Simultaneous in vivo monitoring of multiple brain metals using an online microdialysis-in-loop solid phase extraction-inductively coupled plasma mass spectrometry system†
Cheng-Kuan
Su
a,
Yan-Ling
Lin
a and
Yuh-Chang
Sun
*ab
aDepartment of Biomedical Engineering and Environmental Sciences, National Tsing-Hua University, 30013, Hsinchu, Taiwan. E-mail: ycsun@mx.nthu.edu.tw; Fax: +886-3-5723883; Tel: +886-3-5727309
bNuclear Science and Technology Development Center, National Tsing-Hua University, 30013, Hsinchu, Taiwan
First published on 25th October 2011
Abstract
Using a non-functionalized small-bore polytetrafluoroethylene (PTFE) sample loop as a preconcentrator, a novel hyphenated system—comprising microdialysis sampling, online automatic in-loop solid phase extraction (SPE), and inductively coupled plasma mass spectrometry (ICP-MS)—for monitoring the levels of trace metal ions in living rat brains was developed. Taking advantage of the selective polymer–ion interaction to online separate metal ions from highly saline operating in the in-loop PTFE SPE system, it could online remove the salt matrix selectively from the microdialysate of rat brain extracellular fluid after slightly adjusting the pH; the next analytical cycle could be performed without the need for a pre-conditioning procedure after the prior elution with HNO3. Owing to the simplicity and convenience of the developed operation sequence, this method combines extremely low blank level and detection limits (0.003–0.5 μg L−1) and acceptable spike recoveries (90–98%) with the ability to analyze multiple elements. To demonstrate analytical reliability and compatibility, the analysis of standard reference material NIST 1643e (trace elements in water) and 2670a (trace elements in human urine) as well as continuous long-term monitoring of the variations of multiple trace metal ions in rat brain were performed in this work. According to the analytical results, it showed that the developed hyphenation system had the ability to analyze the SRM samples accurately, and the basal values of Ni (0.36 ± 0.16 μg L−1), Zn (3.09 ± 0.31 μg L−1), and Mn (1.31 ± 0.06 μg L−1) in rat brain extracellular fluid could also be measured.
1. Introduction
In vivo monitoring of the dynamic variations in the concentrations of trace metals is an essential component of toxicokinetics and pharmacokinetics studies. Although trace metals play essential roles in brain functions and connections,1–3 the transfer mechanisms of metal ions in the brain remain unclear because of the lack of adequate analytical strategies.4 The development of microdialysis (MD) sampling techniques has enabled the continuous and dynamic measurement of endogenous substances in brain extracellular fluids, providing a glimpse into how the brain functions—and can be manipulated—in living subjects.5,6Inductively coupled plasma mass spectrometry (ICP-MS) is broadly considered to be the most powerful tool for trace elemental analysis because of its extremely high analytical sensitivity and ability to monitor multiple elements. Ensuring the in situ, in vivo, and continuous measurement of trace brain metals through the combination of MD and ICP-MS requires several analytical challenges to be addressed. First, the removal of salt matrix from microdialysate samples was necessary to avoid consequent signal drift and matrix effects.7 In addition, a highly sensitive hyphenated technique that would allow the analysis of trace elements in microdialysate samples having volumes on the order of tens of microlitres was also required. Furthermore, because of the extremely low concentrations of analyte ions, strict control over the blank levels resulting from labware and added chemicals would be critical.To improve the sensitivity and selectivity of trace analysis in saline samples, several preconcentration procedures using solid adsorbents (e.g., co-precipitation and resin-based extraction) have been developed to facilitate removal of the matrix.7 In view of the demands to minimize blank levels and to overcome the limitations of low microdialysate volumes, the use of common SPE methods was very restricted because of possible contamination during analysis and their inadaptability to handle minute quantities of materials.8 The development9 of the knotted reactor (KR) in 1991 led to techniques forming the basis of filterless collectors applicable to a wide range of applications, from total analysis to chemical speciation10–12 analysis of trace metals. Generally, KR is performed in an open-tubular mode that necessitates neither preconditioning nor lengthy sample loading procedures. Because KRs are mainly used as collectors of metal hydroxide precipitates and as sorption surfaces for soluble metallo-organic complexes, knotted polytetrafluoroethylene (PTFE) tubing that is several meters long is necessary to generate centrifugal force and large amounts of alkaline or complexing agents are necessary to convert the analyte ions into extractable species. Therefore, this conventional approach is impracticable for the determination of trace elements in volume-limited microdialysate samples, where the essential requisites are high temporal resolution and strict contamination control. To take advantage of KRs' unique preconcentration properties while avoiding its limitations, a new separation process using open tubing to preconcentrate trace metal ions from microlitre-scale saline microdialysate samples was explored.
