A beginner's guide to uranium chronometry in nuclear forensics and safeguards
F. E.
Stanley
*
Los Alamos National Laboratory, TA-3, Bldg 29, Diamond Dr., Los Alamos, NM 87545, USA. E-mail: floyd@lanl.gov; Tel: +1 505-606-0287
First published on 11th September 2012
Abstract
Nuclear forensics and international safeguards are rising in importance in the face of increasing concerns over the illicit production and trafficking of nuclear materials worldwide. One of the most valuable approaches in such efforts is the chronometric investigation (“age-dating”) of collected/interdicted materials; well-designed chronometric schemes may simultaneously provide insight into a sample's composition, enrichment history, and the time elapsed since last purification. Given the importance of this analytical approach in obtaining valuable material signatures, a thorough understanding of age-dating principles and special concerns will be of significant value to the rising generation of scientists interrogating nuclear materials collected around the world. In this work, the fundamental concepts of uranium chronometry are discussed, along with several unique concerns and recent applications associated with the use of age-dating approaches in this manner. As significant work remains to be done in these fields, several potential concepts for future development are also highlighted.
 F.E. Stanley | Dr. Floyd E. Stanley is a recent graduate of the Department of Chemistry at the University of Cincinnati, and received both his M.S. and B.S. at Furman University in Greenville, SC. Currently, Dr Stanley works as a mass spectrometrist within the Actinide Analytical Chemistry Group at Los Alamos National Laboratory in New Mexico. Research interests include the use of mass spectrometry approaches in the characterization and age-dating of various nuclear materials, as well as the development and application of novel mass spectrometry ionization sources of value in the measurement of trace analytes. |
1. Introduction
Nuclear forensics materials characterization has been defined as “the technical means by which nuclear materials, whether intercepted intact or retrieved from post-explosion debris, are characterized (as to composition, physical condition, age, provenance, history) and interpreted (as to provenance, industrial history, and implications for nuclear device design).”1 This work, along with safeguards materials research, is critical in current international undertakings to identify illicit materials trafficking, prevent/respond to terrorist activities, and enforce existing international treaties. Accomplishing these goals in a capable and timely fashion, however, tends to require a tremendous range of instrumental approaches and materials characterization strategies (e.g. separations, mass spectrometry, decay counting measurements, microscopy, etc.),2 as well as experts from various fields (e.g. chemistry, geology, physics, etc.). Hence, the preservation of unique knowledge and concepts is critical as a large percentage of the senior members of these communities exit the field.3Chronometric analysis of intercepted (e.g. stolen fuel pellets) or purposefully collected (e.g. cotton swipe samples from a nuclear facility) materials is arguably one of the most valuable methodologies in modern nuclear forensics and will be an essential skill in coming years. Astutely designed approaches can concurrently provide insight into a material's “age” (i.e. time elapsed since last purification), actinide concentrations, and relevant isotopic ratios/enrichment values. These signatures are invaluable in determining the origin, processing history, and intended purpose of a nuclear material and may exclude or incriminate potential participants in its diversion from legitimate use.
Throughout this work, the fundamental concepts and recent applications of uranium chronometry will be discussed; age-dating uranium is arguably more difficult than Pu, because of the larger half-lives involved, and the natural prevalence of many of the radionuclides of potential interest. An understanding of this information will be critical as the importance of nuclear forensics continues to rise throughout the world and a new generation of scientists enters this arena from various technical backgrounds. Presently much of this information: (1) contains various non-intuitive subtleties, (2) can be laborious to retrieve from previous literature or generate independently, and (3) would gain value from further detail in terms of current applications and challenges.
2. Uranium isotopic/chronometric details
Background
Nuclides linked through radioactive decay processes will predictably obey the laws of in-growth, or the Bateman equations4 for more complicated scenarios. Hence, the concentrations of linked species can be calculated at any time following last material purification (i.e. removal of daughter progeny) using minimal information (e.g. half-lives/decay activities and initial quantities).All isotopes of uranium undergo radioactive decay, typically via the ejection of an α-particle (i.e. a helium nucleus) to produce a thorium daughter; other species may be produced via beta (β−) particle emission, though these instances tend to be associated with less valuable chronometric pairs. A valuable example of the α-decay of a uranium isotope and its progeny is as follows:

| 234U:T1/2 = 2.46 × 105 (±0.006 × 105) years |
| 230Th:T1/2 = 7.54 × 104 (±0.03 × 104) years |
Assuming perfect initial purification, the provided information can be used to calculate the expected quantities of 234U and 230Th at some time following processing using the following equations:
| NTU-234 = N0U-234e−λU-234T | (1) |
 | (2) |
| NTU-234 = number of atoms of 234U at time T |
| N0U-234 = number of atoms of 234U at time T0 |
| NTTh-230 = number of atoms of 230Th at time T |
| N0Th-230 = number of atoms of 230Th at time T0 |


In reverse fashion, the experimental measurement of 234U (NTU-234) and 230Th (NTTh-230) in a material, whether obtained from interdiction, archival samples, or even accidentally discovered, can be used to calculate the time elapsed since last purification using the following equation:
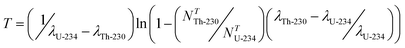 | (3) |
These concepts and equations can theoretically be extended to any uranium–daughter relationship through the proper interchange of variables. However, in practice, the use of many uranium chronometric relationships is hindered by factors such as: (1) overwhelming interferences, (2) exceptionally short lifetimes leading to limited windows of analytical opportunity, (3) exceptionally long lifetimes leading to ultra-trace progeny in-growth, etc.
Uranium chronometric relationships are discussed in greater detail below, along with information pertaining to various considerations and practical subtleties. In following sections, recent applications of these relationships will be highlighted and several special concerns in uranium chronometry are described.
