.93–96 It should be highlighted that ultrasound-promoted cold vapour generation is not free from interferences, since the conversion efficiency is affected by oxidants and complexing agents.
Hydrides can be electrochemically generated, thereby avoiding the use of sodium tetrahydroborate. Thus, electrochemical hydride generation (EC-HG) avoids the use of an expensive chemical reagent that can introduce contamination and is unstable in aqueous solution. Furthermore, the generation media for the hydride-forming elements is similar and the efficiency of HG is not affected by the oxidation state of the analyte. However, some drawbacks have been attributed to EC-HG, including limited applicability, significant interferences from concomitant species, e.g., Cu(II) being the most severe one, and the poor stability of the cathode material.86,97
Dielectric barrier discharge (DBD)-induced CVG has been recently proposed for Hg determination.98 Cold vapour generation in the DBD cell was improved by using formic acid 2% (v/v) as the reaction medium. The mechanism by which Hg0 is formed may be attributed to the decomposition of water and/or low weight organic compounds and formation of radicals and reducing agents by the DBD plasma and UV irradiation. Direct conversion (by oxidation and atomization) of thiomersal into Hg0 has been recently demonstrated by using a DBD cell, hence avoiding pre-treatment steps.99 The authors directly determined thiomersal in commercial vaccine samples by combining DBD-plasma induced vaporization with AFS without the need to use any chemical reagent.
2.6. Advanced oxidation methods
As previously discussed in Section 2.2, conventional sample preparation methods for organic matter removal prior to trace element determination involve the use of concentrated mineral acids and high temperatures. Thus, conventional methods such as acid digestion and/or mineralization of the sample by dry ashing are prone to contamination, being furthermore time- and energy-consuming. Advanced oxidation processes (AOPs) have been developed in recent years, nowadays being considered as greener sample preparation techniques in analytical chemistry. UV-photo-oxidation, ozonolysis and US irradiation, as well as the combination of these three AOPs, have been exploited for trace-element analysis and speciation.11,100 AOPs are based on the use of clean energies and/or chemicals with oxidizing properties and in situ formation of potent oxidizing radicals, such as the hydroxyl radical. Relatively low concentrations of well-known chemical oxidizing agents, such as hydrogen peroxide, potassium persulfate, or the less green potassium dichromate, are commonly added to facilitate the oxidation process. AOPs are used for oxidizing the organic matter of the sample prior to elemental determination, extraction of analytes from solids present in an aqueous medium, and for destroying organometallic compounds, thereby allowing speciation analysis.Among the benefits of AOPs, the reduced amount of chemical reagents needed can be highlighted, with the subsequent advantages in terms of reduced contamination risks, waste generation and economy, the possibility of performing the sample pre-treatment at room temperature, and the low cost of equipment. In addition, AOPs can be performed on-line.
Fernández-Costas et al.101 reported the combination of US-assisted extraction with ozonation for determination of As in biological and environmental solid samples by HG-AFS. Ozonation proved to be highly efficient in the removal of organic matter prior to the elemental analysis, avoiding foam formation and, as a consequence, flame instability.
Sonolysis has been proposed as an efficient oxidation method for the conversion of organomercurials (e.g., methyl-, phenyl-mercury) into inorganic mercury for the subsequent determination of total Hg in waters by FI-CV-AAS. Complete oxidation can be accomplished within 3 min by ultrasound irradiation, hence avoiding chemical oxidants and strong reaction conditions (i.e., high temperature and pressure).102
The combined use of focused US irradiation with ozone (sonozone) was first proposed by Bendicho's group103 as sample treatment for determining reactive arsenic toward sodium tetrahydroborate. Sonozone has been used also for oxidizing methylmercury into inorganic Hg, aimed at determining inorganic and total Hg in undiluted urine samples by FI-CV-AAS.104
A novel high-efficiency photooxidation (HEPO) reactor was introduced by Nakazato and Tao for the efficient and expeditive conversion of organoarsenic species into As(V) prior to the determination of arsenic species in human urine by liquid chromatography (LC)-HEPO-ICP-MS.105 The use of a highly UV transmitting reaction tube extended within a low-pressure Hg lamp allowing irradiation at both 185 and 254 nm was key to the rapid photooxidation of a variety of organoarsenic species. A photooxidation time of 3.5 s was in fact enough for the efficient conversion of organoarsenic species without the addition of any oxidizing agent.
The combination of AOPs with green sample preparation and derivatization procedures prior to the elemental analysis has demonstrated to be a powerful alternative for greening analytical methods. For instance, we reported the use of photooxidation in the presence of H2O2 (UV/H2O2) of thiomersal (sodium ethylmercurithiosalicylate) in ophthalmic solutions prior to the sono-induced cold vapour generation technique.96 Thus, the whole method involves the use of 0.1 mL of H2O2 and 0.35 mL of HCOOH for degradation of thiomersal and reduction/vaporization, respectively.
2.7. Thermal desorption
Desorption of the analytes once they have been preconcentrated is generally carried out by liquid or thermal desorption. Organic solvents and acidic aqueous solutions are commonly used when combining the preconcentration technique with HPLC and/or detection by atomic spectrometry. In spite of being convenient for appropriate desorption of target analytes, the use of such eluting agents is generally considered as a non-green process, especially when relatively large volumes are employed and, in consequence, large volumes of wastes are produced.Thermal desorption is considered as a greener alternative to liquid desorption. In fact, the use of a solventless sample preparation technique with thermal desorption provides the greenest way towards the development of sustainable analytical procedures. Analytes can be thermally desorbed by insertion of the SPME fiber into the injection port of a gas chromatograph.106,107 Thermal desorption can also be performed after SBSE by using a modified thermal desorption unit.32
Nevertheless, certain designs have been reported in the literature that allow the thermal desorption for direct detection by atomic spectrometric techniques. For instance, we have reported the combination of headspace (HS)-SPME with quartz furnace atomic absorption spectrometry (QFAAS) for the determination of tetraethyllead in gasoline and water samples108 and methylmercury in seafood samples by HG and chloride generation.109 A variety of volatilizators were tested, the tube-shaped design being the preferred option due to the higher sensitivity and reproducibility achieved, presumably as a result of its lower inner volume.
Dietz et al.110 employed a home-made thermal desorption unit containing a gas chromatographic stationary phase that allowed the separation in less than two minutes of at least two organoselenium compounds by coupling SPME with QFAAS and ICP-MS.
Metallic surfaces have also been employed for adsorption of target analytes and subsequent thermal desorption. For instance, Hashemi and Rahimi111 proposed the use of a gold wire for the preconcentration of mercury by amalgamation and subsequent determination by ETAAS. Mercury was desorbed at 600 °C by directly inserting the gold wire in the sample introduction hole of the ETAAS instrument. Zierhut et al.112 employed active gold collectors for the preconcentration of mercury species in natural water samples. Amalgamation allowed the adsorption of Hg0, while the adsorption of Hg2+ and MeHg+ was attributed to the catalytic activity of gold nanostructures. Mercury species were released as Hg0 from the gold collectors by thermal desorption at 550 °C, thus allowing the analysis by AFS. More recently, Romero et al.113 used a variety of Pd-based collectors (Pd wire, Pd-coated stainless steel wire and Pd-coated SiO2) for microextraction and preconcentration of mercury. Thermal desorption of mercury amalgamated onto the Pd wires was performed by insertion into a modified quartz-T cell for AAS measurement.
3. Flow analysis in atomic spectrometry
Although flow systems have been proposed in order to automate many analytical methods and improve sample throughput as well as other analytical characteristics, these systems can also be considered from the point of view of green chemistry.1 Thus, according to the first principle, reagent consumption and waste generation are usually lower than in batch procedures.114 In agreement with the fifth and the eighth principles, sample treatment can be simpler and faster, thus avoiding the use of auxiliary reagents and the generation of derivatives.115 On-line monitoring of processes is also achievable to control and prevent pollution,116 thus fulfilling the eleventh principle. In addition, according to the sixth and twelfth principles, automation allows high sample throughput with lower energy consumption and a reduction of potential risks to the analyst.Different flow systems typically used in combination with atomic spectrometry are shown in Fig. 5. As can be seen, these systems extend from the FIA introduced by Ruzicka and Hansen in 1975117 up to more recent flow-based miniaturized systems such as the lab-on-a-chip.118 The evolution of these systems shows a clear trend towards an increase in automation and miniaturization levels. Sample pre-treatment, derivatization, separation and even waste recycling or in-line waste detoxification can be carried out in current flow systems. In addition, these versatile systems are not particularly expensive.
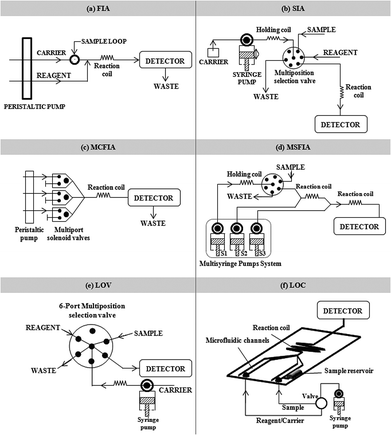 | ||
| Fig. 5 Flow injection systems (a) FIA (flow injection analysis); (b) SIA (sequential injection analysis); (c) MCFIA (multicommutated flow injection analysis); (d) MSFIA (multisyringe flow injection analysis); (e) LOV (lab-on-valve); and (f) LOC (lab-on-a-chip). | ||
In this section we consider the possibilities offered by several flow analysis systems and their role in the development of GAC, including flow and microflow-injection analysis, sequential injection analysis, multicommutation, lab-on-valve and microfluidics chips also named as lab-on-a-chip (LOC).
3.1. Flow and microflow-injection analysis
Flow injection systems, based on the injection of a liquid sample into a moving, non-segmented continuous carrier stream of a suitable liquid, can be easily coupled to continuous atomic spectrometric detectors, such as, flame atomic absorption spectrometry (FAAS), ICP-OES and ICP-MS. On the contrary, for discontinuous techniques, such as ETAAS, the coupling of continuous flow systems is limited. It is possible to achieve a semi-on-line coupling allowing different interesting approaches, i.e. on-line sample treatment.119In the initial stage of development, single-line manifolds with continuous flow were used. Although this configuration provides simple procedures and certain grade of automation, it displays an important drawback from the standpoint of GAC, i.e., reagents are consumed even when a sample is not processed, thereby contributing to increased reagent consumption and waste generation. The use of merging zones with intermittent flow systems minimized this problem, also improving automation, sensitivity and precision. This configuration has been mainly applied in HG or CV. For example, Shao et al.120 developed an intermittent flow reactor for the determination of total mercury by AFS. With this configuration, 700 μL sample, ca. 900 μL reductant (flow rate 4.0 mL min−1 for 14 s) and 1.4 mL carrier (flow rate 4.0 mL min−1 for 21 s) were consumed per replicate. Compared to continuous flow systems, this intermittent configuration reduces the consumption significantly. For instance, Vermier et al.121 proposed a procedure based on a continuous flow system coupled to AFS for mercury determination in biological samples. The volume of the sample and carrier consumed was 35 mL (flow rate 7 mL min−1 for 5 min) and that of the reductant was 15 mL (flow rate 3 mL min−1 for 5 min) per replicate.
Waste generation remains high in FI systems because they involve the use of one or more carrier streams to transport the sample and reagents to the detector. So, new designs related to waste recycling and/or treatments have arisen.122 However, they are not easy to implement, because usually the waste is highly diversified, and therefore, there are no general strategies. Thus, waste recycling has been implemented mainly in procedures involving solvent extraction and/or analyte preconcentration. Generally, phase separation is used to recover solvents in order to reuse them. For instance, a back extraction column has been proposed for recycling benzene using an automated on-line solvent extraction for the determination of As and Sn by ETAAS.123
For on-line waste treatment, different strategies and reagents are used in order to destroy or passivate the toxic species in the effluent. Metal ions must be passivized by adding a reagent in the waste stream. For instance, different trace heavy metals (Cr, Cd, Ba…) were on-line deactivated by co-precipitation with NaOH and Fe(OH)3 in a system developed for the determination of Hg in milk by AFS.71 For the detoxification of degradable wastes, different strategies have been suggested, such as, thermal degradation, oxidative detoxification, photodegradation and biodegradation.4,10
FI systems have allowed automation and acceleration of different sample treatments such as solvent extraction,124 acid digestion,120 SPE125 or SPME.126
On-line digestion results in greener procedures than off-line digestion. In general, lower volumes and concentrations of acids as well as temperatures and pressures are used. For instance, Shao et al.120 developed an on-line acid digestion for Hg determination in mainstream cigarette smoke using 1.4 mL of 4% (v/v) sulphuric acid. In addition, with on-line digestion, a reduced exposure of lab staff to potential risks is achieved (e.g., risks from explosion and emission of vapours).
The use of natural reagents and their immobilization in FI systems is an interesting alternative for the development of environmentally friendly procedures. Maquieira et al.127 proposed the on-line preconcentration of heavy metals using immobilized cyanobacteria as the biosorbent and detection by FAAS. 20 mg of cyanobacteria were immobilized on controlled pore glass retaining the activity for 3 months.
Greening FIA by the miniaturization way has been achieved with micro-FIA systems (μFIA). In atomic spectrometry, μFIA is mainly coupled with ICP-MS since it can be coupled to micronebulizers.128–131 Therefore, the consumption of sample and reagents is only a few microliters, and consequently, waste generation is very low. For instance, the microflow injection system proposed by Takasaki et al.131 for the determination of metals in some mice organs consumes only 20 μL of sample with a flow rate of 10 μL min−1 for 2 minutes.
3.2. Sequential-injection analysis
Sequential injection analysis was developed by Ruzicka et al.132 in 1990 with the aim of overcoming some disadvantages of FIA, mainly high consumption of samples and reagents and complexity of the manifolds. SIA has been considered as the second generation of FIA-based techniques.The main feature of SIA manifolds is the multiposition selection valve providing more simple and robust designs. Fluids are manipulated within the manifold by means of a bi-directional pump. The process is computer-controlled and both the sampling sequence and the volume of each aliquot are defined by software. The sample and reagents are sequentially aspirated. Then, the volume inserted into the analytical path is only that necessary for reaction development. Therefore, a drastic reduction in the consumption of reagents and sample can be achieved, of the order of 10–20 times in relation to FIA.133 For example, the consumption of reagents for CV can be reduced 10 times134 in comparison with FIA methodology based on continuous reagent addition.135
SIA is especially interesting for complete automation of sample treatment. Thus, automation of LLE using FIA has some drawbacks such as low extraction efficiency and limited range of sample-extractant volume ratios.124 These drawbacks can be overcome by SIA, because the extraction step takes place on a thin pseudo-stationary organic film formed on the surface of the tube. It allows a precise control of sample-to-extractant volume ratios in a wide range with higher extraction efficiency, lower reagent consumption and waste generation.136–138
SIA is widely used to automate SPE procedures using atomic detectors, e.g., Wang and Hansen139 designed an automatic sequential injection on-line solid phase extraction system for cadmium determination by ETAAS using a microcolumn packed with PTFE beads. The sample consumption was 3 mL per analysis and the analyte was eluted with only 50 μL of ethanol. After each analysis the microcolumn was washed and regenerated with 400 μL of a mixture of ethanol–nitric acid.