Because open-tube reactors, such as KRs, have eliminated many of the drawbacks associated with flow resistance during packed column-based SPE, it was expected that non-functionalized small-bore PTFE tubing would allow online preconcentration of trace elements in samples featuring high salt contents. To couple MD sampling with ICP-MS determination, a PTFE loop-based reactor in a straight/untangled geometry was developed and positioned between the MD and ICP-MS systems in flow injection mode to remove the salt matrix and enrich the levels of copper, zinc, manganese, nickel, cobalt, cadmium and lead ions in the microdialysate samples. For in vivo monitoring of the transfer kinetics of trace metals in rat brains, a novel automatic micro-trace analytical system using a MD sampling probe, a new online in-loop solid phase extraction (SPE) device, and ICP-MS was hyphenated and applied to the in vivo monitoring of dynamic variations in the levels of trace metals in the extracellular fluids of rat brains.
2. Experimental section
2.1. Chemicals
Given the low concentration of trace metal ions in small-volume microdialysate, all laboratory works were conducted in a class 10000 cleanroom. All laboratory materials, including pipet tips, bottles, vials, were cleaned by immersion in concentrated HNO3 for at least 1 day and then washed with ultrapure water to remove remaining HNO3 before experiments. The ultrapure water used in this study was purified through a Milli-Q Integral water purification system (Millipore Corporation, Billerica, MA). The perfusion solution for MD sampling was prepared by sodium chloride (0.9 g, ultrapure grade, E. Merck, Darmstadt, Germany) dissolved in ultrapure water (100 mL). The buffer solution for online mixing with microdialysate was prepared by dissolving maleic acid disodium (Sigma-Aldrich Corporation, St. Louis, MO, USA) in ultrapure water and adjusting the pH using ultrapure grade nitric acid (J.T. Baker, NJ, USA). Before each day's experiments, the maleic buffer and perfusion solution were further purified through a chelex-100 packed clean-up pre-column to remove any background contributions. 0.5% HNO3 for elution was prepared by ultrapure grade nitric acid. Stock solutions (1000 mg L−1) of analytes were purchased from E. Merck Company and prepared afresh daily using 0.9% NaCl.2.2. FTIR spectroscopy
Polymer–metal complexes were prepared by mixing PTFE powder with aqueous solutions containing 2.5 g L−1 of Cu2+ ions in maleic buffer solution at pH 8. The mixtures were stirred for 2 h. The resulting solids were filtered, washed with ultrapure water, and dried at 65 °C. For comparison, pure PTFE powder was also treated using the same procedure, but in the absence of the metal ions. After preparation of the polymer–metal complexes, an attenuated total reflectance device (Harrick Scientific Corp., New York, USA) coupled with an FT-IR spectrometer (DA8.3, Bomem, Canada) was employed to acquire spectra with the chamber evacuated to pressures of lower than 102 Pa. Several milligrams of PTFE or PTFE–Cu complex powder was evenly sprinkled on a window of zinc selenite and then scanned from 600 to 1000 cm−1.2.3. Apparatus