U-238: low chronometric value
Uranium-238 (238.050788247 amu) is by far the most common isotope of natural uranium, with an isotopic abundance of ∼99.2745%.5 The parent of the 4n + 2 (division of mass number by 4 leaves a remainder of 2) “Uranium Series” decay chain, 238U is a long-lived species (T1/2: ∼ 4.47 × 109 years) that produces the much shorter-lived 234Th (T1/2: ∼ 24.10 days) via the emission of an α-particle (4198.3 keV).6 The relationship between 238U and 234Th as a function of time elapsed since material purification is illustrated in Fig. 1, assuming complete removal of thorium at T0. As shown, this system quickly approaches secular equilibrium† due to the high activity constant of 234Th and minimal change is observed in the 234Th/238U isotopic ratio after ∼150 days following purification;4 this provides a limited window of chronometric value and limits this age-dating approach to materials of less than about 5–6 months old. In addition to the limited window of use, this chronometric relationship is hindered by the natural occurrence of 238U and will likely produce biased age-dating results. Hence, the 234Th/238U chronometer is of minimal use in nuclear forensics and safeguards investigations. | ||
| Fig. 1 A plot of the relationship between the 234Th/238U as determined using standard decay equations. | ||
It is worth noting that uranium-238 decay eventually yields 234U according to the following scheme:

The 234U/238U ratio is not of chronometric value because the progeny species is retained during radiochemical purification, but this relationship is of use in terms of materials characterization (e.g. enrichment determination) as 234U abundance increases proportionally faster than 235U during the enrichment process. However, the full value of this relationship remains a point of contention in light of naturally high variations in the 234U/238U ratio.7
U-236: low chronometric value
Uranium-236 (236.045568006 amu) is the product of 235U (n,γ) reactions occurring both in reactor environments and in nature and may also be produced via the decay of 240Pu.8,9 Though frequently not accounted for, 236U was recently shown to be present naturally at abundances of ∼10−10% using accelerator mass spectrometry (AMS) analysis;10,11 an overview of AMS general principles is provided in recent work by Hellborg and Skog.12 A member of the 4n “Thorium Series” decay chain, 236U is a long-lived radionuclide (T1/2: ∼ 2.342 × 107 years) that produces 232Th, a separate long-lived species (T1/2: ∼ 1.4 × 1010 years), via the emission of an α-particle (4494.3 keV).6 The chronometric relationship between 236U and 232Th is illustrated in Fig. 2, assuming complete removal of thorium at T0 within a hypothetical material. As shown, the 232Th/236U ratio increases in a highly linear fashion (R2 = 0.9995) for millions of years following purification, theoretically providing a large window of analytical opportunity for age-dating. However, the 232Th/236U pair is of no chronometric value in light of the following considerations: (1) this relationship contains a radionuclide (i.e.232Th) that is naturally ubiquitous and capable of interfering with the accurate determination of an age; obtained results would be biased to appear much older than appropriate. (2) The parent species in the chronometer is long-lived, leading to minimal in-growth of the daughter following purification and challenging the limits of modern analytical instrumentation. | ||
| Fig. 2 A plot of the relationship between the 232Th/236U as determined using standard decay equations. | ||
While the 232Th/236U relationship is of no chronometric value, the detection of 236U is a significant occurrence in nuclear forensics and safeguards investigations. Uranium-236 is the product of neutron capture and 236U/238U ratios above ∼10−9 can serve as a critical indicator that a sample has been in the presence of significant neutron flux or plutonium materials.11,13 Hence, 236U is a key signature in determining the processing history associated with a material and the potential activities of previously involved parties.
U-235: chronometric value dependent on relationship of interest
Uranium-235 (235.043929918 amu) is the second most common isotope of natural uranium, with an abundance of ∼0.72%, and the primary fissile radionuclide of interest in the production of uranium-based nuclear power/weapons fuels of various enrichment.5 The parent of the 4n + 3 “Actinium Series” decay chain, 235U is a long-lived radionuclide (T1/2: ∼ 7.04 × 108 years) that produces the much shorter lived 231Th (T1/2: ∼ 25.52 hours) via the emission of an α-particle (4397.8 keV).6 The chronometric relationship between 235U and 231Th is illustrated in Fig. 3A, assuming complete removal of thorium at T0 within a hypothetical material. As shown, this system quickly approaches secular equilibrium due to the high activity value of the daughter species and maximum 231Th concentration is obtained within days. Indeed, 231Th is too short lived to allow this chronometer to be of any practical value.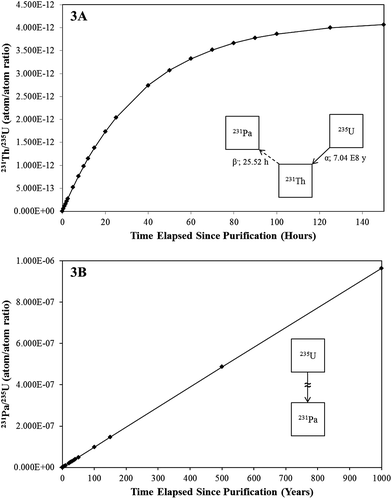 | ||
| Fig. 3 (A) A plot of the relationship between the 231Th/235U as determined using standard decay equations. (B) A plot of the relationship between the 231Pa/235U as determined using standard decay equations. | ||
As 235U and its progeny are less common in nature, relative to 238U for example, the low value of the 231Th/235U chronometer may be circumvented by extending the age-dating approach beyond the simple parent–daughter relationship to include additional generations of decay progeny. In general, if daughter species are very short-lived, then grand-daughter isotopes may serve as highly valuable alternatives for age-dating. For example, 235U-based investigations can be extended to the long-lived 231Pa granddaughter (T1/2: ∼3.276 × 104 years) produced by the β− decay of 231Th.14 The chronometric relationship between 235U and the granddaughter 231Pa is illustrated in Fig. 3B, which shows a highly linear increase in the 231Pa/235U for hundreds of years following purification. Measuring 231Pa in-growth may challenge some modern analytical techniques and methods with high chemical recovery/decontamination15 are needed, especially for young materials or low enrichment materials. Such measurements are also complicated by the lack of a long-lived Pa spike of value in quantitative analysis. However, despite these concerns, this approach has been shown to represent a viable option for a variety of materials.14
While arguably underutilized in modern nuclear forensics and safeguards investigations, the general strategy of exploiting extended chronometric pairs offers highly desirable features. These include: (1) increased confidence in determined age values via the measurement of additional chronometric relationships. (2) A means of detecting efforts to alter radionuclide ratios within a material (e.g. purposeful alteration of a critical ratio that produces anomalous results when compared with separate relationships); this is sometimes called “spoof detection”.16,17 (3) The availability of multiple age-dating windows of different scales (i.e. months to years).