Although SIA can be considered as a greener alternative to FIA, the choice of one or the other depends on the specific analysis and features associated, such as sampling rate, automation grade, sample availability and cost and toxicity of reagents.
3.3. Multicommutation
Multicommutation was developed in 1994 by Reis et al.140 as a flow-analysis option (multicommutated flow injection analysis, MCFIA) in order to increase the versatility of flow systems, reduce reagent consumption, improve mixing and facilitate automation. Multicommutation is fully computer-controlled and utilizes multiple solenoid valves as separate switching devices to create a more flexible flow path that is able to use significantly less reagents than FIA since they are recirculated to their containers when not used.141MCFIA and FIA have recently been compared for the determination of total Se in infant formulas by HG-AAS.142 Although the sample consumption is similar in both systems, consumption of reagents using the multicommutated system is ca. five times lesser than in FIA. It also provides better detection and quantification limits with higher sampling frequency (160 samples per hour with MCFIA vs. 60 samples per hour with FIA).
Multicommutation principles have also been implemented in modified SIA (multisyringe flow injection analysis, MSFIA).133 Conventional rotatory valves used in SIA are replaced by solenoid valves. Although most applications of MSFIA rely on UV-spectrophotometry detection there are some applications for the analysis of trace metals by atomic spectrometric detection, such as AFS143 or ICP-OES.144
Like FIA and SIA techniques, multicommutation systems can incorporate on-line sample treatment. SPE is also the most popular sample treatment, e.g., Leal et al.143 used a column for the preconcentration of arsenic prior to the analysis by AFS. The work demonstrated that MSFIA decreases the consumption of reagents and sample in comparison with FIA. In addition, compared with a SIA system, MSFIA with preconcentration provides higher sample throughput (36 vs. 6 sample h−1) and a detection limit 22 times better.
In spite of the advantages of MSFIA systems, it should be noted that most of the published works do not involve treatment steps, because solenoid pumps do not work properly under backpressure, thus, when a sample treatment step is coupled to the system, the sample throughput can decrease considerably.
Following the trends towards cost-effective procedures, with lower reagent and sample consumption and waste generation as well as fast analysis, the use of multipumping flow systems (MPFSs) has been proposed.145 These systems are based on the combined use of solenoid valves and pumps operated in the form of pulses. The number of pulses defines the total injected volume, while the frequency of pulses defines the liquid flow-rate. MPFSs have been used for trace metal determination by atomic spectrometry.146–148 For example, Pérez-Sirvent et al.147 determined Hg by AFS. When the MPFS-based procedure is compared with conventional flow systems,135 a substantial improvement in sample throughput is obtained (82 samples per hour, instead of 30 samples per hour with conventional peristaltic pumps). In addition, 90% of reagent volume and 95% of sample volume are preserved, so waste generation is considerably diminished.
3.4. Lab-on-valve
Lab-on-valve was introduced by Ruzicka149 in 2000, and has been accepted as the third-generation of FIA. LOV has been developed by implementation of SIA principles in a modified system where all components are integrated on a six-port selection valve. LOV systems involve significant progress in miniaturization, automation and integration of on-line sample treatments. Hence, reagent consumption and effluent generation have been drastically reduced.LOV has been coupled with different atomic spectrometry techniques for the determination of metal species. For instance, CV in a LOV system has been applied for Hg determination by AFS.150 With this configuration the sample and reagent consumption is only 500 μL and 400 μL respectively. Also, they achieved a high sample throughput, i.e., 90 samples per hour. The low sample and reagent consumption, as well as low waste generation, makes these systems suitable for achieving greener analytical approaches.
LOV is used to automate sample treatment, e.g., SPE. There are two strategies for performing SPE, namely, packing the solid material in a micro-column and using the so-called bead-injection (BI) principle. The latter option is widely used in LOV platforms for routine analysis because it avoids the main problems associated with conventional on-line column preconcentration systems, such as high backpressure or deactivation of the surface of the solid material. In LOV-BI, a micro-column is generated in situ by aspiration of the beads. Usually, the solid material is renewed after each analytical cycle. This is the main drawback of this strategy from a standpoint of GAC. In spite of this, when the combination of LOV-BI and ETAAS is used, trace metals can be determined without analyte elution since beads are transferred into the graphite furnace, e.g., Ampan et al.151 developed a LOV-BI procedure for the determination of lead by ETAAS with Sephadex G-25 impregnated with dithizone. It must be emphasized that the lifetime of graphite tubes is short due to the residues deposited onto the graphite tubes after pyrolysis of the beads.
3.5. Lab-on-a-chip
Since the introduction of flow analysis in the 70s, the clearest trend in its evolution is without doubt miniaturization. In this sense, Manz et al.152 developed in 1990 the concept of micro-total analytical systems. This analytical concept has led to the development of the so-called lab-on-a-chip devices, allowing a high miniaturization of the analytical systems.LOC devices have been an active subject of research in the last decade.153 The interest in these devices arises from the incorporation of sample processing steps onto a small chip, thus reducing the required sample volumes, reagent consumption, waste generation and time required for the analysis. Also, these microfluidics systems can enhance the portability compared to traditional instrumentation and they can be fabricated in a cheaper way.
Microfluidic chips are constituted by micrometer-scale channels and reservoirs in a substrate material (usually silica, quartz or plastic). Although the chip material depends on the application, plastic is the most employed today because it is the cheapest. Three different propulsion mechanisms are used in LOC devices: electroosmotic flow, hydrodynamic and centrifugal.154
Chips have been mainly used for metal determination by ICP-MS.155–157 A microchip-based nanolitre sample introducing system for ICP-MS allows designing greener procedures than conventional sampling systems. For instance, Cheng et al.157 developed a microfluidic chip with hydrodynamic flow (20 μL min−1) for the determination of cisplatin in human serum. With this system the sample consumption is very low, only 1.8 nL are needed per analysis, ca. 105 times less than that of the conventional sampling systems. Also, a high sample throughput up to 112 samples per hour was achieved.
As with LOV systems, in LOC devices, different units for sample treatment can be implemented.158–160 As an example, Chen et al.159 developed a microfluidic device for magnetic nanoparticle solid phase microextraction (MSPME). It was applied to the determination of Cd, Pb and Hg by electrothermal vaporization-inductively coupled plasma mass spectrometry (ETV-ICP-MS). For the preparation of the magnetic solid phase column on the chip, 25 mg of the solid phase were aspired and introduced into the magnetic zone. The magnetic nanoparticles were used almost 12 times. Sample consumption was 500 μL per analysis which is lower than that required with conventional procedures involving ETV-ICP-MS.
The use of microfluidic chips allows a great miniaturization of the systems, in addition to a complete automation of the procedure. The main disadvantage of LOC devices is that they are systems of fixed structure, and therefore, it is necessary to design specific chips for each application, hence being less versatile than LOV systems.
4. Solid sampling by atomic spectrometry
Sample preparation typically consumes reagents, solvents, energy and is generally labour-intensive. So, it is not surprising that one of the paradigms of GAC is the elimination of this stage. The ideal analytical method from the point of view of GAC is based on direct measurements without sampling, transport, addition of reagents and waste generation. In this sense, remote and on-site measurements can be considered the best strategies to approach the philosophy of GAC.3 This is not always possible, and therefore, when a sample is analysed in the lab, replacement of traditional ‘wet chemistry’ for sample pre-treatment is considered an important goal.2 In fact, developments made in order to introduce solid samples directly into analytical instruments have been long pursued by analytical researchers.Sample decomposition by acid digestion is common when atomic spectrometric techniques are used. It implies the application of concentrated acids and high temperatures and pressures. Nowadays, microwave energy operating with closed vessels involves an efficient way of saving energy and acids. In spite of this, it continues to be difficult to meet the GAC principles when acid digestion is involved. Therefore, solid sample analysis without or with minimal sample pre-treatment should be an interesting alternative. This includes direct solid sampling and slurry sampling (a suspension of a finely powdered sample).
Atomic spectrometry techniques can resort to different systems in order to cope with solid samples, such as electrothermal vaporization, arc/spark ablation, laser ablation (LA) or glow discharge (GD) sources. It is important to lay emphasis on those atomic spectrometric techniques that allow performing green analysis in a non-destructive way due to their ability to directly analyse solid samples such as X-ray fluorescence techniques. Solid sampling using different atomic spectrometric techniques has been thoroughly reviewed, which points out the interest in this topic.161–180Table 3 shows some characteristics in order to compare the greenness of different atomic spectrometries using solid sampling.181,182
| Electrothermal vaporization/atomization | Arc/spark ablation | Laser ablation | Glow discharge | X-ray | |
|---|---|---|---|---|---|
| a Ref. 181. b Ref. 169. c Ref. 182. | |||||
| Considered techniques | ETAAS | Arc/spark-OES | LA-ICP-OES | GD-OES | μ-XRF |
| ETV-ICP-OES | Arc/spark-ICP-OES | LA-ICP-MS | GD-MS | TXRF | |
| ETV-ICP-MS | ICP-MS | LIBS | EDXRF | ||
| WDXRF | |||||
| Multielemental capability | ETAAS/No | Yes | Yes | Yes | Yes |
| ETV-ICP-OES/Yes | |||||
| ETV-ICP-MS/Yes | |||||
| Sample quantity | 0.1–1 mga | 1–10 mga | 0.25 ng–2 mgbquasi non-destructive | 10–100 mga | Non-destructive 0.1 μg–500 mgc |
| Usual sample pretreatment | Homogenization | No | No (LAM) | No | No |
| Temperature–pressure conditions | ETAAS/temperature program (up to 3000 °C) in an Ar atmosphere | High potential difference between the electrodes in an Ar atmosphere | 10–100 mJb | High potential difference in an inert gas atmosphere (usually Ar) at reduced or atmospheric pressure | Ambient temperature. Atmospheric pressure or vacuum |
| ETV-ICP/(up to 10000 °C), transport by an Ar flow |
|||||
| Portability | No | Yes | LA/ICP-OES/No | No | μ-XRF/Yes |
| LA/ICP-MS/No | TXRF/Semi-portable | ||||
| LIBS/Yes | EDXRF/Yes | ||||
| WDXRF/No | |||||
| Automation | Yesa | Yesa | Noa | Noa | Certain grade of automation (sample changer motorized) |
| Sample throughput | ETAAS/15 min per analytea | 1–2 min per samplea | 2–5 min per samplea | 10 min per samplea | 4–120 samples per hourc |
| ETV-ICP-MS/15 min per samplea | |||||
The advantages of solid sampling from the perspective of GAC over acid digestion or other sample pre-treatments (e.g., dry ashing, fusion) can be summarized as follows.
(a) It avoids the use of corrosive and hazardous chemicals.
(b) It reduces energy consumption by elimination of heating.
(c) It minimizes waste generation.
(d) It reduces the exposure of laboratory staff to acid vapours and improves lab safety.
(e) It reduces labour and improves sample throughput.
(f) It is possible to analyse a very small sample mass [i.e., analysis at micro- (10−2 to 10−3 g), submicro- (10−3 to 10−4 g) or even ultramicro-scale (<10−4 g)].183
Solid sampling can be considered especially green for samples that are difficult to digest even with very drastic conditions, and for elements whose levels are usually very low. For instance, Qi et al.184 have recently reported a digestion method for determination of the platinum group elements in geological samples by ICP-MS. Due to their chemical characteristics and low concentration, both laborious and tedious dissolution procedures are required. Thus, between 2 and 10 g of the sample are first digested with 15–30 mL of HF in a PTFE beaker on a hot plate to remove silicates up to dryness. The residue is then digested with 5 mL of HF + 15 mL of HNO3 at 190 °C for about 24–48 h in a stainless steel pressure bomb. After cooling, 2 mL of HCl are added and evaporated to dryness on a hot plate. In order to remove most of the residual acids, 5 mL of concentrated HCl are added to the bomb. After drying, the residue is dissolved with 40 mL of 2 N HCl. The resultant solution is then decanted to a 50 mL tube for centrifuging at 2800 rpm for 5 min. The upper portion is used to preconcentrate platinum group elements by coprecipitation. The precipitate is dissolved with aqua regia, the solution is evaporated to near dryness, redissolved again with 0.3 mL of aqua regia and diluted to 10 mL. An ion exchange resin is necessary to remove some interfering elements, such as Cu, Ni, Zr and Hf.
The last procedure can be compared with solid sampling by laser ablation and ICP-MS. For instance, Boulyga and Heumann185 suspended 1 g of sample in 0.2–5 mL of different isotope spike solutions. This suspension is dried at 75 °C, homogenized by mixing with a Teflon pestle and pressed into pellets for direct laser ablation. 3 mg of a rock sample are ablated within one analytical run (1 min ablation time). 50–67 successive runs are performed for each rock sample. Undoubtedly, solid sampling provides clear advantages with regard to the digestion procedure: it eliminates the use of HF, HNO3 and HCl; it reduces steps, time and labour and reduces the temperature of operation (190 vs. 75 °C).
4.1. Electrothermal vaporization/atomization for solid sampling
Trace metals can be directly vaporized and atomized from a solid sample introduced into an electrothermal atomizer/vaporizer, e.g., graphite furnace. It is an interesting alternative to sample dissolution in AAS, ICP-OES and ICP-MS. Although direct solid insertion (DSI) into a flame or plasma is somehow possible, ETV systems have successfully extended solid sampling to ICP-OES and ICP-MS. The latter approach provides matrix removal during the pyrolysis step, for which it can be considered as a thermochemical reactor allowing in situ sample treatment.165From 1971, the year in which the first application of solid sampling by ETAAS with a commercial atomizer was published,186 until today, important progress regarding instrumentation for solid sampling has been achieved. Spectrometers equipped with autosamplers that allow weighing, introduction of the solid sample automatically and calibration with aqueous standards as well as the addition of a matrix modifier are now commercially available (e.g., SSA 600L-fully automated solid sampler with a liquid dosing unit and up to 84 samples from Analytik Jena).187
Although ETV was used for the first time in 1974,188 applications of solid sampling with this system were performed much later. Probably, this fact can be associated with the commercialization of more robust ETVs at the end of the 90s. A degree of automation analogous to ETAAS can be achieved with ETV systems. An autosampler and a microbalance can be coupled to an ETV-unit for solid sampling by ICP-OES or ICP-MS (e.g., autosampler AWD-50 from Spectral Systems Advanced Technologies).189 The main green advantage of ETV-ICP-OES and ETV-ICP-MS with respect to ETAAS for solid sampling is their multielemental capability.
In spite of that, solid sampling with these atomic spectrometric techniques is unusual in routine labs. Though the state-of-the-art of the instrumentation has eliminated the difficulties related with the insertion of the solid sample into the atomizer, i.e., the risk of sample losses and contamination during the weighing and transfer from the balance to the atomizer,161 important analytical problems (e.g., precision continues to be worse than for liquid samples and calibration with certified reference materials is still the most reliable approach, the use of aqueous standards and the standard addition method being only possible sometimes) and drawbacks from the viewpoint of GAC continue to be present.
(a) Chemical modifiers interact less effectively with the analyte occluded within the sample matrix, and hence, a higher quantity is used in comparison with liquid samples.