The MD/in-loop PTFE SPE/ICP-MS hyphenation system (Fig. 1) consisted of three main parts: the MD probe, the in-loop PTFE SPE device, and the ICP-MS system. The MD sampling system is composed of a syringe pump (KDS 260, KD Scientific, Holliston, MA, USA) and a 24-mm-long MD probe (CMA/20, CMA, Solna, Sweden) and a 4-mm-long, 0.5-mm-diameter, metal-free polycarbonate (PC) membrane that had a molecular weight cut-off (MWCO) of 20 kDa. The connections of the syringe pump to the inlet of the MD probe and of the outlet of the MD probe to a commercial polyetheretherketone tee (ZT.5LFPK, Valco, Lucerne, Switzerland) were both accomplished using fluorinated ethylene propylene (FEP) tubing (internal volume: 1.2 L per 100 mm length; length: 10 cm; i.d.: 0.12 mm; CMA, Solna, Sweden). The microdialysate was online mixed with the maleic buffer solution carried by another channel of the same syringe pump; the mixture was immediately delivered to an eight-port valve (C2-2348D, Valco) via a handmade modified tubing adopter (3409500, CMA).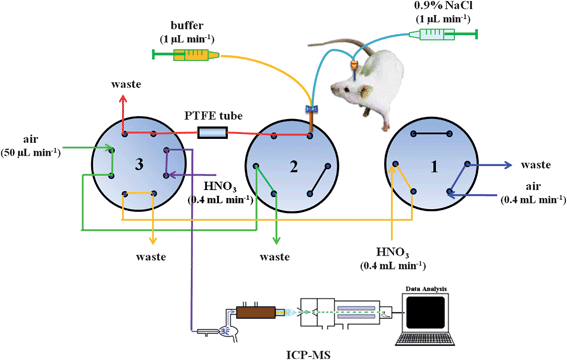 | ||
| Fig. 1 Schematic representation of the flow injection laboratory-on-valve system for trace element preconcentration through complexation with a PTFE tubing. Valve sequence: (a) 1A2A3A in the stage of sample loading for 10 min, (b) 1A2B3A in the stage of removing matrix for 90 s, (c) 1A2B3B in the stage of elution for 120 s, (d) 1B2B3B in the stage of replacing HNO3 with air for next-round sample loading for 30 s. V1 and V2: six-port rotary valves. V3: eight-port rotary valves. | ||
The online automated flow-injection pretreatment system contained two six-port valves (C22Z-3186E, Valco) and an eight-port valve. All the valves were synchronized and programmed by a single laptop via a serial valve interface (SIV-110, Valco). The operational sequence of the online pretreatment system is given in Table 1. The in-loop PTFE SPE device merely consisted of a PTFE tubing (80 cm long × 0.018 cm i.d., Alltech Associates, Inc., Deerfield, IL, USA). The peristaltic tubing for driving carrier stream and flow manifolds connecting all of the pieces of apparatus was washed with 0.5% HNO3 until the contaminants were substantially eliminated; moreover, to avoid contamination of the analyte ions, Norm-Ject plastic perfusion syringes (Henke Sass Wolf, GmbH; Tufflingen, Germany) were used throughout this study.
| step | function | valve position | time, s | medium pumped | flow rate, μL min−1 |
|---|---|---|---|---|---|
| A | Fill buffered dialysate into PTFE tubing | V1: load | 600 | Dialysate | 1.0 |
| V2: load | Buffer | 1.0 | |||
| V3: load | |||||
| B | Remove waste | V1: load | 90 | Buffered dialysate | 50 |
| V2: injection | |||||
| V3: load | |||||
| C | Detach the analyte and deliver to ICP-MS | V1: load | 120 | HNO3 | 400 |
| V2: injection | |||||
| V3: injection | |||||
| D | Replace HNO3 with air carrier stream | V1: injection | 30 | Air | 400 |
| V2: injection | |||||
| V3: injection |
The microdialysate was online mixed with a stream of maleic buffer at the same flow rate (1 μL min−1) and then directly introduced to a PTFE tubing for the separation and preconcentration; in order to greatly avoid any possible contamination, an air carrier stream (flow rate: 50 μL min−1) driven via a conventional peristaltic pump was used to remove the waste from the PTFE tubing. After removing the waste, the adsorbed analyte ions from the interior of PTFE tubing were detached using 0.5% HNO3 and transported to the ICP-MS system through another carrier stream (flow rate: 400 μL min−1) for final determination. Linear calibration curves for analyte ions were obtained using external standards from the ion intensities at m/z 55 (Mn), m/z 59 (Co), m/z 60 (Ni), m/z 64 (Zn), m/z 65 (Cu), m/z 114 (Cd) and m/z 208 (Pb).