U-234: high chronometric value
Uranium-234 (234.040952088 amu) is a natural uranium isotope, with an abundance of ∼0.0055%.5 A member of the 4n + 2 decay chain, 234U is a long-lived radionuclide (T1/2: ∼ 2.46 × 105 years) that produces 230Th (T1/2: ∼ 7.54 × 104 years) via the emission of an α-particle (4774.6 keV).6 This decay scheme yields one of the most valued and widely employed uranium chronometers in modern nuclear forensics and safeguards research and will be discussed in the applications section of this work.18,19 The chronometric relationship between 234U and 230Th is illustrated in Fig. 4, assuming complete removal of thorium at T0 within a hypothetical material. As shown, the 230Th/234U ratio increases in a highly linear fashion for thousands of years following purification (R2 = 0.9992 between 0 and 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years), before approaching equilibrium. This behavior affords a wide window of opportunity for detailed age-dating research, and in-growth of the daughter product, even in many relatively low enrichment materials, that is appreciable to the point of being amenable to analysis by various techniques.15,18 Furthermore, high levels of natural interference and activity constant “mismatches” are a reduced concern for this chronometer.
000 years), before approaching equilibrium. This behavior affords a wide window of opportunity for detailed age-dating research, and in-growth of the daughter product, even in many relatively low enrichment materials, that is appreciable to the point of being amenable to analysis by various techniques.15,18 Furthermore, high levels of natural interference and activity constant “mismatches” are a reduced concern for this chronometer.
 | ||
| Fig. 4 A plot of the relationship between the 230Th/234U as determined using standard decay equations. | ||
As introduced above, age-dating strategies based on 234U can be extended to multiple generations of progeny. For example, previous literature has examined the use of the 214Bi/234U relationship in the chronometric analysis of uranium bearing materials.20 This strategy, however, is limited to the analysis of highly enriched and/or old materials.
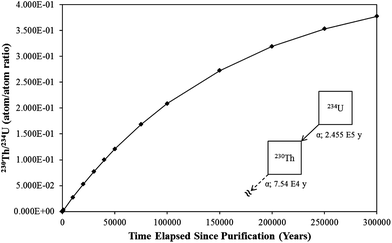
U-233: mid-value, special applications chronometer
Uranium-233 (233.039635207 amu) is an anthropogenic, fissile uranium isotope produced via the neutron irradiation of 232Th;6 this is a multi-step process shown below:
A member of the 4n + 1 decay chain, 233U (T1/2: ∼ 1.592 × 105 years) decays to 229Th (T1/2: ∼ 7.880 × 103 years) via the emission of an α-particle (4824.2 keV).6 The chronometric relationship between 233U and 229Th is illustrated in Fig. 5, assuming complete removal of 229Th at T0 within a hypothetical material. As shown, the 229Th/233U ratio increases in a linear fashion for thousands of years following purification (R2 = 0.9991 between 0 and 2500 years), before approaching equilibrium. Similar to 230Th/234U, this behavior provides a significant window of chronometric opportunity and significant in-growth of the daughter species for the use of both mass spectrometry and counting techniques.
 | ||
| Fig. 5 A plot of the relationship between the 229Th/233U as determined using standard decay equations. | ||
The first completely non-natural system discussed in this work, the 229Th/233U relationship can serve as a special applications chronometer of significant value; however, substantial room exists for expanding the application of this relationship. Specific points that support the value of the 229Th/233U chronometer include: (1) the value of each non-natural radionuclide as an analytical spike/tracer.18,21 (2) The capacity of 233U to act as a critical nuclear materials signature that can provide insight into user activities.22 (3) The existence and rising interest in thorium-based reactors that involve the use of fissile 233U.
U-232: low to mid-value, special applications chronometer
Uranium-232 (232.037156152 amu) is a non-natural uranium isotope produced by neutron capture reactions involving 232Th; examples of formation routes include:and


A member of the 4n decay chain, 232U decays relatively quickly (T1/2: ∼ 68.9 years) to the shorter lived 228Th (T1/2: ∼ 1.913 years) via the emission of an α-particle (5320.12 keV).6 The chronometric relationship between 232U and 228Th is illustrated in Fig. 6 as a function of time elapsed since material purification, assuming complete removal of thorium at T0. As shown, the 228Th/232U ratio will increase in a linear fashion for only a relatively short period of time (R2 = 0.9964 between 0 and 1 year) before approaching transient equilibrium.4 This behavior provides a limit window of chronometric value and age-dating with this system must be restricted to young materials. In practice, however, the 228Th/232U ratio is rarely directly employed in chronometric studies; the major value of this system arises from its anthropogenic nature. As with 229Th/233U, the 228Th/232U relationship is a potential materials signature and, given the relatively short half-lives of these radionuclides, they are excellent tracer/spike23 candidates for analytical counting measurements (e.g. alpha spectrometry).