(b) Additional reagents may be needed (e.g., hydrogen, methane or oxygen in the ashing step) in order to remove interferences.
(c) Higher atomization temperatures are required to release the analyte in comparison with liquid samples, especially when refractory elements are determined.
(d) Deterioration of the atomizer is a critical issue and additional cleaning-out steps can be necessary.
The expertise of the analyst is the key in the success of solid sampling. Paradoxically, the labour increases, especially along method development, validation and control. Every analyte and matrix requires a complete evaluation, i.e., optimization of the temperature program, matrix chemical modifier, establishment of a suitable mass interval, etc. This is especially remarkable for ETV-ICP-MS due to the large number of potential matrix interferences. Data treatment is also relevant because outliers frequently appear. So, in order to minimize their influence, the number of replicates required in solid sampling is higher.190
On the other hand, the corrosion of the graphite atomizer has been reported by Ortner et al.191 as ‘often disastrous in solid sampling into graphite boats’ and it can be considered an important disadvantage in GAC. In addition, release of gaseous and aerosol products from the matrix is enhanced in solid sampling. A series of relatively simple strategies such as the use of modifiers and coatings in graphite atomizers can be used in order to avoid this problem, i.e., the lifetime of carbide-modified graphite atomizers is longer than that of standard graphite tubes for solid sampling.191 The design of alternative atomizers for solid sampling still continues, e.g., a recent model of an atomizer named ‘crucible with separated zones’.192 It consists of a graphite tube placed in the atomization zone and heated independently of the evaporation zone so that the filtration of sample vapours through porous walls takes place.
Tungsten based atomizers, mainly W-tubes, have been proposed as an alternative to graphite furnaces for solid sampling, especially for slurry sampling. Green characteristics such as simplicity, low cost and low power requirements have been achieved with these atomizers. Tungsten is available at low cost, heating rates are high and a simple power supply is sufficient. In addition, no water circulating system is required as a coolant. Hou and Jones have reviewed these atomizers in analytical atomic spectrometry, including their potential in ETAAS, ETV-ICP-OES and ETV-ICP-MS with slurry sampling.193 However, not everything is green in this type of atomizers. These are usually purged with up to 10% of hydrogen mixed with argon and have a relatively short life.194
Although sample pre-treatment should be avoided whenever possible in order to retain the green advantages of solid sampling, it is not always possible. Drying and grinding up to a suitable particle size are usually performed to improve precision. In those cases, slurry sampling can be considered more advantageous since it shares the advantages of direct solid sampling (minimum sample pre-treatment) and liquid sampling (better precision, introduction with a conventional autosampler, calibration with an aqueous standard, efficiency of chemical modifiers, temperature programs and atomizer corrosion similar to liquid samples). Therefore, slurry sampling is more suitable for routine analysis than direct solid analysis.190
Miniaturization, automation and acceleration can be simultaneously achieved with slurry sampling (SS). For preparing slurries, a few milligrams of the powdered sample (typically 10–20 mg) and a volume of the liquid diluent in the range of 1–1.5 mL are added into an autosampler cup. In general, this liquid medium consists of a diluted solution of nitric acid (e.g., 3% v/v) and, in some cases, a very small amount of a stabilizing agent such as Triton X-100 or glycerol. Homogenization of the slurry is critical and, probably, this is the weakest point of the slurry technique from the perspective of GAC. Although it can be carried out by magnetic stirring, ultrasound agitation can be certainly considered as a greener option. In contrast to direct solid sampling, additional equipment only for the ultrasonic homogenization of the slurry is required. In this sense, commercial systems developed in the 90's incorporate an ultrasonic probe into the ETAAS autosampler for automating the procedure, i.e., USS-100 Ultrasonic Slurry Sampler from Perkin-Elmer. Only a few seconds of application of this energy is sufficient in order to obtain a homogeneous slurry.
SS can be used with ETV for multielemental determinations by ICP-MS and ICP-OES. For instance, Lin and Jiang194 used ultrasonic slurry sampling (USS)-ETV-ICP-MS for determining Cr, Mo, Pd, Cd, Pt and Pb in drug tablets. Amberger and Broekaert195 proposed the direct determination of trace elements in boron carbide powders by SS-ETV-ICP-OES. Slurries can be also nebulized directly into flames and plasmas. A comparison of SS-ETV and slurry nebulization (SN) for determination of trace impurities in titanium dioxide powder by ICP-MS has been carried out by Xiang et al.196 In both cases, the particle size is critical, though SS-ETV-ICP-MS has a lower particle size effect compared to SN-ICP-MS (particle diameter <50 vs. 1 μm respectively). Then, it is necessary to consider the increased operation time that reduction of particle size implies.
4.2. Arc/spark ablation
Arc/spark optical emission spectrometry has been used since long for direct analysis of solid samples. Ablation is carried out by an electrical discharge between an electrode and the conducting sample. In spite of the remarkable features (e.g., a multielemental determination could be performed in ca. 30 s), this classical technique has not garnered interest as an investigation topic due to the availability of more advanced techniques such as ICP-OES or ICP-MS, with better analytical characteristics for liquid sampling but with more difficulties as regards solid sampling.197 In 99% of cases, this technique is used in routine labs of the metallurgical industry. The remaining 1% corresponds mainly to the analysis of refractories, precious metals, steel-making slags and geological materials.198 These last samples must be powdered and mixed with high purity graphite, which diminishes the scope of green chemistry.Some technological advances incorporated into instruments can be considered as decisive in order to green this routine technique. Nowadays, more compact and lower-cost instruments requiring little maintenance can be acquired. In particular, this has been achieved with the incorporation of charge-coupled devices (CCD), gas-filled vacuum ultraviolet (VUV) optical chambers, digitally controlled arc/spark excitation sources, advanced electronic time-resolved spectroscopy (TRS), fiber optics and multiple optical cells.197 Portable systems for a variety of applications including routine sorting, positive material identification (PMI) or metal certification can be commercially obtained.199 New developments according to GAC principles have been recently patented, e.g., in order to conserve argon200 or to use low current spark generators.201
Today arc/spark ablation can be used for sample introduction in ICP-OES and ICP-MS, although its use is limited in comparison with lasers or glow discharge sources. A commercial spark ablation accessory known as ‘Separate Sampling and Excitation Accessory (SSEA)’ is used for this purpose. It improves the analytical characteristics of arc/spark spectroscopy and allows extending the solid sampling capability to routine laboratories.202–204 Both cost and maintenance are lower as compared to other systems such as laser ablation.
4.3. Laser ablation
Laser ablation is a powerful tool for solid sampling that has been increasingly applied in several fields. In contrast to arc/spark ablation, laser ablation has greater versatility. All types of materials can be used for laser ablation, i.e., conducting and non-conducting, organic and inorganic samples. In addition, LA can be used as a spatial and depth-resolved high resolution sampling technique.169 Very small amounts of samples, in the order of ng or even pg, can be analysed using laser ablation microprobes (LAMs). So it can be considered as an essentially non-destructive or minimally destructive technique from the viewpoint of GAC.3LA systems can be used as sampling and excitation sources (e.g., laser induced breakdown spectroscopy, LIBS) and coupled with ICP (LA-ICP-OES and, in particular, LA-ICP-MS). This last technique combines the green advantages of solid sampling discussed above with multielemental and isotopic analysis capability and high sensitivity. The sample is ablated in an airtight cell and the formed aerosol is carried in a continuous flow of an inert gas to the ICP where it is excited and ionized for quantification by MS.169 In the case of LIBS, the plasma plume generated on the sample surface is used for optical emission measurements. Incident laser light and emission lines are resolved both spectrally and temporally. The instrumentation is simple and includes an ablation laser, an optic collection system, a monochromator or an echelle spectrometer and a multi-element photodiode array (typically a CCD).171 In spite of the good characteristics of LA for solid sampling, matrix effects must be considered in all cases.
The formed aerosol by ablation of the sample depends not only on the sample but also on laser characteristics. First, lasers of ruby and later lasers of neodymium:yttrium aluminium garnet (Nd:YAG) and excimer lasers have been used for LA. There is a relationship between laser wavelength, laser irradiance, optical penetration depth, particle size, and analytical precision and accuracy. In general, it is accepted that shorter UV wavelengths improve ablation characteristics in respect to infrared and visible wavelengths, especially for highly transparent samples. Both, excimer and Nd:YAG lasers can produce UV light pulses. In general, excimer lasers offer the shortest wavelength and a precise sampling due to their spatial coherence. In contrast, Nd:YAG lasers are simpler, more cost-effective, require little maintenance and produce higher repetition rates.205
LA procedures can be divided according to the mass sample and analysis goal into bulk sample analysis and microprobe analysis. In bulk sample analysis, there is a large amount of sample and the difficulty lies in obtaining a representative analysis. The spot diameter is generally >100 μm corresponding to 2 μg–2 mg of ablated mass and then, in many cases, samples must be homogenized.169 Pulverization and even fusion to form a glass might be necessary. For instance, a meta-tetraborate fusion procedure was used with powdered rock by Leite et al.206 in LA-ICP-MS. Obviously, when a fusion procedure is necessary, the green advantages of LA are clearly diminished. Therefore, homogeneous materials such as alloys retain all the advantages of analysis by LA from the point of view of GAC.
LAM is especially interesting for applications where a spatial resolution is necessary (lateral and in-depth in the low-μm and nm range, respectively), i.e., microanalysis, in-depth profiling, and surface mapping.175 Though LAMs can reduce sensitivity since a small amount of sample is ablated, it can be considered a green tool. The spot diameter is generally <100 μm corresponding to 0.25 ng–2 μg of ablated mass. Pulsed laser energy used in LAMs is small (few mJ) compared with that used for bulk sampling (10–100 mJ).169 Usually, excimer lasers with short UV wavelengths are used. Shorter pulses improve LA localization; in particular ultra-short laser pulses (<1 ps) and more concretely femtosecond lasers are applied in order to improve the analytical characteristics.175
LAMs may require sample treatment only in some cases, for instance, for bioimaging of elements in biological tissues by LA-ICP-MS. Soft tissues are paraffin-embedded or frozen, then samples are sliced (usual thickness: 20–200 μm) and deposited on a glass slide. Both the thickness of the tissue and the laser parameters should be optimized. Cooled laser ablation cells are preferably used for this purpose.207 Usually, soft biological tissues are easy to ablate from a glass substrate and then, a Nd:YAG laser with a wavelength of 266 nm is sufficient to obtain highly spatially resolved images.208
In regard to LIBS, green intrinsic characteristics can be identified in this technique, since in situ analysis and remote sensing with no sample preparation are suitable. Fieldable laser-induced breakdown spectrometers have been developed in recent years. Fortes and Laserna209 have recently revised trends and applications with these instruments. Portable, remote and stand-off spectrometers are considered. In general, portability requires compact lasers with portable power supply and miniaturized spectrometers. Usually, Nd:YAG lasers are used due to size requirements, reliability and ruggedness. A calibration procedure without standard reference materials and based on the calculation of temperature and the electron density in the plasma (calibration-free) can be used with portable LIBS.210 Spectral libraries and different data processing algorithms can improve the green characteristics of LIBS for field measurements. In addition, automation of portable instruments is possible. A fully automated portable LIBS instrument has been developed by Palanco et al.211 When a fiber optic cable is used in a LIBS system, stand-off (both the laser and the signal are transmitted along an open path configuration) and remote (laser and/or the signal are transmitted through a fiber optic cable) analyses of samples for long distances can be made. It has allowed extending LIBS application to areas contaminated by toxic or radioactive materials without danger to the analyst. Very interesting information about applications of these systems can be found in the above-mentioned review.209
4.4. Glow discharge sources
Glow discharge sources with optical emission spectrometry (OES) or mass spectrometry (MS) are considered powerful and versatile for bulk, surface and interface analysis of different types of materials.177 Nowadays, GDs have gained interest due to the development of radio frequency (rf) sources, since it has allowed extending its application to non-conductive samples such as organic coatings, glasses, ceramics, etc. Although with very similar performance in both depth resolution and sensitivity, direct current (dc) sources continue to be used most frequently, only conducting materials can be analyzed.212In a Grimm-type GD, sputtering is used to generate atoms and ions directly from the solid sample surface ‘layer by layer’. Low-pressure plasmas are initiated by applying a high potential (kV) between two electrodes, one of which is formed by the sample (usually the cathode). Electrons and positive ions from a discharge gas (e.g., Ar or mixtures of Ar with N2, O2, H2 or He) are accelerated towards the cathode surface and when these have sufficient energy, result in releasing the material (sputtering process). This process depends strongly on the sample material and its surface properties. On the contrary, atomization, excitation and ionization processes (separated in space and time of the sputtering process) are practically independent of the surface sample.176 For this reason, little matrix effects have been observed and quantification by MS or OES is usually simpler than in other techniques for solid sampling.212 Without doubt, this question can be considered interesting in order to achieve greener procedures.
New developments in instrumentation have been decisive in order to extend the application of this technique and improve both its green and analytical characteristics. In particular, efforts have been focused on the design of new GDs, but also on improvements of vacuum technology and interface and on the implementation of CCDs and ToF (time-of-flight) detectors for OES and MS, respectively, in order to acquire spectra faster.177 For instance, pulsed rf/dc GDs have represented an important advance in trace analysis since they allow separating elemental and molecular excitations. This mode provides high instantaneous power, increasing the atomization, excitation, and ionization processes without thermal sample degradation. Numerous applications (e.g., polymers) are now possible with these sources.171,213 Magnetically boosted GD sources that incorporate a magnetic field to improve sputtering rates and ionization/excitation efficiencies have been also proposed.213,214 Atmospheric sampling GDs with untreated atmospheric gases were firstly introduced as an ionization source for organic compounds, though it has been also used for metal determination.215 The miniaturization of GD sources has also been considered, although it has been used so far for liquid and gas samples. A small glow discharge electron source has been used for miniaturized mass spectrometers.216 Low power requirements, mechanical ruggedness, and quality of the data produced make these sources suitable with a portable handheld mass spectrometer.
Solid sampling with GDs is not without problems. Non-flat samples (e.g., screws and tubes) cannot directly be mounted on a GD and special accessories are required. Porous samples (e.g., foams and certain ceramics) are also difficult to handle because they are not vacuum tight since usually the sample seals the source.212 Particulate solids can be analysed using rf-GD-OES. For this, sol–gel thin films or glasses are synthesized by acid-catalysed condensation. Slurries of powdered samples are incorporated into the films and analysed for both main and trace element components.217,218
GD-OES and GD-MS can be compared with other techniques used for surface analysis, such as SIMS (secondary ion mass spectrometry) and SEM (scanning electron microscopy with X-ray analysis). GD-OES and GD-MS are considered cheaper, faster and easier for quantification than SIMS, and faster and with better depth resolution than SEM. For bulk analysis, GD-OES can compete with spark/arc emission and with X-ray techniques.3,212 Obviously, GD-OES and GD-MS have some drawbacks such as limited lateral resolution and inability for micro-spot analysis. Recently, the LA-GD combination has been proposed for solid sampling. Laser ablates the sample and GD ionizes the ablated aerosol.219,220
4.5. X-ray fluorescence spectrometry
X-ray fluorescence spectrometry (XRF) can be considered as a quantitative and qualitative analytical tool that can fulfil GAC principles. Different techniques are available: wavelength dispersive X-ray fluorescence (WDXRF), energy dispersive X-ray fluorescence (EDXRF), total reflection X-ray fluorescence (TXRF) and micro-X-ray fluorescence (μ-XRF). All offer fast and non-destructive analysis using clean procedures that can routinely be applied to solid samples. In general, analysis can be carried out within a time in the range of 30–1000 s with a good precision.221–224In spite of this, it is not usual to consider XRF as a set of green techniques due to risks that this energy involves. The X-ray beam can be generated with a ceramic tube or from radionuclides.225,226 Radionuclides are more dangerous for the analyst because they generate γ rays, are always “switch on” and have shorter lifetimes. Their use is subject to strict regulations and can only be handled by authorized personnel.227 X-ray tubes are mostly used in the analytical instrument since they are safe. In addition, miniature X-ray tubes have made possible the development of portable instruments for in situ analysis. This can contribute to change the negative perspective on these techniques from the GAC standpoint.