The ICP mass spectrometer was an Agilent 7500a system (Agilent, Palo Alto, CA, USA). A Micromist nebulizer (AR35-1-EM04EX, Glass Expansion, Victoria, Australia) was fitted to a Scott-type quartz double-pass spray chamber. The instrumental operating conditions selected for optimal sensitivity and stability are presented in Table 2.
| Microdialysis sampling | |
| Probe | 4 mm × 0.5 mm membrane, metal-free; MWCO = 20 kDa |
| Perfusate | 0.9% NaCl solution (pH 7.4) |
| Perfusion flow rate | 1 μL min−1 |
| Sampling time | 600 s |
| In-loop extraction | |
| Tube material | PTFE |
| Length of tubing | 80 cm |
| Inner diameter of tubing | 0.018 cm |
| Buffer solution | 20 mM maleic buffer, pH 8.0 |
| Sample loading | 2 μL min−1 |
| Removal of waste | 50 μL min−1 |
| Eluent | 0.5% HNO3 |
| Elution flow rate | 400 μL min−1 |
| ICP-MS | |
| ICP-MS parameters | Agilent 7500a |
| Ar flow rates | |
| Plasma | 15 L min−1 |
| Auxiliary | 0.9 L min−1 |
| Nebulizer | 1.01 L min−1 |
| Makeup | 0.11 L min−1 |
| Plasma forward power | 1500 W |
| Sample cone | Nickel, 1 mm orifice |
| Skimmer cone | Nickel, 0.4 mm orifice |
2.4. Samples digestion procedure
To digest the standard reference materials, 1 mL HNO3 (65%) was added to 1 mL NIST 1643e (trace elements in water) and 2670a (trace elements in human urine), respectively. Afterwards, vessels were introduced into the microwave oven, where one sample vessel was monitored by pressure and temperature control during all the operation. The sample digestion was performed using a MARS 5 microwave digestion system (CEM Corporation, Matthews, NC, USA). When the digestion ended, the samples were diluted to 100 mL with ultrapure water and adjusted to the optimized sample acidity for the extraction of trace metal ions.2.5. In vivo experiments
By placing the MD probe in the solution of a known concentration (1 μg L−1) of analyte ions and perfusing at 1 μL min−1, it was able to determine the recovery of the used MD probe. To offer the sufficient sensitivity, the probe recovery was considered as an index to evaluate the degree of membrane fouling; in most cases, it was confined within the range 10–20%.Adult male Sprague–Dawley rats (450–550 g) were obtained from the Laboratory Animal Center of the National Science Council of the Republic of China (Taipei, Taiwan). These animals, which were specifically pathogen-free, were acclimatized to their environmentally controlled quarters (25 °C; 12 h light/12 h dark cycle) for at least 5 days before experimentation and then fasted overnight prior to sacrifice. The rats were fed with a standard diet and water and were treated under the regulations of the “Principles of Laboratory Animal Care” (NIH publication no. 86-23, revised, 1985). The study was approved by the committee of experimental animals of National Tsing-Hua University.