 | ||
| Fig. 6 A plot of the relationship between the 228Th/232U as determined using standard decay equations. | ||
Other uranium isotopes
Including non-natural species, dozens of uranium isotopes are now known; a summary of these radionuclides, excluding those discussed above, and their relevant properties is provided in Table 1. The vast majority of these analytes are of little to no interest in chronometric investigations because of their relative rarity and short half-lives, as low as a microsecond in some cases. However, one species of potential interest is 237U; uranium-237 is not sufficiently long-lived to provide a useful, universal age-dating strategy, but is of importance as a decay product of 241Pu and a significant contributor to the production of 237Np, in light of its short-lived nature, in some plutonium-bearing materials. Its detection, therefore, during uranium isotopic measurements may serve as a useful materials signature.| Nuclide | T 1/2 | Radiation (%) | Energy (keV) | Intensity (%) | Progeny |
|---|---|---|---|---|---|
| 242U | 16.8 (±0.5) min | β−(100%) | 395 | 79.6 | Np-242 |
| 370 | 13.4 | ||||
| 166 | 4.3 | ||||
| 374 | 3.519 | ||||
| 274 | 1.3 | ||||
| 240U | 14.1 (±0.1) h | β−(100%) | 108.7 | 75.0 | Np-240 |
| 94.7 | 25.0 | ||||
| 51.7 | 1.818 | ||||
| 74.7 | 1.313 | ||||
| 239U | 23.45 (±0.02) min | β−(100%) | 391.16 | 69.014 | Np-239 |
| 419.46 | 18.724 | ||||
| 407.66 | 9.419 | ||||
| 374.96 | 1.9624 | ||||
| 237U | 6.752 (±0.002) days | β−(100%) | 64.5418 | 51.4 | Np-237 |
| 68.5918 | 42.3 | Np-237 | |||
| 137.5719 | 3.023 | Np-237 | |||
| 231U | 4.2 (±0.1) days | β+(100%) | 381.65 | 100 | Pa-231 |
| 230U | 20.8 (±n/a) days | α(100%) | 5888.47 | 67.44 | Th-226 |
| 5817.57 | 32.0020 | ||||
| 229U | 58 (±3) min | EC(∼80%) | 290.41 | — | Pa-229 |
| α(∼20%) | 6360.6 | 12.812 | Th-225 | ||
| 6332.3 | 4.04 | ||||
| 6297.3 | 2.2020 | ||||
| 228U | 9.1 (±0.2) min | α(>95%) | 6680.10 | 68.5 | Th-224 |
| 6590.10 | 28.4 | ||||
| 227U | 1.1 (±0.1) min | α(100%) | 6860.30 | 50.6 | Th-223 |
| 7060.60 | 20.00 | ||||
| 6740.50 | 16.4 | ||||
| 6905.60 | 14.3 | ||||
| 226U | 0.35 (±0.15) s | α(100%) | 7570.20 | 85.5 | Th-222 |
| 7420.20 | 15.5 | ||||
| 225U | 9.5 × 10−2 (±1.5 × 10−2) s | α(100%) | 8014.38682 | 100 | Th-221 |
| 224U | 9 × 10−4 (±3 × 10−4) s | α(100%) | 8619.821222 | 100 | Th-220 |
| 223U | 1.8 × 10−5 (±1 × 10−5) s | α(100%) | 8780.40 | 100 | Th-219 |
| 222U | 1.0 × 10−6 (±1 × 10−4) s | α(100%) | 9500.100 | 100 | Th-218 |
| 221U | 7 × 10−7 (±n/a) s | α(−) | 9950 | — | Th-217 |
| β+(−) | 3190 | — | Pa-221 | ||
| 219U | 4.2 × 10−5 (±3.4 × 10−12) s | α(100%) | 9680.40 | 100 | Th-215 |
| 218U | 5.1 × 10−4 (±1.7 × 10−9) s | α(100%) | 8774.43863 | 100 | Th-214 |
| 217U | 1.6 × 10−2 (±2.1 × 10−5) s | α(≤100%) | 8018.11 | 100 | Th-213 |
3. Special considerations
Numerous unique and frequently non-intuitive considerations, beyond those routinely associated with conducting analytical investigations (e.g. measurement uncertainty), are currently associated with conducting chronometric investigations of uranium-bearing materials. Examples of such considerations include the impact of initial purification impurities, high uncertainties for current decay information, and more. These challenges are introduced below along with brief discussions of recent, relevant literature.Incomplete initial purification
A key assumption in modern uranium chronometry is that initial purification is essentially perfect, such that remaining daughter concentrations are negligible at T0. However, this assumption is invalid for many materials, including uranium ores, processing intermediates, and scrap, and determined ages will be of reduced/minimal value. This point is highlighted dramatically in Table 2, which compares well known ages and experimentally determined ages for materials produced in varied locations.24 As shown, significant positive bias was noted in each case as a function of progeny leftover at last purification, and obtained age-values were impossibly large.| Production mill | Known age (years) | Age determined by 230Th/234U chronometer (years) | Difference (years) |
|---|---|---|---|
| Denison (Canada) | 37.8 | ∼3370 | +3332 |
| Rio Algom (Canada) | 27.1 | ∼2241 | +2214 |
| Ranstad (Sweden) | 43 | ∼63.7 | +20.7 |
| Beverley-1 (Australia) | 8.7 | ∼558 | +549 |
| Ranger-3 (Australia) | 9.9 | ∼679 | +670 |
Recent work by Williams and Gaffney21 showed that perfect purification is also a questionable assumption even for many materials produced and validated using vigorous methods, such as regularly employed uranium standard reference materials. Of seven reference materials investigated in this work, only one (NBL CRM U100) returned an accurate age value; all other samples were negatively impacted by residual 230Th and returned values exceeding expectations. A comparison of the expected and determined ages for several of these standards is illustrated in Fig. 7, and clearly illustrates recurring positive bias. Hence, this work clearly reinforces the idea that, in many cases, ages determined by modern uranium chronometry do not accurately represent production dates, but rather point to a maximum possible material age.