Although X-ray instruments could potentially be dangerous, current instruments pose few risks to users when used properly. The manufacture and use of X-ray instruments is highly regulated. In fact, the design of the instruments limits even accidental exposure. Spectrometers have a lead shielded, closed design that avoids the scattering of the X-ray beam. In addition, the surrounding of the equipment should be frequently checked to ensure that there is no scattering of the X-ray radiation and there is no potential risk for the analyst by radiation exposure (the maximum permissible whole-body dose from occupational exposure is 5 ‘Roentgen Equivalent Man, REM’, per year. When portable equipments are used, wearing heavy gloves is advisable.228
The main green advantage of XRF is without doubt its ability for direct analysis of the sample without sample treatment or with a little preparation such as grinding, cutting, etc. Bulk materials can be layered with a thickness ≤0.5 mm by cutting, sawing or shredding.229 For samples such as metals or ceramics, it is necessary to polish for creating a smooth and clean surface when WDRXF is used. Grinding to grain sizes of lower than 75 μm is used to obtain a homogeneous powder. Then, for higher density solids only a pressing step is necessary in most XRF techniques. For low-density solids, the addition of a binding agent is needed before pressing to obtain a stable and homogeneous pellet. Natural substances such as starch, wax or cellulose can be used for this purpose being desirable for GAC in contrast to others such as polyvinyl.230
These non-destructive techniques can be used directly even with fresh samples. The fresh sample is homogenized and frozen and then, a portion is pressed into a pellet and analysed. For instance, the elemental content of the edible part of lobsters has been established by EDXRF.231
When the sample available is very small, fusion is often used as the sample treatment in EDXRF and WDXRF.232 Usually, borate is used as the fusion reagent (above 1000 °C). Fusion melt is a homogeneous glass with a defined matrix.
TXRF allows analysis of powdered samples without pressing or forming pellets, only placing on a cleaned carrier, usually quartz glass. Different strategies can be used with powdered samples in this technique. For example, a cotton-wool tip has been used for analysing inorganic pigments in oil paintings. For sampling, the surface of oil paintings is wiped off by means of a cotton-wool tip. A few micrograms of the cotton-wool tip are sufficient for the analysis.233 Recently, the use of slurry sampling has been proposed in TXRF. For example, biological samples224 using an ultrasonic probe for slurry homogenization have been analysed. A 10 μL aliquot of the slurry is deposited onto the quartz glass carrier.
μ-XRF has been developed very rapidly in the last decade, mainly due to its versatility. It uses rotatory X-ray tubes which provide synchrotron radiation, resulting in a very sensitive technique with limits of detection (LODs) at the ppt level.178 Not only is the elemental composition of a sample easy to obtain, but also the related spatial distribution without sample treatment. Commercial portable equipments (e.g., ARTAX® equipment from Bruker)234 are available. They allow in situ measurements within 30–60 s, and then, sampling and transport to the lab are not necessary. Portable systems are mainly applied for archeometry and restoration since they avoid contact and damage, whatever the objects under investigation.235
TXRF can be applied to micro, trace, and surface analyses. This technique can provide detection limits at the part per billion level.236 Commercially available TXRF instruments (e.g., S2 Picofox® from Bruker)237 are considered semi-portable systems. This technique does not need inert gases and/or a vacuum system. Certain grade of automation can be reached using a motorized sample changer.
EDXRF is widely used for the determination of major, minor and trace elements (LODs at ppm level).180 An increased automation level as in TXRF is reached by using an automatic sample changer. Commercial portable EDXRF instruments are air-cooled systems and they can operate in vacuum or in air (e.g., Tracer® family from Bruker).238 This equipment has been mainly designed for in situ measurements in art, archeometry, geological research, environment and also for specific industrial applications, i.e., sorting scrap, alloy analysis and sorting for alloy verification. The use of this portable equipment can have some drawbacks since instruments are less robust and quantitative analysis can be troublesome. Forster et al.239 pointed out that surface morphology, surface coatings and grain size of materials can cause attenuation of incident and fluorescent X-rays, yet appropriate mathematical corrections can provide a high level of accuracy and precision. Qualitative analysis with portable systems has been applied for a more efficient sampling, i.e., soil sampling,222 in order to perform a fast general screening of the soil composition and detect the points of contamination. It allows selecting representative points, thereby saving time in the sampling stage.
WDXRF is mainly applied for direct solid analysis (soils, metals, ceramics, etc.) in routine labs. WDXRF analysis splits the characteristic wavelengths with a high resolution, which is especially relevant for light elements (LODs at the ppm level).180 It is faster than EDXRF and TXRF (i.e., 100 s vs. 500 s). In contrast to μXRF and EDXRF, portable systems for WDXRF are not available. So in WDXRF analysis, sampling and transport of the sample to the lab are needed. In general, sample consumption is higher compared to other X-ray techniques, i.e., only a few micrograms of sample are necessary in TXRF and EDXRF vs. 0.1–5.0 g in WDXRF.221,240–242 The analysis is usually carried out under vacuum, and for the analysis of loose powders, an inert atmosphere (He or N2) is needed to prevent air from absorbing the fluorescence X-rays. As in TXRF and EDXRF, the use of an automatic sample changer is possible.
5. Greening instrumentation for atomic spectrometry
Nowadays, remarkable greenness in atomic spectrometry has been reached through miniaturization, automation and design of cost-effective systems, which allow lower consumption of gases (e.g., fuel, plasmogen, carrier), samples and reagents, and/or simplified sample pre-treatment. Improvements made in instrumentation for atomic spectrometry (including inorganic mass spectrometry) from the early prototypes to the modern instruments have also contributed to reach higher levels of greenness. As an example, implementation of efficient background correction systems in the early atomic absorption spectrometers made it possible to carry out many applications with a significant saving in operations such as matrix elimination by typical procedures, which in turn, brought about a decrease in errors.243 More recently, the development of efficient collision and reaction cells for elimination of some interferences caused by polyatomic ions has simplified the application of quadrupolar ICP-MS to complex matrices.244Undoubtedly, major developments made in most atomic spectrometric techniques have run in parallel with improvements of green features. For instance, automation provided by modern autosamplers in techniques such as ETAAS, allowing intelligent dilution, injection of sample, diluents and matrix modifiers at any sequence, performing calibration curve from one single standard, recalibration, etc. has dramatically changed method development with this technique, hence saving time, efforts and decreasing errors.
A component that is commonly used for sample introduction in atomic spectrometry is the pneumatic nebulizer. Development of miniaturized nebulizers that allow minimum consumption of sample is one of the goals of GAC. In contrast to conventional nebulizers, micronebulizers allow the generation of a stable aerosol at liquid flow-rates on the order of several microliters per minute.245 Additional advantages of micronebulizers are the very low dead volumes and lesser memory effects. Micronebulizers originate finer primary aerosols, higher solution transport rates and higher sensitivity as compared to conventional pneumatic nebulizers, yet some prototypes are prone to blocking.246 The suitability of several micronebulizers for analysis of microsamples has been shown for techniques such as ICP-OES247 and ICP-MS.248
The design of efficient sample introduction systems such as those based on thermospray interfaces has allowed an improvement in LODs apart from decreasing the sample consumption. In the thermospray technique, the liquid is pumped through a capillary and heated uniformly along a length of a few cm. The heating causes the liquid to partially vaporize, giving rise to a blast of vapour that converts the remaining liquid to droplets. This interface has been used in conjunction with several techniques such as ICP-OES, ICP-MS, FAAS and ETAAS.249 As a result of the increased sensitivity achieved with the thermospray technique, this interface can also benefit the coupling between HPLC and atomic spectrometry for speciation studies.250
Greening atomizers for atomic spectrometry is another area of research. Efforts have been made in decreasing the power and gas consumption as well as the size of atomizers in order to approach the concept of ‘portable instrument’.
With the arrival of plasma techniques (i.e., ICP-OES, ICP-MS), multielement analysis at a trace level has been facilitated. However, the high operating cost represented by the Ar consumption can limit their application in smaller labs. Thus, conventional ICP torches have been modified so that the Ar and power consumption can be reduced. One way to reduce Ar consumption is the design of minitorches.251 Nevertheless, those systems are somewhat more prone to interferences in comparison with conventional torches.
Microplasmas for atomic emission have been described, e.g., high-frequency plasmas, dc-discharges, MW plasmas.252 However, the heat capacity of these systems is limited and sample introduction in the vapour phase instead of liquid phase is mandatory. In this way, typical derivatization techniques such as hydride generation are useful. These approaches are not free from drawbacks due to the unstabilizing effect caused by the excess hydrogen generated during chemical hydride generation. Alternative HG procedures such as those based on electrochemical hydride generation or UV reduction are promising.253,254 Other strategies for spreading the applicability of microplasmas are their use as detectors in gas chromatography255 or the generation of dry vapours using electrothermal vaporization.256
Miniaturized flames have been also developed for atomization of volatile compounds. Thus, miniaturized diffusion flames such as Ar/H2 have found application for AFS after HG.257 These systems offer low consumption of gases, low background emission and lesser interferences as compared with other atomizers.258 Multiatomizers configured inside a quartz tube allow achieving improved sensitivity as compared with diffusion flames apart from enhanced linearity and resistance against interferences in comparison with quartz tubes.259 The multiatomizer is similar to conventional quartz tube atomizers, but it is punctured over the horizontal arm so that air can be dosed through the orifices to the optical inner volume. In this way, multiple microflames arise inside the tube as a result of the multiple hydrogen radical clouds formed.
Efficient atomic fluorescence detectors have been marketed using the hydrogen generated in the reaction with sodium tetrahydroborate to feed a miniaturized diffusion flame of Ar/H2 with application in total trace analysis (Hg, As, Se, Te, Sb, Bi) and as specific detectors for speciation when interfaced with liquid or gas chromatography.260
Miniaturization has also spread to hyphenated techniques for speciation in biological systems. Thus, for metallome analysis in small amounts of tissues and cells, new tools based on miniaturized HPLC techniques (e.g., narrow bore, capillary and nano-HPLC) coupled to ICP-MS have been developed, which are expected to clarify the physiological and biological roles of metalloproteins.261 The coupling between capillary and, especially, nano-flow HPLC with ICP-MS demands for the development of suitable interfaces so that the sample uptake is compatible with the mobile phase flow-rate. For this, micronebulizers discussed above are required.262
Other green couplings between HPLC and ICP-MS for speciation of hydride forming elements are based on on-line UV/TiO2 photocatalysis, thus eliminating conventional hydride generation with the NaBH4–NaOH system.263
An interesting technique avoiding time-consuming procedures for preconcentration is the use of ‘in-atomizer’ trapping. This approach has been tried in different ways.
First systems were based on trapping in a graphite tube for electrothermal atomization.264 Apart from improving sensitivity, in situ trapping eliminates the effect of kinetic interferences in HG. Moreover, these systems can be easily automated. More recently, trapping onto quartz surfaces in an excess oxygen and further atomization in multiatomizers or conventional quartz tubes have also been proposed to achieve very low detection limits.258 Trapping of volatile hydrides has also been performed in miniature electrothermal devices, e.g., tungsten coil265 and bare and modified strips with noble metals (Pt, Ir, Rh).266 W-coil devices can be used as atomizers for ETAAS, electrothermal atomizer laser-excited atomic fluorescence spectrometry (ETA-LEAFS) and ETV-ICP-OES, being ideal for the design of low-cost, compact and portable instrumentation for field environmental and clinical analysis.267 W-coil atomizers along with a simple CCD-based spectrometer make the development of AAS instrumentation for field analysis possible. A few representative examples of miniaturized atomizers for AAS are shown in Fig. 6.
 | ||
| Fig. 6 Examples of miniaturized and low consumption atomizers for atomic absorption spectrometry (Ref. 250, 255, 265, 267 and 268). | ||
The recent implementation of fast multielemental sequential analysis in AAS through novel instrumentation based on continuous radiation sources (Xe-arc lamp), echelle monochromators and CCD detectors can be considered a step forward in the achievement of greener AAS methods.269 The observation of the analytical line at high resolution facilitates method development and helps avoiding spectral interferences.
As indicated above, most successful achievements so far toward the achievement of portable instruments have been reported in the miniaturization of techniques such as XRF and LIBS. These techniques are essentially non-destructive, fast and they provide instantaneous multi-element analysis.2
6. Chemometrics for greening atomic spectrometry methods
Can chemometrics be a tool for greening atomic spectrometry methods? According to Namieśnik,270 the reduction of labour-intensive procedures, energy and reagent consumption is essential for the implementation of GAC principles. Automation and robotization, multianalyte determination in a single analytical cycle and wider utilization of hyphenated techniques are considered by this author as the main ways for greening the analytical laboratory. There is no doubt that chemometrics is of paramount importance to achieve the aforementioned developments, hence being an indirect way of saving time, labour, energy and reagents. In general, chemometrics helps extracting information with a smaller number of experiments and preventing errors in an environmentally friendly way. In spite of this, chemometrics is not usually considered from the point of view of GAC.In this regard, Armenta et al.4 as well as Koel and Kaljurand2 recognize the importance of chemometrics in the development of solvent free methodologies based on direct measurements without sample pre-treatment. Particularly, this conception has been focused on molecular spectroscopy techniques that involve large amounts of spectral data, such as UV-vis spectrometry, fluorescence, near infrared (NIR), mid-infrared (MIR) and nuclear magnetic resonance (NMR). Some examples taken into consideration by these authors are the use of calibration by partial least squares (PLS) in NIR, MIR or Raman spectroscopy, thereby enhancing the application of these techniques in the analytical lab on a routine basis4 or the use of parallel factor analysis (PARAFAC) in the resolution of mixtures using excitation–emission spectroscopy (EEM).2
Chemometrics (including exploring data, optimization, calibration, signal processing, pattern recognition and artificial intelligence) can be considered also as an interesting tool for greening atomic spectrometry methods. As mentioned above, chemometrics become particularly significant to reduce the number of measurements or to obtain much simpler and efficient analytical procedures with atomic spectrometry techniques.