The rats were initially anaesthetized with urethane (1.5 g kg−1 body weight, intraperitoneal injection); they remained anaesthetized throughout the experimental period. After mounting the head of the rat on a stereotaxic apparatus (Davis Kopf Instruments, Tujunga, CA), a midline incision of the skull was executed. The microdialysis probe, which was perfused with saline solution (flow rate: 1.0 μL min−1) was implanted into the exposed brain (5 mm anterior, 5 mm lateral to the bregma, and 5 mm from the brain surface). After implanting MD probe and 15 measurements to reach equilibrium, the basal values of the analytes were monitored continuously. Basal Mn, Co, Ni, Cu, Zn, and Pb levels were monitored for at least 1 h prior to administration of MnSO4. After intraperitoneal injection (1 g MnSO4 kg−1 body weight), the levels of Mn were monitored continuously every 14 min. The rats were euthanatized as the experiments were terminated.
3. Results and discussion
3.1. PTFE–metal complexation
To investigate the retention behavior of metal ions toward the interior wall of the PTFE tubing, PTFE powder and Cu2+ ions were mixed together at pH 8 and then evaporated the solvent prior to Fourier transform infrared (FTIR) spectroscopic analysis. Fig. 2 reveals that a new characteristic absorption appeared at 858 cm−1, associated with the interactions between fluorine atoms and hydrated Cu2+ ions,13 only when Cu2+ ions were mixed with the PTFE suspension. Furthermore, because Cu, Zn, Ni, Co, Cd, and Pb ions are all positively charged species at pH 8 (isoelectric points: Cu, 9.4; Zn, 10.3; Ni, 11.1; Co, 11.4; Cd, 10.4; Pb, 11.0),14 metal precipitation did not occur, suggesting that the polymer matrix coordinated the Cu2+ ions through its fluorine atoms without the need to add alkaline or complexing agents.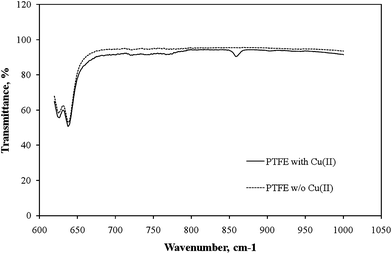 | ||
| Fig. 2 FTIR spectra of pure PTFE and PTFE that had been reacted with copper ions. A new characteristic absorption, associated with the interactions between fluorine atoms and hydrated Cu2+ ions, appeared at 858 cm−1. | ||
In this work, the interaction between the C–F moiety and positive metal ions was proposed to be the main mechanism to facilitate the complexation retention of transition metal ions when the pH of the sample solution was set to 8. In addition, compared to the traditional chelating-resin SPE procedure, because the washing and conditioning steps have been proven to be unnecessary and no apparent changes in the concentration levels of the elements of interest occurred after passing through the proposed online non-resin-based in-tube SPE system, it is considered as a promising sorbent for use in the online determination of trace metal ions in small-volume microdialysate samples.
3.2. Optimization of in-loop PTFE SPE procedure
To optimize the analytical performance of using PTFE tubing as a sorbent medium for collecting analyte ions from saline solutions, the effects of two main parameters on the extraction efficiency were studied: the pH of the sample solution and the sample flow rate.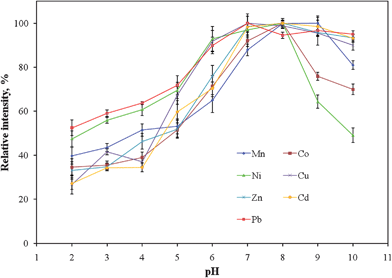 | ||
| Fig. 3 Recoveries of analyte ions plotted with respect to pH. All the data were normalized to the values of their respective maximum intensities. Error bars represent standard deviations (n = 5). | ||
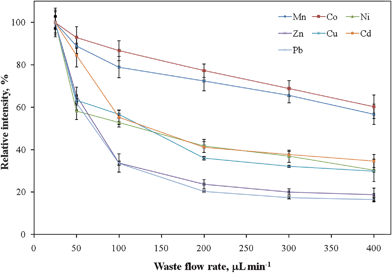 | ||
| Fig. 4 Signal intensities of analyte ions with respect to the flow rate of sample loading. All the data were normalized to the maximum value. Error bars represent standard deviations (n = 5). | ||
3.3. Analytical characteristics
After optimizing the in-loop PTFE SPE procedure, the effect of Na+ ions present in 0.9% NaCl solution, which has the same salinity as microdialysate, on the adsorption of analytes toward the PTFE tubing was investigated. The experimental results indicated that 95% or more analyte ions could be extracted from a saline solution in which the concentration of Na+ ions was 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times that of the analyte ions. These satisfactory analytical results suggested that the developed procedure could be used to accurately determine the concentrations of trace metal ions of interest in complicated microdialysate samples.