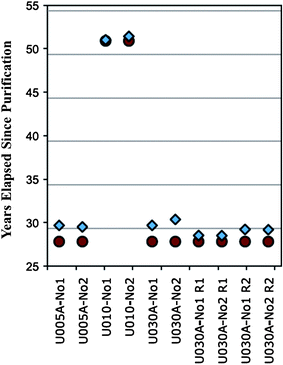 | ||
| Fig. 7 Comparison of the known and determined, via the 230Th/234U chronometer, production ages for several commonly employed uranium enrichment standards. Adapted from ref. 20. | ||
Varga and coworkers (Institute of Transuranium Elements; Karlsruhe, Germany) recently introduced and applied a complementary uranium age-dating method that circumvents the challenge of incomplete initial purification when interrogating uranium ore concentrates (a.k.a. yellow cakes).24 The proposed strategy is based on the measurement of trace thorium isotopics, using ICP-MS, gamma spectrometry, and alpha counting strategies, to determine a material 228Th/232Th ratio and monitor its variation with time; hence, this method advantageously exploits Th remaining after material purification. Several significant limitations, however, exist that hinder this strategy, including: (1) the proposed method is only applicable to materials of less than ∼40 years of age. (2) Relatively large errors were noted for many obtained age-values. (3) The pattern of 228Th/232Th variation with time may yield two distinctly different ages for a given ratio. Additional detail for each of these limitations is provided in ref. 24.
Accounting for incomplete material purification clearly requires the use of multiple, orthogonal strategies and chronometers; resulting data, however, may still be undetectably biased and novel approaches to residual thorium accountancy are needed.
Uncertainty in half-life values
Under normal circumstances, the isotopic composition of a previously processed, closed system uranium material will be dictated solely by initial purity (discussed above) and the half-lives of the radionuclides present. The uncertainties associated with currently accepted half-life values can, therefore, significantly impact the determination a material's age and the legal defensibility of resulting data. In fact, many relevant uncertainties currently drastically exceed the levels desired for conducting confident nuclear forensics and safeguards research.A recent report by Inn and coworkers22 explored the current levels of uncertainty associated with many radionuclides of interest in nuclear forensics in the context of desired legal targets outlined by the Daubert Standard for scientific evidence.25 Even ignoring all instrumental considerations, the half-life uncertainties of interest in uranium chronometry were universally calculated to significantly exceed target levels; these finding are highlighted in Table 3. Alarmingly, the most extreme case listed (232Th) misses the current target level by more than 40 million years.
| Nuclide | Current half-life uncertainty (years) | Target half-life uncertainty (years) | Difference (years) |
|---|---|---|---|
| 231Pa | 260 | 44 | 216 |
| 229Th | 120 | 11 | 109 |
| 230Th | 300 | 53 | 247 |
| 232Th | 60000000 |
19000000 |
41000000 |
| 232U | 1.1 | 0.11 | 0.99 |
| 234U | 600 | 290 | 310 |
| 235U | 1000000 |
960000 |
40000 |
| 236U | 60000 |
32000 |
28000 |
Given the discrepancies introduced above, substantial future efforts will be required to refine the decay information for all uranium isotopes and progeny radionuclides. Until completion of such work, poorly defined uncertainties must be considered a major challenge to age-dating uranium materials with high accuracy and legal defensibility.
Closed versus open system behavior
Uranium age-dating investigations frequently assume closed system behavior; in this scenario, uranium progeny are completely retained by the system. For closed system materials, measured radionuclide ratios should agree well with those calculated using standard equations and relatively accurate age values will likely be obtained. While this assumption may be reasonable for highly controlled (e.g. reference standards) and/or young materials, closed system behavior is not guaranteed for samples exposed to significant environmental conditions/weathering. In these open-system samples, uranium progeny may be transported out of or into the material of interest at different rates, relative to both the parent and other decay products.26,27Open system behavior may lead to inaccurate age-dating results of little or no value; these results may differ between the distinct chronometers (e.g.230Th/234U vs.231Pa/235U) present in a single material, further complicating research efforts. As little can be done to retroactively prevent the impact of environmental processes, new sampling strategies are needed that selectively recover materials that have experienced minimal weathering. These methods would likely be of greatest value in safeguards research, but may find application in a range of fields.
Available reference materials
Well-validated references materials are invaluable in conducting confident measurements of a sample's elemental makeup, isotopic composition, and age. However, there is a unique and severe shortage of reference materials addressing the needs of modern nuclear forensic and safeguards investigations. For example, as of 2008, no reference materials were available to serve as either uranium or plutonium chronometry standards;28 this largely remains true today. Most age-dating measurements, therefore, still rely on the use of separate standards to measure parents and daughters in chronometric relationships. Additional details on the dearth of useful nuclear forensics and safeguards reference standards can be found in ref. 28, which also discusses the lack of standards in related radiobioassay and environmental research.Predominant strategies for addressing the current lack of chronometry standards include expanding certification of current isotopic reference materials for age-dating use28 and developing novel methods to allow for more independent production of standards by research groups around the world. Independent, in-house production of a uranium radiochronometry reference material was recently reported by the Institute for Transuranium Elements (ITU).29 This work yielded a means of producing uranium chronometry standards in a straightforward and rapid manner, but was only applied to the 230Th/234U relationship in a single highly enriched uranium material. Hence, this strategy requires significant further research, involving additional chronometers and materials, to ensure legal defensibility of any data obtained in conjunction with its use.