To a greater or lesser extent, chemometrics is involved in all steps of analytical methods, from development and validation up to data evaluation. For instance, principal component analysis (PCA) and a multi-criteria target function have been used in ICP-MS for 83 isotopes and 21 operating parameters.271 Chemometrics has been also used to evaluate the combined uncertainty for mercury speciation by GC-AFS in the validation step.272 Multivariate techniques for pattern recognition are being increasingly applied to trace metals data in order to discriminate between samples. In general, well-established chemometric tools are used, e.g., PCA, cluster analysis (CA), linear discriminant analysis (LDA), soft independent modelling of class analogy (SIMCA) or artificial neural networks (ANNs). Practically, we can find applications in all fields of interest such as clinical273 and environmental,274 but in particular in food science for establishing geographical origins, e.g., honeys,275 onions,276 alcoholic beverages,277 coffee,278 mussels,279etc.
Though optimization is fundamental in the development of analytical methods, the most usual approach for this purpose in atomic spectrometry continues to be the non-systematic way, based on the subjective experience of the analytical chemist (trial-and-error approach). If this experience is less, the workload in the laboratory is large and, in addition, optimization is not guaranteed.280 In most cases, optimization is carried out using a univariate strategy. It implies a large number of experiments, which increase the consumption of reagents and are labour-intensive. In order to overcome these disadvantages, multivariate optimization is a more desirable option from the point of view of GAC. Different strategies have been used in atomic spectrometry, in particular experimental design and response surface methods.281 In those methods, different variables are simultaneously and systematically studied. In addition, they provide more information about variables (e.g., interaction between them) with fewer experiments, which is especially important for optimizing sample preparation stages, e.g. microwave-assisted digestion,282 preconcentration using different sorbents,283,284etc. Optimization of measurement conditions in different atomic spectrometric techniques is also carried out with multivariate strategies, e.g., application of the experimental design for optimization of lead determination by high-resolution continuum source hydride generation atomic absorption spectrometry.285 Experimental design for method optimization using spectrometric techniques has been reviewed by Bianchi and Careri.286 Hibbert and Armstrong287 have reviewed the applications of Bayesian methods, including spectroscopy and mass spectrometry. The use of response surface methodology for optimization in analytical chemistry has been reviewed by Bezerra et al.288
Multivariate calibration methods represent a useful strategy to fight analyte interferences, so these can be considered as important tools from the standpoint of GAC. A tutorial review on multivariate calibration in atomic spectrometry techniques has been published by Andrade et al.289 These authors include among the advantages of multivariate calibration ‘lower workloads, increased laboratory turnarounds, economy, higher efficiency in method development, and relatively simple ways to take account of complex interferences’, all of which are directly related to GAC. Application of multiple ordinary least squares regression (MOLSR), principal components regression (PCR), PLS and ANN on FAAS, HG-AAS, ETAAS, ICP-OES and LIBS are presented in a practice-oriented way. These multivariate calibration methods may be applied to monoelemental techniques considering the transient signals (absorbance vs. time) as a spectrum (absorbance at multiple wavelengths).
On the other hand, signal processing can be considered as a simple way for improving instrumental performances without hardware component changes and/or replacement by expensive systems.290 Signal processing is used to enhance signal vs. noise, to improve peak resolution, to decompose complex signals, etc. Undoubtedly, it allows extracting more information from analytical data with a lower number of experiments. For example, chemical interference correction, including separation, can be replaced or minimized by chemometric interference correction. Mathematical algorithms for background modelling and correction have been designed and applied to different atomic techniques in order to improve their analytical characteristics.291–296 At present, signal processing is an active field especially when transient signals are obtained. For instance, Chaves et al.297 proposed the processing of fast transient signals provided by different sample introduction systems (FI, LA, GC, HPLC) with modern simultaneous ICP-OES instruments. Prikler et al.298 compared different strategies as convolution with Gaussian distribution curves, Fourier transform, and wavelet transform for improving the detection power in HPLC-ICP-MS. A total signal integration method has been proposed as an alternative to quantify transient signals with LA and ICP-MS.299
In general, application of artificial intelligence (AI), especially expert systems (ES), intelligent analysers and robot systems, constitutes an efficient way so as to turn the analytical lab green, since lab efficiency is increased and the occupational exposure of the personnel and reagent consumption are decreased.280,300 An intelligentized analytical lab results in a more cost-effective use of staff and resources. AI can be used from the selection and optimization of an analytical method up to the final report, including detection of malfunctions and validation. During the 90s, a large number of ESs were developed for different atomic spectrometry applications. ESs are programs with a heuristic knowledge based on the experience of experts. ESs have been included in FAAS spectrometers for full automation (in particular for error detection and correction).301 In ICP-OES, ESs are used for prediction and correction of spectral interferences,302,303 spectral line simulation and selection,304,305 system diagnosis306 or as a hybrid expert-database system for sample preparation by microwave-assisted dissolution.307,308 ES has been also used in ICP-MS for controlling the spectrometer,309,310 the whole sample pre-treatment process311 and developing a simple diagnostic procedure to automatically ensure the quality of results.312 In XRF, it has been applied for the evaluation of different possibilities in the resolution of a given analytical problem.313,314 Intelligent analysers have also been designed with different atomic spectometers,315–317i.e., an intelligent flow system has been proposed for the on-line speciation of metal ions at a wide range of concentrations without requiring manifold reconfiguration.316
7. Conclusions
A remarkable greenness can be achieved when GAC principles are applied to Atomic Spectrometry. For this, every stage of the analytical process should be focused. Advances in instrumentation, sample preparation techniques, chemometric treatment of data, etc. can drive significant improvements not only in analytical characteristics but also in green issues of the whole methodology. Labs involved in trace metal analysis can benefit from the concepts of green chemistry, since apart from advantages inherent to the implementation of automated, simplified, accelerated and miniaturized systems, there is also a great conservation of reagents, solvents and energy, less risks to the analyst, and less production of wastes. More progress is expected over the next years concerning this topic with the downsizing of instruments, implementation of new materials (e.g., nanomaterials) and development of analytical devices for on-site determination.List of abbreviations
| AAS | Atomic absorption spectrometry |
| AFS | Atomic fluorescence spectrometry |
| AI | Artificial intelligence |
| ANNs | Artificial neural networks |
| AOPs | Advanced oxidation processes |
| ASE | Accelerated solvent extraction |
| BI | Bead-injection |
| CA | Cluster analysis |
| CCD | Charge-coupled devices |
| CPE | Cloud point extraction |
| CV | Cold vapour |
| CVG | Chemical vapour generation |
| DBD | Dielectric barrier discharge |
| dc | Direct current |
| DSI | Direct solid insertion |
| EC-HG | Electrochemical hydride generation |
| EDXRF | Energy dispersive X-ray fluorescence |
| EEM | Excitation-emission spectroscopy |
| ES | Expert systems |
| ETAAS | Electrothermal atomic absorption spectrometry |
| ETA-LEAFS | Electrothermal atomizer laser-excited atomic fluorescence spectrometry |
| ETV | Electrothermal vaporization |
| FAAS | Flame atomic absorption spectrometry |
| FI | Flow injection |
| FIA | Flow injection analysis |
| FMAE | Focused microwave-assisted extraction |
| GAC | Green analytical chemistry |
| GC | Gas chromatography |
| GD | Glow discharge |
| GSK | GlaxoSmithKline |
| HEPO | High-efficiency photooxidation |
| HF | Hollow fiber |
| HG | Hydride generation |
| HIFU | High-intensity focused ultrasound |
| HPLC | High performance liquid chromatography |
| HS | Headspace |
| ICP-MS | Inductively coupled plasma-mass spectrometry |
| ICP-OES | Inductively coupled plasma-optical emission spectrometry |
| IL | Ionic liquid |
| LA | Laser ablation |
| LAM | Laser ablation microprobe |
| LC | Liquid chromatography |
| LDA | Linear discriminant analysis |
| LIBS | Laser induced breakdown spectroscopy |
| LLE | Liquid–liquid extraction |
| LOC | Lab-on-a-chip |
| LODs | Limits of detection |
| LOV | Lab-on-valve |
| LPME | Liquid-phase microextraction |
| MAD | Microwave-assisted digestion |
| MAE | Microwave-assisted extraction |
| MCFIA | Multicommutated flow injection analysis |
| μ-FIA | Micro-flow injection analysis |
| μ-TAS | Micro-total analytical system |
| μ-XRF | Micro-X-ray fluorescence |
| MIR | Mid-infrared |
| MOLSR | Multiple ordinary least squares regression |
| MPFS | Multipumping flow system |
| MS | Mass spectrometry |
| MSFIA | Multisyringe flow injection analysis |
| MSPME | Magnetic nanoparticle solid phase microextraction |
| MW | Microwave |
| Nd:YAG | Neodymium:yttrium aluminium garnet |
| NEMI | National environment methods index |
| NIR | Near infrared |
| NmimCl | 1-Chlorovinyl-3-methylimidazolium chloride |
| NMR | Nuclear magnetic resonance |
| OES | Optical emission spectrometry |
| PARAFAC | Parallel factor analysis |
| PCA | Principal component analysis |
| PCR | Principal components regression |
| PLE | Pressurized liquid extraction |
| PLS | Partial least squares |
| PMAE | Pressurized microwave-assisted extraction |
| PMI | Positive material identification |
| PTFE | Polytetrafluoroethylene |
| PVC | Polyvinyl chloride |
| QFAAS | Quartz furnace atomic absorption spectrometry |
| REM | Roentgen Equivalent Man |
| rf | Radio frequency |
| SBSE | Stir bar sorptive extraction |
| SDS | Sodium dodecyl sulphate |
| SEM | Scanning electron microscopy |
| SFE | Supercritical fluid extraction |
| SIA | Sequential injection analysis |
| SIMCA | Soft independent modelling of class analogy |
| SIMS | Secondary ion mass spectrometry |
| SLM | Supported liquid membrane |
| SN | Slurry nebulization |
| SPE | Solid-phase extraction |
| SPME | Solid-phase microextraction |
| SS | Slurry sampling |
| SSEA | Separate sampling and excitation accessory |
| ToF | Time-of-flight |
| TRS | Time-resolved spectroscopy |
| TXRF | Total reflection X-ray fluorescence |
| UAE | Ultrasound-assisted extraction |
| US | Ultrasound |
| USAED | Ultrasound-assisted enzymatic digestion |
| USS | Ultrasonic slurry sampling |
| VUV | Vacuum ultraviolet |
| WDXRF | Wavelength dispersive X-ray fluorescence |
| XRF | X-ray fluorescence |
Acknowledgements
Financial support from the Spanish Ministry of Economy and Competitiveness (Project CTQ2009-06956/BQU), the Xunta de Galicia (project 10PXIB 314030 PR) and the Vigo University (Contract for Reference Research Groups 09VIA08) is gratefully acknowledged. The Spanish Ministry of Education, Culture and Sport is acknowledged for financial support through a FPU pre-doctoral grant to Vanesa Romero. The Portuguese Foundation for Science and Technology is acknowledged for financial support through a Post-Doctoral grant to Francisco Pena-Pereira.References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- M. Koel and M. Kaljurand, Green Analytical Chemistry, Royal Society of Chemistry, Cambridge, 2010 Search PubMed.
- M. de la Guardia and S. Garriges, Challenges in Green Analytical Chemistry, Royal Society of Chemistry, Cambridge, 2011 Search PubMed.
- S. Armenta, S. Garrigues and M. de la Guardia, TrAC, Trends Anal. Chem., 2008, 27, 497–511 CrossRef CAS.
- M. Tobiszewski, A. Mechlińska, B. Zygmunt and J. Namieśnik, TrAC, Trends Anal. Chem., 2009, 28, 943–951 CrossRef CAS.
- M. Tobiszewski, A. Mechlinska and J. Namieśnik, Chem. Soc. Rev., 2010, 39, 2869–2878 RSC.
- M. de la Guardia, TrAC, Trends Anal. Chem., 2010, 29, 577 CrossRef CAS.
- C. J. Welch, M. Biba, R. Hartman, T. Brkovic, X. Gong, R. Helmy, W. Schafer, J. Cuff, Z. Pirzada and L. Zhou, TrAC, Trends Anal. Chem., 2010, 29, 667–680 CrossRef CAS.
- F. Pena-Pereira, I. Lavilla and C. Bendicho, TrAC, Trends Anal. Chem., 2010, 29, 617–628 CrossRef CAS.
- S. Garrigues, S. Armenta and M. de la Guardia, TrAC, Trends Anal. Chem., 2010, 29, 592–601 CrossRef CAS.
- C. Bendicho, F. Pena, M. Costas, S. Gil and I. Lavilla, TrAC, Trends Anal. Chem., 2010, 29, 681–691 CrossRef CAS.
- A. Molina-Diaz, J. F. García-Reyes and B. Gilbert-López, TrAC, Trends Anal. Chem., 2010, 29, 654–666 CrossRef CAS.
- M. Farré, S. Pérez, C. Gonçalves, M. F. Alpendurada and D. Barceló, TrAC, Trends Anal. Chem., 2010, 29, 1347–1362 CrossRef.
- M. Tobiszewski and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 25, 67–73 CrossRef.
- C. Bendicho, I. de La Calle, F. Pena, N. Cabaleiro and I. Lavilla, TrAC, Trends Anal. Chem., 2012, 31, 50–60 CrossRef CAS.
- http://www.nemi.gov, last accessed, June 2012.
- A. Galuszka, P. Konieczka, Z. M. Migaszewski and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef CAS.
- V. Camel, Spectrochim. Acta, Part B, 2003, 58, 1177–1233 CrossRef.
- C. Bendicho, I. Lavilla, F. Pena and M. Costas, in Challenges in Green Analytical Chemistry, ed. M. de la Guardia and S. Garrigues, Royal Society of chemistry, Cambridge, 2011, pp. 63–99 Search PubMed.
- V. A. Lemos, L. S. G. Teixeira, M. A. Bezerra, A. C. S. Costa, J. T. Castro, L. A. M. Cardoso, D. S. de Jesus, E. S. Santos, P. X. Baliza and L. N. Santos, Appl. Spectrosc. Rev., 2008, 43, 303–334 CrossRef CAS.
- M. L. Chen, Y. N. Zhao, D. W. Zhang, Y. Tian and J. H. Wang, J. Anal. At. Spectrom., 2010, 25, 1688–1694 RSC.
- Y. Tian, Z. M. Xie, M. L. Chen and J. H. Wang, J. Anal. At. Spectrom., 2011, 26, 1408–1413 RSC.
- Z. Mester, R. Sturgeon and J. Pawliszyn, Spectrochim. Acta, Part B, 2001, 56, 233–260 CrossRef.
- Z. Mester and R. Sturgeon, Spectrochim. Acta, Part B, 2005, 60, 1243–1269 CrossRef.
- S. Risticevic, V. H. Niri, D. Vuckovic and J. Pawliszyn, Anal. Bioanal. Chem., 2009, 393, 781–785 CrossRef CAS.
- F. Pena-Pereira, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2009, 64, 1–15 CrossRef.
- S. Dadfarnia and A. M. Haji Shabani, Anal. Chim. Acta, 2010, 658, 107–119 CrossRef CAS.