Pu-bearing materials
While perhaps apparent, it should be noted that many of the concepts and concerns in uranium chronometry are further complicated by the presence of plutonium in a material of interest. Plutonium isotopes produce uranium species via radioactive decay and will alter many obtained isotopic ratios/age values. Hence, an understanding of Pu contributions to uranium isotopics can be critical in nuclear forensic and safeguard investigations of uranium. If needed, plutonium chronometry is discussed more thoroughly in ref. 16.Uncertainty in methodology
Arguably a concern in any analytical investigation, sources of methodological uncertainties must be meticulously accounted for in age-dating and nuclear forensics investigations. The difficulty associated with this consideration is compounded by the variety of techniques and applications potential of interest. For example, the separations strategies used in preparing relatively pure solutions of actinides routinely provide decontamination factors of approximately 106; however, these methods differ significantly based on the actinides of interest, sample matrix, separations media, etc. and may complicate analyses.In addition to sample preparation differences, sources of instrumental uncertainty vary significantly between the techniques of greatest interest in age-dating nuclear materials. Such techniques include a variety of mass spectrometry (TIMS, MC-ICP-MS) and decay counting (α, β, γ) methods that operate on drastically different principles, which encompass distinct causes/magnitudes of uncertainty.2
4. Some recent applications
The principles of uranium chronometry have been applied to a variety of fields and extended to nuclear forensics research. Today, the most commonly applied uranium chronometer in nuclear forensics and safeguards is undeniably the 230Th/234U relationship, though other candidates are used with varying levels of interest. Examples of recent uses of such chronometers are introduced below and some of the findings/challenges noted in these works are highlighted.230Th/234U
The 230Th/234U chronometer has been employed for decades in geological and environmental investigations and is a relatively well understood age-dating strategy.30–32 In recent years, this approach has been extensively applied to nuclear forensics and safeguards research interested in much younger materials (70 years or less) than those considered in environmental/geological investigations. Several reports now detail the use of the 230Th/234U relationship to age-date uranium standards and practical considerations associated with conducting such measurements using orthogonal methods (e.g. TIMS, MC-ICP-MS,33 and alpha spectrometry).17 Recent literature out of Germany also highlights 230Th/234U chronometry in the interrogation of interdicted nuclear materials (discussed more in Section 5);34 more case studies, if desired, are detailed in ref. 16.In general, available research supports the stated limitations of 230Th/234U chronometry. Specifically, reported findings reinforce the importance of sufficient material enrichment and age in successfully conducting chronometry research based on 234U progeny. Furthermore, these reports tend to support the use of more laborious analytical methods (e.g. isotope dilution alpha spectrometry and TIMS) to obtain high confidence age values.18,35 Methods commonly considered to be more “user friendly”, such as ICP-MS, have shown significant potential, but still require significant efforts to minimize error.36,37 Additional discussion on the current coexistence of TIMS and ICP-MS methods can be found in a 2004 report by Walczyk.38
231Pa/235U
The 231Pa/235U ratio has also been used extensively in environmental and geological studies,26,39 and been applied to the measurement of nuclear forensics materials. An example of such work has demonstrated the use of this approach to determine the age of relatively young uranium materials (e.g. reference materials <50 years).14 Results obtained in this report generally agreed with expected values; absolute biases, however, ranged as high as 4.4% (∼1.9 years) for one material that was 43.5 years old. Age values were comparable to those observed for 230Th/234U measurements, though the 231Pa/235U results were associated with somewhat larger uncertainties. One likely reason for this is the distinction in parent half-lives (i.e.235UT1/2 > 234UT1/2) leading to drastically different levels of progeny in-growth between the two decay relationships; hence, a means of more accurately measuring trace Pa concentrations would bolster some modern uranium chronometry investigations.Despite the potential of the 231Pa/235U chronometer, this approach is not routinely exploited in the analysis of real-world materials. Indeed, significant opportunities remain in further researching/establishing this age-dating strategy in conjunction with enriched uranium materials.
229Th/233U
Of the chronometers discussed in this section, the 229Th/233U relationship is arguably the least exploited age-dating strategy in the interrogation of nuclear materials. The rarity of 233U in most uranium materials is certainly a contributing reason for this fact. Additionally, though, the relatively large error associated with the half-lives of the radionuclides in this relationship (e.g.229Th T1/2: ∼ 7.880 × 103 ± 120 years)6 is a complicating factor in obtaining quality data and a deterrent to expanded use of the 229Th/233U chronometer.A recent report out of the University of Cincinnati, in conjunction with Savannah River National Laboratory, explored the use of the 229Th/233U chronometer as a potential material “double spike” based on the predictable in-growth of the daughter species and the anthropogenic nature of the system.40 While this work focused on a 233U standard reference material (SRM-995), rather than its application to other samples, the resulting data reinforces many of the special considerations outlined above. For example, the results obtained in ref. 40 provided age values similar to expectations, but measurement uncertainties were quite large; this is no doubt impacted by large half-life uncertainties. Some possible biases in the obtained results were also discussed in this work and support the potential negative impact of incomplete material purification at T0.