- F. Pena-Pereira, I. Lavilla and C. Bendicho, Anal. Chim. Acta, 2010, 669, 1–16 CrossRef CAS.
- E. Baltussen, P. Sandra, F. David and C. Cramers, J. Microcolumn Sep., 1999, 11, 737–747 CrossRef CAS.
- F. David and P. Sandra, J. Chromatogr., A, 2007, 1152, 54–69 CrossRef CAS.
- A. Prieto, O. Basauri, R. Rodil, A. Usobiaga, L. A. Fernández, N. Etxebarría and O. Zuloaga, J. Chromatogr., A, 2010, 1217, 2642–2666 CrossRef CAS.
- J. Vercauteren, C. Pérès, C. Devos, P. Sandra, F. Vanhaecke and L. Moens, Anal. Chem., 2001, 73, 1509–1514 CrossRef CAS.
- J. Millos, M. Costas-Rodríguez, I. Lavilla and C. Bendicho, Anal. Chim. Acta, 2008, 622, 77–84 CrossRef CAS.
- J. Pacheco-Arjona, P. Rodriguez-Gonzalez, M. Valiente, D. Barclay and O. F. X. Donard, Int. J. Environ. Anal. Chem., 2008, 88, 923–932 CrossRef CAS.
- I. de la Calle, N. Cabaleiro, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2009, 64, 874–883 CrossRef.
- J. L. Gómez-Ariza, V. Bernal-Daza and M. J. Villegas-Portero, Anal. Chim. Acta, 2004, 520, 229–235 CrossRef.
- A. Moreda-Piñeiro, J. Moreda-Piñeiro, P. Herbello-Hermelo, P. Bermejo-Barrera, S. Muniategui-Lorenzo, P. López-Mahía and D. Prada-Rodríguez, J. Chromatogr., A, 2011, 1218, 6970–6980 CrossRef.
- M. Costas, I. Lavilla, S. Gil, F. Pena, I. de la Calle, N. Cabaleiro and C. Bendicho, Anal. Chim. Acta, 2010, 679, 49–55 CrossRef CAS.
- M. Zougagh, M. Valcárcel and A. Ríos, TrAC, Trends Anal. Chem., 2004, 23, 399–405 CrossRef CAS.
- M. D. Luque de Castro and M. T. Tena, TrAC, Trends Anal. Chem., 1996, 15, 32–37 CrossRef CAS.
- J. Kronholm, K. Hartonen and M. L. Riekkola, TrAC, Trends Anal. Chem., 2007, 26, 396–412 CrossRef CAS.
- V. Camel, Analyst, 2001, 126, 1182–1193 RSC.
- L. H. Keith, L. U. Gron and J. L. Young, Chem. Rev., 2007, 107, 2695–2708 CrossRef CAS.
- L. Chimuka and R. Majors, LC·GC Eur., 2004, 17, 396–401 CAS.
- J. A. López-López, C. Mendiguchía, J. J. Pinto and C. Moreno, TrAC, Trends Anal. Chem., 2010, 29, 645–653 CrossRef.
- K. Chakrabarty, P. Saha and A. K. Ghoshal, J. Membr. Sci., 2010, 350, 395–401 CrossRef CAS.
- J. F. Peng, R. Liu, J. F. Liu, B. He, X. L. Hu and G. B. Jiang, Spectrochim. Acta, Part B, 2007, 62, 499–503 CrossRef.
- L. Xia, Y. Wu and B. Hu, J. Mass Spectrom., 2007, 42, 803–810 CrossRef CAS.
- C. Bosch Ojeda and F. Sánchez Rojas, Anal. Bioanal. Chem., 2009, 394, 759–782 CrossRef CAS.
- C. Ortega, M. R. Gomez, R. A. Olsina, M. F. Silva and L. D. Martinez, J. Anal. At. Spectrom., 2002, 17, 530–533 RSC.
- Y. Yamini, M. Faraji, S. Shariati, R. Hassani and M. Ghambarian, Anal. Chim. Acta, 2008, 612, 144–151 CrossRef CAS.
- K. Simitchiev, V. Stefanova, V. Kmetov, G. Andreev, N. Kovachev and A. Canals, J. Anal. At. Spectrom., 2008, 23, 717–726 RSC.
- N. N. Meeravali and S. J. Jiang, J. Anal. At. Spectrom., 2008, 23, 1365–1371 RSC.
- M. Faraji, Y. Yamini and M. Rezaee, Talanta, 2010, 81, 831–836 CrossRef CAS.
- A. E. Karatapanis, Y. Fiamegos and C. D. Stalikas, Talanta, 2011, 84, 834–839 CrossRef CAS.
- R. Q. Aucélio, R. M. de Souza, R. C. de Campos, N. Miekeley and C. L. P. da Silveira, Spectrochim. Acta, Part B, 2007, 62, 952–961 CrossRef.
- I. Lavilla, N. Cabaleiro, M. Costas, I. de la Calle and C. Bendicho, Talanta, 2009, 80, 109–116 CrossRef CAS.
- P. R. Aranda, P. H. Pacheco, R. A. Olsina, L. D. Martinez and R. A. Gil, J. Anal. At. Spectrom., 2009, 24, 1441–1445 RSC.
- R. J. Cassella, D. M. Brum, C. E. R. de Paula and C. F. Lima, J. Anal. At. Spectrom., 2010, 25, 1704–1711 RSC.
- P. Tundo, P. Anastas, D. StC. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff and W. Tumas, Pure Appl. Chem., 2000, 72, 1207–1228 CrossRef CAS.
- E. M. Martinis, P. Berton, R. P. Monasterio and R. G. Wuilloud, TrAC, Trends Anal. Chem., 2010, 29, 1184–1201 CrossRef CAS.
- S. Jafarvand and F. Shemirani, Microchim. Acta, 2011, 173, 353–359 CrossRef CAS.
- C. Erkey, J. Supercrit. Fluids, 2000, 17, 259–287 CrossRef CAS.
- L. Xia, B. Hu, Z. Jiang, Y. Wu and Y. Liang, Anal. Chem., 2004, 76, 2910–2915 CrossRef CAS.
- X. Jia, Y. Han, X. Liu, T. Duan and H. Chen, Spectrochim. Acta, Part B, 2011, 66, 88–92 CrossRef.
- P. T. Anastas, in Clean Solvents: Alternative Media for Chemical Reactions and Processing, ed. M.A. Abraham and L. Moens, American Chemical Society, Washington, DC, 2002, pp. 1–9 Search PubMed.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- http://www.rsc.org/suppdata/gc/c0/c0gc00918k/c0gc00918k.pdf, last accessed June 2012.
- P. Cava-Montesinos, E. Ródenas-Torralba, A. Morales-Rubio, M. L. Cervera and M. de la Guardia, Anal. Chim. Acta, 2004, 506, 145–153 CrossRef CAS.
- E. Ródenas-Torralba, P. Cava-Montesinos, A. Morales-Rubio, M. L. Cervera and M. de la Guardia, Anal. Bioanal. Chem., 2004, 379, 83–89 CrossRef.
- R. B. R. Mesquita and A. O. S. S. Rangel, Anal. Chim. Acta, 2009, 648, 7–22 CrossRef CAS.
- Y. L. Yu, Z. Du, M. L. Chen and J. H. Wang, J. Anal. At. Spectrom., 2007, 22, 800–806 RSC.
- Y. L. Yu, Y. Jiang, M. L. Chen and J. H. Wang, TrAC, Trends Anal. Chem., 2011, 30, 1649–1658 CrossRef CAS.
- L. Xia, X. Li, Y. Wu, B. Hu and R. Chen, Spectrochim. Acta, Part B, 2008, 63, 1290–1296 CrossRef.
- Y. Li, C. Zheng, Q. Ma, L. Wu, C. Hu and X. Hou, J. Anal. At. Spectrom., 2006, 21, 82–85 RSC.
- S. Z. Mohammadi, D. Afzali and Y. M. Baghelani, Anal. Chim. Acta, 2009, 653, 173–177 CrossRef CAS.
- H. Sereshti, V. Khojeh, M. Karimi and S. Samadi, Anal. Methods, 2012, 4, 236–241 RSC.
- S. Z. Mohammadi, D. Afzali, M. A. Taher and Y. M. Baghelani, Talanta, 2009, 80, 875–879 CrossRef CAS.
- K. Grudpan, S. K. Hartwell, S. Lapanantnoppakhuna and I. McKelvie, Anal. Methods, 2010, 2, 1651–1661 RSC.
- M. Tuzen, O. D. Uluozlu and M. Soylak, J. Hazard. Mater., 2007, 144, 549–555 CrossRef CAS.
- M. Tuzen, O. D. Uluozlu, I. Karaman and M. Soylak, J. Hazard. Mater., 2009, 169, 345–350 CrossRef CAS.
- C. Zhang, Y. Li, X. Y. Cui, Y. Jiang and X. P. Yan, J. Anal. At. Spectrom., 2008, 23, 1372–1377 RSC.
- C. Zeng, N. Zhou and J. Luo, J. Anal. At. Spectrom., 2012, 27, 120–125 RSC.
- P. Wu, L. He, C. Zheng, X. Hou and R. E. Sturgeon, J. Anal. At. Spectrom., 2010, 25, 1217–1246 RSC.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, Anal. Chem., 2003, 75, 2092–2099 CrossRef CAS.
- C. Zheng, Y. Li, Y. He, Q. Ma and X. Hou, J. Anal. At. Spectrom., 2005, 20, 746–750 RSC.
- Y. He, X. Hou, C. Zheng and R. E. Sturgeon, Anal. Bioanal. Chem., 2007, 388, 769–774 CrossRef CAS.
- M. A. Vieira, A. S. Ribeiro, A. J. Curtius and R. E. Sturgeon, Anal. Bioanal. Chem., 2007, 388, 837–847 CrossRef CAS.
- C. Zheng, R. E. Sturgeon and X. Hou, J. Anal. At. Spectrom., 2009, 24, 1452–1458 RSC.
- C. Han, C. Zheng, J. Wang, G. Cheng, Y. Lv and X. Hou, Anal. Bioanal. Chem., 2007, 388, 825–830 CrossRef CAS.
- S. Gil, I. Lavilla and C. Bendicho, Anal. Chem., 2006, 78, 6260–6264 CrossRef CAS.
- S. Gil, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2007, 62, 69–75 CrossRef.
- A. S. Ribeiro, M. A. Vieira, S. Willie and R. E. Sturgeon, Anal. Bioanal. Chem., 2007, 388, 849–857 CrossRef CAS.
- S. Gil, I. Lavilla and C. Bendicho, J. Anal. At. Spectrom., 2007, 22, 569–572 RSC.
- F. Laborda, E. Bolea and J. R. Castillo, Anal. Bioanal. Chem., 2007, 388, 743–751 CrossRef CAS.
- X. Wu, W. Yang, M. Liu, X. Hou and C. Zheng, J. Anal. At. Spectrom., 2011, 26, 1204–1209 RSC.
- Q. Wu, Z. Zhu, Z. Liu, H. Zheng, S. Hu and L. Li, J. Anal. At. Spectrom., 2012, 27, 496–500 RSC.
- J. L. Capelo-Martínez, P. Ximénez-Embún, Y. Madrid and C. Cámara, TrAC, Trends Anal. Chem., 2004, 23, 331–340 CrossRef.
- F. Fernández-Costas, I. Lavilla and C. Bendicho, Spectrosc. Lett., 2006, 39, 713–725 CrossRef.
- J. L. Capelo, I. Lavilla and C. Bendicho, Anal. Chem., 2000, 72, 4979–4984 CrossRef CAS.
- J. L. Capelo, I. Lavilla and C. Bendicho, Anal. Chem., 2001, 73, 3732–3736 CrossRef CAS.
- J. L. Capelo, C. Maduro and A. M. Mota, J. Anal. At. Spectrom., 2004, 19, 414–416 RSC.
- T. Nakazato and H. Tao, Anal. Chem., 2006, 78, 1665–1672 CrossRef CAS.
- L. Abrankó, L. Yang, R. E. Sturgeon, P. Fodor and Z. Mester, J. Anal. At. Spectrom., 2004, 19, 1098–1103 RSC.
- R. Peñalver, N. Campillo and M. Hernández-Córdoba, Talanta, 2011, 87, 268–275 CrossRef.
- M. S. Fragueiro, F. Alava-Moreno, I. Lavilla and C. Bendicho, J. Anal. At. Spectrom., 2000, 15, 705–709 RSC.
- S. Fragueiro, I. Lavilla and C. Bendicho, J. Anal. At. Spectrom., 2004, 19, 250–254 RSC.
- C. Dietz, T. Pérez-Corona, Y. Madrid-Albarrán and C. Cámara, J. Anal. At. Spectrom., 2003, 18, 467–473 RSC.
- P. Hashemi and A. Rahimi, Spectrochim. Acta, Part B, 2007, 62, 423–428 CrossRef.
- A. Zierhut, K. Leopold, L. Harwardt, P. Worsfold and M. Schuster, J. Anal. At. Spectrom., 2009, 24, 767–774 RSC.
- V. Romero, I. Costas-Mora, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2011, 66, 156–162 CrossRef.
- C. Pons, R. Forteza, A. O. S. S. Rangel and V. Cerdà, TrAC, Trends Anal. Chem., 2006, 25, 583–588 CrossRef CAS.
- A. Economou, TrAC, Trends Anal. Chem., 2005, 24, 416–425 CrossRef CAS.
- W. C. Tseng, P. H. Chen, T. S. Tsay, B. H. Chen and Y. L. Huang, Anal. Chim. Acta, 2006, 576, 2–8 CrossRef CAS.
- J. Ruzicka and E. H. Hansen, Anal. Chim. Acta, 1975, 78, 145–157 CrossRef CAS.
- M. Tokeshi and T. Kitamori, in Advances in Flow Analysis, ed. M. Trojanowicz, Wiley VCH, Weinheim, 2008, pp. 149–165 Search PubMed.
- R. Pereiro, in Flow Analysis with Atomic Spectrometric Detectors, ed. A. Sanz-Medel, Elsevier, Amsterdam, 1999, pp. 34–61 Search PubMed.
- L. J. Shao, W. E. Gan, W. B. Zhang and Q. D Su, J. Anal. At. Spectrom., 2005, 20, 1296–1298 RSC.
- G. Vermeir, C. Vandecasteele and R. Dams, Anal. Chim. Acta, 1991, 242, 203–208 CrossRef CAS.
- S. Armenta and M. de la Guardia, in Challenges in Green Analytical Chemistry, ed. M. de la Guardia and S. Garrigues, Royal Society of Chemistry, Cambridge 2011, pp. 286–298 Search PubMed.
- T. Taniai, A. Sakuragawa and A. Uzawa, ISIJ Int., 2004, 44, 1852–1858 CrossRef CAS.
- G. Tao and Z. Fang, Spectrochim. Acta, Part B, 1995, 50, 1747–1755 CrossRef.
- R. M. Crespón-Romero and M. C. Yebra-Biurrun, Anal. Chim. Acta, 2008, 609, 184–191 CrossRef.
- C. Cui, M. He and B. Hu, J. Hazard. Mater., 2011, 187, 379–385 CrossRef CAS.