5. An international nuclear forensics scenario employing uranium age-dating
The real-world use of nuclear forensics methodologies and the age-dating principles discussed above has previously been demonstrated in the investigation of seized uranium samples. Two such case studies, both detailed in ref. 34, involve materials from Lithuania (uranium dioxide pellets) and the Czech Republic (uranium powders), as well as collaborations with scientists located in Germany. The former of these two scenarios is discussed below to highlight the myriad of techniques that may be called upon in the characterization of nuclear materials, the role of age-dating within the larger setting of nuclear forensics, and the international nature of current concerns regarding materials trafficking.Uranium pellets from Lithuania
In mid-2003, scientists at the ITU received four uranium dioxide pellets (Fig. 8) from Lithuania for nuclear forensics characterization; seizure details were not provided. The interrogation scheme used for these samples can be applied to a variety of materials and progressed from relatively superficial analysis to more detailed measurements designed to determine material content, impurities, and age. The general analytical processing scheme associated with this case study was as follows: | ||
| Fig. 8 Image of a Lithuanian uranium dioxide pellet analyzed as part of an international nuclear forensics investigation. Adapted from ref. 34. | ||
• Initial characterizations focused on fundamental observations, including height, weight, geometry/dimensions; the pellets were found to be highly similar in these properties.
• Following preliminary characterization, the samples were interrogated for uranium content using hybrid K-edge densitometry, potentiometric titration, and isotope dilution mass spectrometry using 233U as a tracer radionuclide of known concentration. Uranium content was determined to be between 87.43 and 87.99% by mass.
• Sample chemical impurities were determined using ICP-MS techniques and were found to include Al, Ca, Cr, Cu, Fe, K, Mg, and others.
• Pellet uranium isotopic composition was determined using a three-fold approach for high confidence; the employed techniques included high resolution gamma spectrometry, TIMS and MC-ICP-MS. The isotopic make-up was determined to be approximately 234U – 0.014%; 235U – 2.0005%; 236U – 0.0071%; 238U – 97.978%.
• Isotopic measurements were expanded to determine the age of the sample, as calculated from the material 230Th/234U ratio derived from TIMS and MC-ICP-MS analyses. The ratio was determined through isotope dilution techniques using 228Th and 233U as tracer radionuclides, in a standard addition fashion. The age of the material was determined to be ∼12.6 years; efforts were made to confirm this value using the 231Pa/235U chronometer, but proved unsuccessful due to minimal progeny in-growth over such a limited material lifetime.
Following completion of the above analyses, the collected results were compared to reference information available within a custom database established by the ITU.41 In contrast to many investigations, the background and intended purpose of the investigated pellets was able to be determined with an extremely high level of certainty. The details pertaining to material attribution are provided below.
The dimensions, uranium content, and uranium isotopics of the seized pellets immediately suggested the involvement of a Russian type water-cooled, graphite-moderated reactor, either the RBMK-1000 or the RBMK-1500. The RBMK-1000 is an older and more common reactor, but there is only one RBMK-1500 reactor in the world (Ignalina Unit 2) and it is operating within Lithuanian territory.
Ignalina Unit 2, a relatively new reactor, relies upon fuel provided exclusively from a single manufacturer, MZ Electrostal in Moscow. The chemical impurities present in the uranium pellets were compared with this manufacturer production values for confirmation of material providence. In all cases, the measured impurities were in-line with MZ Electrostal product specifications.
The age-dating results determined in this case served as a final confirmatory parameter of pellets' the origin. Manufacturer records state that Ignalina fuel production commenced in December of 1989. Therefore, the age value determined experimentally is this work strongly suggests that the seized pellets were manufactured for specific use at Ignalina Unit 2 in the early months of reactor fuel production (1990).
Interestingly, the International Atomic Energy Agency (Vienna, Austria) databases contain records of a fuel assembly theft from the Ignalina facility in 1992.42 Given the age of the seized material and the confident identification of the pellets' manufacturer, the analyzed samples were likely part of this assembly; the intact assembly was ∼110 kg in total and its theft represents a exceptional diversion of nuclear materials.
6. Conclusions
Nuclear forensics and safeguards research efforts are of rising importance in the face of increasing concerns over the illicit production and trafficking of nuclear materials (e.g. smuggled yellow cakes, stolen fuel pellets, etc.). Well-designed age-dating capabilities will, therefore, represent a crucial skill set for the rising generation of investigators in these arenas, many of whom will enter with widely differing scientific backgrounds. This work provides a straightforward and centralized introduction to many of the fundamental concepts, unique concerns, and recent applications of modern uranium chronometry, and attempts to point out several areas for potential future advancement/refinement. From this introduction, it is clear there are various potential strategies for interrogating uranium-bearing nuclear materials, but that significant challenges exist in further validating/developing these approaches and ensuring their legal defensibility. Particular opportunities for future contributions that were mentioned in this work include: expanded investigation of extended chronometric relationships, development of novel methods of producing needed references materials, refinement of currently accepted half-life values, and design of novel sampling strategies that yield accurate age-dating results regardless of sample environment.Acknowledgements
The author would like to thank Dr Khalil Spencer of Los Alamos National Laboratory for his assistance in preparing this work. The institutional number for this document is LA-UR-12-22288.References
- Joint Working Group of the American Physical Society and the American Association for the Advancement of Science, Nuclear Forensics: Role, State of the Art, and Program Needs, AAAS Publication Services, USA, 2008 Search PubMed.
- F. E. Stanley, A. M. Stalcup, H. B. Spitz and J. Rad, Nucl. Chem., 2012 DOI:10.1007/s10967-012-1927-3.
- Committee on Nuclear Forensics, National Research Council, Nuclear Forensics: A Capability at Risk, The National Academy Press, USA, 2010 Search PubMed.
- E. D. Ehmann and D. E. Vance, in Radiochemistry and Nuclear Methods of Analysis, ed. J. D. Windefordner and I. M. Kolthoff, John Wiley and Sons, Inc, NY, 1991 Search PubMed.
- “Uranium Isotope Data”, http://periodictable.com/Isotopes/092.238/index2.p.full.dm.html, accessed 10 May 2012.
- “U-238”, http://www-nds.iaea.org/relnsd/vcharthtml/VChartHTML.html, accessed 14 May 2012.
- J. K. Osmond and M. Ivanovich, in Uranium-Series Mobilization and Surface Hydrology. In: Uranium Series Disequilibrium: Applications to Earth, Marine, and Environmental Sciences, ed. M. Ivanovish and R. S. Harmon, Clarendon Press, Oxford, 2nd edn, 1992 Search PubMed.
- K. M. Wilcken, Accelerator Mass Spectrometry of Natural 236U and 239Pu with Emphasis on Nucleogenic Isotope Production, Ph.D. dissertation, Australian National University, 2006.
- J. T. Fabryka-Martin, Production of Radionuclides in the Earth and Their Hydrogeologic Significance, with Emphasis on Chlorine-36 and Iodine-129, Ph.D. dissertation, University of Arizona, 1988.
- X.-L. Zhao, M.-J. Nadeau, L. R. Kilius and A. E. Litherland, Nucl. Instrum. Methods Phys. Res., Sect. B, 1994, 92, 249 CrossRef CAS.
- B. A. Buchholz, T. A. Brown and T. F. Hamilton, et al. , Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 259, 773 CrossRef.
- R. Hellborg and G. Skog, Mass Spectrom. Rev., 2008, 27, 398 CrossRef CAS.
- S. Richter, A. Alonso, W. De Bolle, R. Wellum and P. D. P. Taylor, Int. J. Mass Spectrom., 1999, 193, 9 CrossRef CAS.
- A. Morgenstern, C. Apostolidis and K. Mayer, Anal. Chem., 2002, 74, 5513 CrossRef.
- N. Vajda and C.-K. Kim, Anal. Chem., 2011, 83, 4688 CrossRef CAS.
- K. J. Moody, I. D. Hutcheon and P. M. Grant, Nuclear Forensic Analysis, CRC Press Taylor and Francis Group, FL, 2005 Search PubMed.
- M. Miller, The Nonproliferation Review, 2007, 14, 33 CrossRef.
- M. Wallenius, A. Morgenstern, C. Apostolidis and K. Mayer, Anal. Bioanal. Chem., 2002, 374, 379 CrossRef CAS.
- Z. Varga and G. Suranyi, Anal. Chim. Acta, 2007, 599, 16 CrossRef CAS.
- C. T. Nguyen and J. Zsigrai, Nucl. Instrum. Methods Phys. Res., Sect. B, 2006, 243, 187 CrossRef CAS.
- R. W. Williams and A. M. Gaffney, Proc. Radiochim. Acta, 2011, 1, 31 Search PubMed.
- K. Inn, S. Jerome, J. Keightley and J. Mann, Appl. Radiat. Isot., 2012 DOI:10.1016/j.apradiso.2012.02.019.
- K. G. Heumann, Int. J. Mass Spectrom. Ion Processes, 1992, 118/119, 575 CrossRef.
- Z. Varga, M. Wallenius, K. Mayer, E. Hrnecek and J. Rad, Nucl. Chem., 2011, 290, 485 CrossRef CAS.
- United States Supreme Court, Daubert v. Merrel Dow Pharmaceuticals, Inc., USA, 1993, vol. 509, p. 589.
- B. J. Szabo and J. N. Rosholt, J. Geophys. Res., 1969, 74, 3253 CrossRef CAS.
- P. Hille, Earth Planet. Sci. Lett., 1979, 42, 138 CrossRef CAS.
- K. G. W. Inn, H. Kurosaki and C. Frechou, et al. , Appl. Radiat. Isot., 2008, 66, 835 CrossRef CAS.
- Z. Varga, A. Nicholl, M. Wallenius and K. Mayer, Anal. Chim. Acta, 2012, 718, 25 CrossRef CAS.
- R. S. Harmon, D. C. Ford and H. P. Schwarcz, Can. J. Earth Sci., 1977, 14, 2543 CrossRef CAS.
- M. Ikeya and K. Ohmura, Earth Planet. Sci. Lett., 1983, 65, 34 CrossRef CAS.
- R. A. Mortlock, R. G. Fairbanks, T.-C. Chiu and J. Rubenstone, Geochim. Cosmochim. Acta, 2005, 69, 649 CrossRef CAS.
- R. Thomas, Spectroscopy, 2002, 16, 38 Search PubMed.
- M. Wallenius, K. Mayer and I. Ray, Forensic Sci. Int., 2006, 156, 55 CrossRef CAS.
- Canberra, A Practical Guide to Successful Alpha Spectroscopy, http://www.canberra.com/literature/953.asp, accessed June 14.
- Z. Varga, M. Wallenius and K. Mayer, J. Anal. At. Spectrom., 2010, 25, 1958 RSC.
- J. Hellstrom, J. Anal. At. Spectrom., 2003, 18, 1346 RSC.
- T. Walczyk, Anal. Bioanal. Chem., 2004, 378, 229 CrossRef CAS.
- H. Zou and A. Zindler, Geochim. Cosmochim. Acta, 2000, 64, 1809 CrossRef CAS.
- F. E. Stanley, S. E. Glover, A. M. Stalcup, H. B. Spitz, D. M. Beals and J. Rad, Nucl. Chem., 2012 DOI:10.1007/s10967-012-1927-3.
- Y. Dolgov, Y. Bibilashvili, N. Chorokhov, L. Koch, R. Schenkel and A. Schubert, Case Studies with a Relational Database System for Identification of Nuclear Material of Unknown Origin, in Proceedings of the Russian International Conference on Nuclear Material Protection, Control and Accounting, Obninsk, Russia, 9–14 March, 1997, p. 116 Search PubMed.
- IAEA Database on Illicit Trafficking of Nuclear Materials and Other Radioactive Sources. Open Information April 23, 2002.
Footnote |
| † A case wherein the decay activities of the parent and daughter radionuclides becomes equal after some time. |
| This journal is © The Royal Society of Chemistry 2012 |