- A. Maquieira, H. A. M. Elmahadi and R. Puchades, Anal. Chem., 1994, 66, 3632–3638 CrossRef CAS.
- P. Giusti, Y. N. Ordoñez, C. P. Lienemann, D. Schaumlöffel, B. Bouyssiere and R. Łobiński, J. Anal. At. Spectrom., 2007, 22, 88–92 RSC.
- N. Homazava, A. Ulrich and U. Krähenbühl, Spectrochim. Acta, Part B, 2008, 63, 777–783 CrossRef.
- N. Homazava, T. Suter, P. Schmutz, S. Toggweiler, A. Grimberg, U. Krähenbühl and A. Ulrich, J. Anal. At. Spectrom., 2009, 24, 1161–1169 RSC.
- Y. Takasaki, M. Watanabe, H. Yukawa, A. Sabarudin, K. Inagaki, N. Kaji, Y. Okamoto, M. Tokeshi, Y. Miyamoto, H. Noguchi, T. Umemura, S. Hayashi, Y. Baba and H. Haraguchi, Anal. Chem., 2011, 83, 8552–8558 CrossRef.
- J. Ruzicka, G. D. Marshall and G. D. Christian, Anal. Chem., 1990, 62, 1861–1866 CrossRef CAS.
- M. Miró, V. Cerdá and J. M. Estela, TrAC, Trends Anal. Chem., 2002, 21, 199–210 CrossRef.
- W. E. Doering, R. R. James and R. T. Echols, Fresenius' J. Anal. Chem., 2000, 368, 475–479 CrossRef CAS.
- G. Tao, S. N. Willie and R. E. Sturgeon, Analyst, 1998, 123, 1215–1218 RSC.
- R. C. D. Costa and A. N. Araújo, Anal. Chim. Acta, 2001, 438, 227–233 CrossRef CAS.
- A. N. Anthemidis, Talanta, 2008, 77, 541–545 CrossRef CAS.
- A. N. Anthemidis and K. I. G. Ioannou, Anal. Chim. Acta, 2010, 668, 35–40 CrossRef CAS.
- J. Wang and E. H. Hansen, J. Anal. At. Spectrom., 2002, 17, 248–252 RSC.
- B. F. Reis, M. F. Giné, E. A. G. Zagatto, J. L. F. C. Lima and R. A. S. Lapa, Anal. Chim. Acta, 1994, 293, 129–138 CrossRef CAS.
- V. Cerdá, R. Forteza and J. M. Estela, Anal. Chim. Acta, 2007, 600, 35–45 CrossRef.
- M. Pistón and M. Knochen, Int. J. Anal. Chem., 2012 DOI:10.1155/2012/918292.
- L. O. Leal, N. V. Semenova, R. Forteza and V. Cerdá, Talanta, 2004, 64, 1335–1342 CrossRef CAS.
- Y. Fajardo, E. Gómez, F. Garcias, V. Cerdá and M. Casas, Anal. Chim. Acta, 2005, 539, 189–194 CrossRef CAS.
- R. A. S. Lapa, J. L. F. C. Lima, B. F. Reis, J. L. M. Santos and E. A. G. Zagatto, Anal. Chim. Acta, 2002, 466, 125–132 CrossRef CAS.
- C. M. P. V. Lopes, A. A. Almeida, J. L. M. Santos and J. L. F. C. Lima, Anal. Chim. Acta, 2006, 555, 370–376 CrossRef CAS.
- C. Pérez-Sirvent, M. J. Martínez-Sánchez, M. García-Lorenzo, I. López-García and M. Hernández-Córdoba, Anal. Bioanal. Chem., 2007, 388, 495–498 CrossRef.
- I. López-García, J. Arroyo-Cortez and M. Hernández-Córdoba, Talanta, 2008, 75, 480–485 CrossRef.
- J. Ruzicka, Analyst, 2000, 125, 1053–1060 RSC.
- Y. L. Yu, Z. Du and J. H. Wang, J. Anal. At. Spectrom., 2007, 22, 650–656 RSC.
- P. Ampan, J. Ruzicka, R. Atallah, G. D. Christian, J. Jakmunee and K. Grudpan, Anal. Chim. Acta, 2003, 499, 167–172 CrossRef CAS.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, 1–6, 244–248 CrossRef.
- T. Vilkner, D. Janasek and A. Manz, Anal. Chem., 2004, 76, 3373–3386 CrossRef CAS.
- R. D. Johnson, V. G. Gavalas, S. Daunert and L. G. Bachas, Anal. Chim. Acta, 2008, 613, 20–30 CrossRef CAS.
- Q. J. Song, G. M. Greenway and T. McGreedy, J. Anal. At. Spectrom., 2004, 19, 883–887 RSC.
- G. Pearson and G. Greenway, J. Anal. At. Spectrom., 2007, 22, 657–662 RSC.
- H. Cheng, Z. Xu, J. Liu, X. Wang and X. Yin, J. Anal. At. Spectrom., 2012, 27, 346–353 RSC.
- J. P. Lafleur and E. D. Salin, J. Anal. At. Spectrom., 2009, 24, 1511–1516 RSC.
- B. Chen, S. Heng, H. Peng, B. Hu, X. Yu, Z. Zhang, D. Pang, X. Yue and Y. Zhu, J. Anal. At. Spectrom., 2010, 25, 1931–1938 RSC.
- B. Chen, B. Hu, P. Jiang, M. He, H. Peng and X. Zhang, Analyst, 2011, 136, 3934–3942 RSC.
- C. Bendicho and M. T. C. de Loos-Vollebregt, J. Anal. At. Spectrom., 1991, 6, 353–374 RSC.
- A. Martin-Esteban and B. Slowikowski, Crit. Rev. Anal. Chem., 2003, 33, 43–55 CrossRef CAS.
- M. G. R. Vale, N. Oleszczuk and W. N. L. dos Santos, Appl. Spectrosc. Rev., 2006, 41, 377–400 CrossRef CAS.
- B. Welz, M. G. R. Vale, D. L. G. Borges and U. Heitmann, Anal. Bioanal. Chem., 2007, 389, 2085–2095 CrossRef CAS.
- M. Resano, F. Vanhaecke and M. T. C. de Loos-Vollebregt, J. Anal. At. Spectrom., 2008, 23, 1450–1475 RSC.
- M. Aramendía, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2009, 648, 23–44 CrossRef.
- Z. Zaide, Z. Kaizhong, H. Xiandeng and L. Hong, Appl. Spectrosc. Rev., 2005, 40, 165–184 CrossRef.
- F. L. King, J. Teng and R. E. Steiner, J. Mass Spectrom., 1995, 30, 1061–1075 CrossRef CAS.
- D. Günther, S. E. Jackson and H. P. Longerich, Spectrochim. Acta, Part B, 1999, 54, 381–409 CrossRef.
- J. D. Winefordner, I. B. Gornushkin, D. Pappas, O. I. Matveev and B. W. Smith, J. Anal. At. Spectrom., 2000, 15, 1161–1189 RSC.
- J. M. Vadillo and J. J. Laserna, Spectrochim. Acta, Part B, 2004, 59, 147–161 CrossRef.
- J. D. Winefordner, I. B. Gornushkin, T. Correll, E. Gibb, B. W. Smith and N. Omenetto, J. Anal. At. Spectrom., 2004, 19, 1061–1083 RSC.
- D. Günther and B. Hattendorf, TrAC, Trends Anal. Chem., 2005, 24, 255–265 CrossRef.
- C. Pickhardt, H. J. Dietze and J. S. Becker, Int. J. Mass Spectrom., 2005, 242, 273–280 CrossRef CAS.
- B. Fernandez, F. Claverie, C. Pecheyran and O. F. X. Donard, TrAC, Trends Anal. Chem., 2007, 26, 951–966 CrossRef CAS.
- J. Pisonero, B. Fernandez and D. Günther, J. Anal. At. Spectrom., 2009, 24, 1145–1160 RSC.
- B. Fernández, R. Pereiro and A. Sanz-Medel, Anal. Chim. Acta, 2010, 679, 7–16 CrossRef.
- K. Janssens, W. D. Nolf, G. V. D. Snickt, L. Vincze, B. Vekemans, R. Terzano and F. E. Brenker, TrAC Trends Anal. Chem., 2010, 29, 464–478 CrossRef CAS.
- U. E. A. Fittschen and G. Falkenberg, Spectrochim. Acta, Part B, 2011, 66, 567–580 CrossRef CAS.
- M. West, A. T. Ellis, P. J. Potts, C. Streli, C. Vanhoof, D. Wergrzynek and P. Wobrauschek, J. Anal. At. Spectrom., 2011, 26, 1919–1963 RSC.
- M. Resano, E. García-Ruiz, M. A. Belarra, F. Vanhaecke and K. S. McIntosh, TrAC, Trends Anal. Chem., 2007, 26, 385–395 CrossRef CAS.
- B. Bekhoff, B. Kanngießer, N. Langhoff, R. Wedell and H. Wolff, Handbook of Practical X-Ray Fluorescence Analysis, Springer, New York, 2006 Search PubMed.
- M. A. Zezzi Arruda, Trends in Sample Preparation, Nova Science Publishers, New York, 2007 Search PubMed.
- L. Qi, J. Gao, X. Huang, J. Hu, M. F. Zhou and H. Zhong, J. Anal. At. Spectrom., 2011, 26, 1900–1904 RSC.
- S. F. Boulyga and K. G. Heumann, Anal. Bioanal. Chem., 2005, 383, 442–447 CrossRef CAS.
- J. D. Kerber, At. Absorpt. Newsl., 1971, 10, 104–105 CAS.
- www.analytik-jena.de/en/solid-AA/Systems-4400, last accessed June 2012.
- D. E. Nixon, V. A. Fassel and R. N. Kniseley, Anal. Chem., 1974, 46, 210–213 CrossRef CAS.
- www.spectral-systems.de/english/autosamp.htm, last accessed June 2012.
- M. A. Belarra, M. Resano, F. Vanhaecke and L. Moens, TrAC, Trends Anal. Chem., 2002, 21, 828–839 CrossRef CAS.
- H. M. Ortner, E. Bulskab, U. Rohra, G. Schlemmerc, S. Weinbruchd and B. Welz, Spectrochim. Acta, Part B, 2002, 57, 1835–1853 CrossRef.
- V. N. Oreshkin and G. I. Tsizin, J. Anal. Chem., 2011, 66, 951–954 CrossRef CAS.
- X. Hou and B. T. Jones, Spectrochim. Acta, Part B, 2002, 57, 659–688 CrossRef.
- M. L. Lin and S. J. Jiang, J. Anal. At. Spectrom., 2011, 26, 1813–1818 RSC.
- M. A. Amberger and J. A. C. Broekaert, J. Anal. At. Spectrom., 2010, 25, 1308–1315 RSC.
- G. Xiang, B. Hu and Z. Jiang, J. Mass Spectrom., 2006, 41, 1378–1385 CrossRef CAS.
- V. Thomsen and J. L. Spencer, Spectroscopy, 2011, 26, 18–21 CAS.
- V. Thomsen, Spectroscopy, 2010, 25, 44–45 Search PubMed.
- www.angstrom-inc.com/spectrometers.html, last accessed June 2012.
- P. D. Pant, PCT Int. Appl., WO 2011114340 A2 20110922, 2011 Search PubMed.
- F. Vincent, J. P. Sola and G. Wuethrich, Brit. UK Pat. Appl., GB 2466198 A 20100616, 2010.
- https://www.thermo.com/eThermo/CMA/PDFs/./productPDF_57273.PDF, last accessed June 2012.
- A. F. Kiera, S. Schmidt-Lehr, M. Song, N. Bings and J. A. C. Broekaert, Spectrochim. Acta, Part B, 2008, 63, 287–292 CrossRef.
- G. Buchbinder, N. Verblyudov and A. Clavering, Spectroscopy, 2009 Search PubMed.
- J. Roy and L. Neufeld, Spectroscopy, 2004, 19, 16–28 Search PubMed.
- T. D. F. Leite, R. Escalfoni Jr., T. C. O. da Fonseca and N. Miekeley, Spectrochim. Acta, Part B, 2011, 66, 314–320 CrossRef CAS.
- I. Konz, B. Fernández, M. L. Fernández, R. Pereiro and A. Sanz-Medel, Anal. Bioanal. Chem., 2012, 403, 2113–2125 CrossRef CAS.
- J. S. Becker, Int. J. Mass Spectrom., 2010, 289, 65–75 CrossRef CAS.
- F. J. Fortes and J. J. Laserna, Spectrochim. Acta, Part B, 2010, 65, 975–990 CrossRef.
- A. Ciucci, V. Palleschi, S. Rastelli, A. Salvetti, D. P. Singh and E. Tognoni, Laser Part. Beams, 1999, 17, 793–797 CrossRef CAS.
- S. Palanco, A. Alises, J. Cuñat, J. Baena and J. J. Laserna, J. Anal. At. Spectrom., 2003, 18, 933–938 RSC.
- T. Nelis and J. Pallosi, Appl. Spectrosc. Rev., 2006, 41, 227–258 CrossRef CAS.
- R. Pereiro, A. Solá-Vázquez, L. Lobo, J. Pisonero, N. Bordel, J. M. Costa and A. Sanz-Medel, Spectrochim. Acta, Part B, 2011, 66, 399–412 CrossRef CAS.
- M. J. Heintz and G. M. Hieftje, Spectrochim. Acta, Part B, 1995, 50, 1109–1124 CrossRef.
- G. P. Jackson, R. G. Haire and C. Duckworth, J. Anal. At. Spectrom., 2003, 18, 665–669 RSC.
- L. Gao, Q. Song, R. J. Noll, J. Duncan, R. G. Cooks and Z. Ouyang, J. Mass Spectrom., 2007, 42, 675–680 CrossRef CAS.
- W. C. Davis, B. C. Knippel, J. E. Cooper, B. K. Spraul, J. K. Rice, D. W. Smith, Jr and R. K. Marcus, Anal. Chem., 2003, 75, 2243–2250 CrossRef CAS.
- T. M. Brewer, W. Clay Davis and R. K. Marcus, J. Anal. At. Spectrom., 2006, 21, 126–133 RSC.
- D. Fliegel and D. Günther, Spectrochim. Acta, Part B, 2009, 64, 399–407 CrossRef.
- M. Tarik and D. Günther, J. Anal. At. Spectrom., 2010, 25, 1416–1423 RSC.
- E. Marguí, M. Hidalgo and I. Queralt, Spectrochim. Acta, Part B, 2005, 60, 1363–1372 CrossRef.
- G. Custo, S. Boeykens, L. Dawidowski, L. Fox, D. Gomez, F. Luna and C. Vazquez, Anal. Sci., 2005, 21, 751–756 CrossRef CAS.
- N. Bukowiecki, P. Lienemann, C. N. Zwicky, M. Furger, A. Richard, G. Falkenberg, K. Rickers, D. Grolimund, C. Borca, M. Hill, R. Gehrig and U. Baltensperger, Spectrochim. Acta, Part B, 2008, 63, 929–938 CrossRef.
- I. de la Calle, M. Costas, N. Cabaleiro, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2012, 67, 43–49 CrossRef CAS.
- R. E. A. Jiménez, Spectrochim. Acta, Part B, 2001, 56, 2331–2336 CrossRef.
- http://ecfr.gpoaccess.gov/cgi/t/text/text-idx?c=ecfr&sid=e29fdcb9d258d1ed8c80f521ef9801a4&rgn=div6&view=text&node = 42:5.0.1.1.5.3&idno = 42, last accessed June 2012.
- http://epswww.unm.edu/xrd/xrd_haz.pdf, last accessed June 2012.
- Environmental Protection Agency, USA, Environmental Technology Verification Report: Field Portable X-Ray Fluorescence Analyzer, Spectrace TN 9000 and TN Pb Field Portable X-Ray Fluorescence Analyzers, EPA/600/R-97/145, Washington DC, 1998 Search PubMed.
- J. Injuk, R. Van Grieken, A. Blank, L. Eksperiandova and V. Buhrke, in Handbook of Practical X-Ray Fluorescence Analysis, ed. B. Bekhoff, B. Kanngießer, N. Langhoff, R. Wedell and H. Wolff, Springer, New York, 2006, pp. 415–424 Search PubMed.
- R. B. Hallet and P. R. Kyle, Geostand. Newsl., 1993, 17, 127–133 CrossRef.
- S. Barrento, A. Marques, B. Teixeira, P. Vaz-Pires, M. L. Carvalho and M. L. Nunes, Food Chem., 2008, 111, 862–867 CrossRef CAS.
- M. F. Galluza, S. Vicente, M. Orduña and M. J. Ventura, X-Ray Spectrom., 2012, 41, 176–185 CrossRef.
- C. Vazquez, G. Custo, N. Barrio, J. Burúcua, S. Boeykens and F. Marte, Spectrochim. Acta, Part B, 2010, 65, 852–858 CrossRef.
- http://www.bruker-axs.com/artax.html, last accessed June 2012.
- G. Buzanich, P. Wobrauscheck, C. Streli, A. Markowicz, D. Wegrzynek, E. Chine-Cano and S. Bamford, Spectrochim. Acta, Part B, 2007, 62, 1252–1256 CrossRef.
- A. von Bohlen, Spectrochim. Acta, Part B, 2009, 64, 821–832 CrossRef.
- http://www.bruker-axs.com/s2picofox.html, last accessed June 2012\.
- http://www.bruker-axs.com/traceriiiv.html, last accessed June 2012.
- N. Forster, P. Grave, N. Vickery and L. Kealhofer, X-Ray Spectrom., 2011, 40, 389–398 CrossRef CAS.
- Y. Shibata, J. Suyama, M. Kitano and T. Nakamura, X-Ray Spectrom., 2008, 38, 410–416 CrossRef.
- E. Marguí, C. Fontàs, A. Buendía, M. Hidalgo and I. Queralt, J. Anal. At. Spectrom., 2009, 24, 1253–1257 RSC.
- A. A. Shaltout, B. Welz and M. A. Ibrahim, Microchem. J., 2011, 99, 356–363 CrossRef CAS.
- A. J. Aller, Espectroscopia atómica electrotérmica analítica, Secretariado de Publicaciones y Medios Audiovisuales de la Universidad de León, León, 2003 Search PubMed.
- H. E. Taylor, Inductively Coupled Plasma-Mass Spectrometry Practices and Techniques, Academic Press, San Diego, 2001 Search PubMed.
- S. E. Maestre, J. L. Todolí and J. M. Mermet, Anal. Bioanal. Chem., 2004, 379, 888–899 CrossRef CAS.
- J. L. Todoli, V. Hernandis, A. Canals and J. M. Mermet, J. Anal. At. Spectrom., 1999, 14, 1289–1295 RSC.
- F. Vanhaecke, M. V. Holderbeke, L. Moens and R. Dams, J. Anal. At. Spectrom., 1996, 11, 543–548 RSC.
- S. F. Boulyga and J. S. Becker, Fresenius' J. Anal. Chem., 2001, 370, 612–617 CrossRef CAS.
- J. A. Koropchak and M. Veber, Crit. Rev. Anal. Chem., 1992, 23, 113–141 CrossRef CAS.
- X. Zhang, D. Chen, R. Marquardt and J. A. Koropchak, Microchem. J., 2000, 66, 17–53 CrossRef CAS.
- N. Praphairaksit, D. R. Wiederin and R. S. Houk, Spectrochim. Acta, Part B, 2000, 55, 1279–1293 CrossRef.
- J. A. C. Broekaert and V. Siemens, Anal. Bioanal. Chem., 2004, 380, 185–189 CrossRef CAS.
- B. Özmen, F. M. Matysik, N. H. Bings and J. A. C. Broekaert, Spectrochim. Acta, Part B, 2004, 59, 941–950 CrossRef.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, Anal. Chem., 2004, 76, 2401–2405 CrossRef CAS.
- F. G. Bessoth, O. P. Naji, J. C. T. Eijkel and A. Manz, J. Anal. At. Spectrom., 2002, 17, 794–799 RSC.
- J. A. C. Broekaert and U. Engel, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyer, John Wiley & Sons, Chichester, 2000, pp. 9613–9667 Search PubMed.
- J. Cai, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyer, John Wiley & Sons, Chichester, 2000, pp. 2270–2292 Search PubMed.
- J. Dědina, Spectrochim. Acta, Part B, 2007, 62, 846–872 CrossRef.
- J. Dědina and T. Matoušek, J. Anal. At. Spectrom., 2000, 15, 301–304 RSC.
- W. T. Corns, P. B. Stockwell, L. E. Ebdon and S. J. Hill, J. Anal. At. Spectrom., 1993, 8, 71–77 RSC.
- Y. Ogra, Biomed. Res. Trace Elem., 2008, 19, 34–42 CAS.
- R. Lobinski, D. Schaumlöffel and J. Szpunar, Mass Spectrom. Rev., 2006, 25, 255–289 CrossRef CAS.
- Y. C. Sun, Y. C. Chang and C. K. Su, Anal. Chem., 2006, 78, 2640–2645 CrossRef CAS.
- H. Matusiewicz and R. E. Sturgeon, Spectrochim. Acta, Part B, 1996, 51, 377–397 CrossRef.
- O. Y. Ataman, Spectrochim. Acta, Part B, 2008, 63, 825–834 CrossRef.
- B. Docěkal, S. Gucer and A. Selecká, Spectrochim. Acta, Part B, 2004, 59, 487–495 CrossRef.
- X. D. Hou, K. E. Levine, A. Salido, B. T. Jones, M. Ezer, S. Elwood and J. B. Simeonsson, Anal. Sci., 2001, 17, 175–180 CrossRef CAS.
- B. Welz and M. Sperling, Atomic Absorption Spectrometry, Wiley-VCH, Weinheim, 1999 Search PubMed.
- B. Welz, S. Morés, E. Casarek, M. G. R. Vale, M. Okruss and H. Becker-Ross, Appl. Spectrosc. Rev., 2010, 45, 327–354 CrossRef CAS.
- J. Namieśnik, J. Sep. Sci., 2001, 24, 151–153 CrossRef.
- E. Marengo, M. Aceto, E. Robotti, M. Oddone and M. Bobba, Talanta, 2008, 76, 1224–1232 CrossRef CAS.
- Z. Jokai and P. Fodor, J. Anal. At. Spectrom., 2009, 24, 1229–1236 RSC.
- G. R. Lloyd, S. Ahmad, M. Wasim and R. G. Brereton, Anal. Chim. Acta, 2009, 649, 33–42 CrossRef CAS.
- K. Schaefer, J. W. Einax, V. Simeonov and S. Tsakovski, Anal. Bioanal. Chem., 2010, 396, 2675–2683 CrossRef CAS.
- M. J. Latorre, R. Peña, S. García and C. Herrero, Analyst, 2000, 125, 307–312 RSC.
- K. Ariyama, H. Horita and A. Yasui, Anal. Sci., 2004, 20, 871–877 CrossRef CAS.
- R. Iglesias Rodríguez, M. Fernández Delgado, J. Barciela García, R. M. Peña Crecente, S. García Martín and C. Herrero Latorre, Anal. Bioanal. Chem., 2010, 397, 2603–2614 CrossRef.
- A. P. Fernandes, M. C. Santos, S. G. Lemos, M. M. C. Ferreira, A. R. A. Nogueira and J. A. Nobrega, Spectrochim. Acta, Part B, 2005, 60, 717–724 CrossRef.
- M. Costas-Rodriguez, I. Lavilla and C. Bendicho, Anal. Chim. Acta, 2010, 664, 121–128 CrossRef CAS.
- M. Otto, Chemometrics. Statistical and Computer Application in Analytical Chemistry, Wiley-VCH, Weinheim, 2007 Search PubMed.
- X. T. Morer, L. Gonzalez-Sabaté, L. Fernandez-Ruano and M. P. Gomez-Carracedo, in Basic Chemometric Techniques in Atomic Spectroscopy, ed. J. M. Andrade, Royal Society of Chemistry, Cambridge, 2009, pp. 51–141 Search PubMed.
- M. Cocchi, C. Durante, A. Marchetti, M. Li Vigni, C. Baschieri, L. Bertacchini, S. Sighinolfi, L. Tassi and S. Totaro, in Microwaves: Theoretical Aspects and Practical Applications in Chemistry, ed. A. Marcheti and S. Sighinolfi, Transworld Research Network, T. C., Kerala, 2011, pp. 203–226 Search PubMed.
- J. M. O. Souza and C. R. T. Tarley, Int. J. Environ. Anal. Chem., 2009, 89, 489–502 CrossRef CAS.
- E. Kendüzler, S. Baytak, Ö. Yalçinkaya and A. R. Türker, Can. J. Anal. Sci. Spectrosc., 2007, 52, 91–100 Search PubMed.
- S. L. C. Ferreira, S. M. Macedo, D. C. dos Santos, R. M. de Jesus, W. N. L. dos Santos, A. F. S. Queiroz and J. B. de Andrade, J. Anal. At. Spectrom., 2011, 26, 1887–1891 RSC.
- F. Bianchi and M. Careri, Curr. Anal. Chem., 2008, 4, 142–151 CrossRef CAS.
- D. B. Hibbert and N. Armstrong, Chemom. Intell. Lab. Syst., 2009, 97, 211–220 CrossRef CAS.
- M. A. Bezerra, R. E. Santelli, E. P. Oliveira, L. S. Villar and L. A. Escaleira, Talanta, 2008, 76, 965–977 CrossRef CAS.
- J. M. Andrade, M. J. Cal-Prieto, M. P. Gómez-Carracedo, A. Carlosena and D. Prada, J. Anal. At. Spectrom., 2008, 23, 15–28 RSC.
- C. Y. Fu, L. I. Petrich, P. F. Daley, A. K. Burnham and K. Alan, Anal. Chem., 2005, 77, 4051–4057 CrossRef CAS.
- T. Pettke, C. A. Heinrich, A. C. Ciocan and D. Gunther, J. Anal. At. Spectrom., 2000, 15, 1149–1155 RSC.
- T. Nomizu, H. Hayashi, N. Hoshino, T. Tanaka, H. Kawaguchi, K. Kitagawa, K. Kuniyuki and S. Kaneco, J. Anal. At. Spectrom., 2002, 17, 592–595 RSC.
- D. Q. Zhang, C. M. Li, L. L. Yang and H. W. Sun, Anal. Chim. Acta, 2000, 405, 185–190 CrossRef CAS.
- I. Lopez-García, M. Sánchez-Merlos, P. Viñas and M. Hernández-Cordoba, J. Anal. At. Spectrom., 2001, 16, 1185–1189 RSC.
- M. Felipe-Sotelo, J. M. Andrade, A. Carlosena and D. Prada, Anal. Chem., 2003, 75, 5254–5261 CrossRef CAS.
- X. Ma and Z. Zhang, J. Anal. At. Spectrom., 2004, 19, 738–742 RSC.
- E. S. Chaves, S. Compernolle, M. Aramendia, E. Javierre, E. Tresaco, M. T. C. de Loos-Vollebregt, A. J. Curtius and F. Vanhaecke, J. Anal. At. Spectrom., 2011, 26, 1833–1840 RSC.
- S. Prikler, D. Pick and J. W. Einax, Anal. Bioanal. Chem., 2012, 403, 1109–1116 CrossRef CAS.
- J. M. Cottle, M. S. A. Horstwood and R. R. Parrish, J. Anal. At. Spectrom., 2009, 24, 1355–1363 RSC.
- H. Yihua, T. Li, W. Xi, H. Xiandeng and L. Yong-Ill, Appl. Spectrosc. Rev., 2007, 42, 119–138 CrossRef.
- S. Lahiri and M. J. Stillman, Anal. Chem., 1992, 64, 283A–291A CAS.
- H. Ying, P. Yang, X. Wang and B. Huang, Spectrochim. Acta, Part B, 1996, 51, 877–886 CrossRef.
- Z. Zhang and X. Ma, Curr. Top. Anal. Chem., 2002, 3, 105–123 CAS.
- D. P. Webb and E. D. Salin, J. Anal. At. Spectrom., 1989, 4, 793–796 RSC.
- P. Yang, H. Ying, X. Wang and B. Huang, Spectrochim. Acta, Part B, 1996, 51, 889–896 CrossRef.
- C. Sartoros, J. F. Alary, E. D. Salina and J. M. Mermet, Spectrochim. Acta, Part B, 1997, 52, 1923–1927 CrossRef.
- F. A. Settle Jr, B. I. Diamondstone, H. M. Kingston and M. A. Pleva, J. Chem. Inf. Model., 1989, 29, 11–17 CrossRef.
- F. A. Settle Jr, P. J. Walter, H. M. Kingston, M. A. Pleva, T. Snider and W. Boute, J. Chem. Inf. Model., 1992, 32, 349–353 CrossRef.
- F. Klages and G. Wuensch, J. Prakt. Chem./Chem.-Ztg., 1996, 338, 16–22 CrossRef CAS.
- G. Wuensch and F. Klages, J. Prakt. Chem./Chem.-Ztg., 1996, 338, 593–597 CrossRef CAS.
- C. C. Huang, M. H. Yang and T. S. Shih, Anal. Chem., 1997, 69, 3930–3939 CrossRef CAS.
- H. Ying, J. Murphy, J. W. Trompa, J. M. Mermet and E. D. Salina, Spectrochim. Acta, Part B, 2000, 55, 311–326 CrossRef.
- B. van den Bogaert, J. B. W. Morsink and H. C. Smit, Anal. Chim. Acta, 1992, 270, 107–113 CrossRef.
- B. van den Bogaert, J. B. W. Morsink and H. C. Smit, Anal. Chim. Acta, 1992, 270, 115–130 CrossRef.
- D. Wienke, T. Vijn and L. Buydens, Anal. Chem., 1994, 66, 841–849 CrossRef CAS.
- C. Pons, M. Miró, E. Becerra, J. M. Estela and V. Cerda, Talanta, 2004, 62, 887–895 CrossRef CAS.
- M. I. G. S. Almeida, M. A. Segundo, J. L. F. C. Lima and A. O. S. S. Rangel, J. Anal. At. Spectrom., 2009, 24, 340–346 RSC.
| This journal is © The Royal Society of Chemistry 2012 |