000 times that of the analyte ions. These satisfactory analytical results suggested that the developed procedure could be used to accurately determine the concentrations of trace metal ions of interest in complicated microdialysate samples.
In traditional ion exchange-based SPE procedures, the need for washing and conditioning steps not only ensures a cumbersome and time-consuming procedure but also necessitates the use of large volumes of reagents, thereby tending to result in a high reagent blank.16 By using the polymer–ion interaction described above for metal ion extraction and a clean stream of air for rinsing, the results suggested that correcting for elemental concentrations in the resultant solution would be unnecessary because no apparent changes in the concentrations of the elements of interest would occur after passing the microdialysate through the proposed online system.
To determine the enrichment factors of the analyte ions, the signal intensities of 10 μL standard solution (containing 5 μg L−1 of each element) obtained by the developed system were divided by that obtained by direct injection ICP-MS measurement. The results indicated that the enrichment factors could be from 7 to 10. In addition, the standard deviations of 7 h continuous monitoring were found to be less than 7% for all tested ions. As indicated in Table 3, the detection limits (based on three times the standard deviation of the baseline noise; n = 7) were in the range 0.003–0.523 μg L−1. Because the extracellular concentrations of Mn, Cu, and Zn ions in the brain are in the microgram-per-liter regime, this technique appears uniquely suited for continuous and simultaneous monitoring of the dynamic changes occurring in the concentrations of multiple elements in the brain.
| element | detection limita, μg L−1 | spike recoveryb (%) | 2670a (urine) | 1643e (natural water) | ||
|---|---|---|---|---|---|---|
| certified value, μg L−1 | measured value, μg L−1 | certified value, μg L−1 | measured value, μg L−1 | |||
| a Sample volume: 10 μL. b Spike concentration: 0.5 μg L−1. c Reference value. d Limit of quantification = 0.47 μg L−1. | ||||||
| Mn (m/z 55) | 0.112 | 96 | 330c | 337 ± 1 | 38.97 ± 0.45 | 39.05 ± 2.17 |
| Co (m/z 59) | 0.003 | 98 | — | — | 62.41 ± 0.69 | 63.70 ± 0.50 |
| Ni (m/z 60) | 0.058 | 97 | 300c | 299 ± 4 | 27.06 ± 0.33 | 28.62 ± 1.52 |
| Cu (m/z 65) | 0.144 | 91 | 370 ± 30 | 387 ± 9 | 22.76 ± 0.31 | <LOQd |
| Zn (m/z 66) | 0.523 | 92 | — | — | 78.5 ± 2.2 | 75.61 ± 2.22 |
| Cd (m/z 114) | 0.005 | 98 | 88 ± 3 | 87 ± 2 | 6.57 ± 0.07 | 7.17 ± 0.04 |
| Pb (m/z 208) | 0.012 | 90 | 109 ± 4 | 106 ± 8 | 19.63 ± 0.21 | 18.21 ± 1.15 |
The analytical reliability of this method was confirmed through analyses of the certified reference materials NIST 1643e (trace elements in water) and 2670a (trace elements in human urine) after their decomposition through microwave-assisted digestion. Table 3 shows that the analytical results were in good agreement with the certified values even in the presence of complicated matrices. These satisfactory analytical results indicated that the proposed method accurately determines the concentrations of trace metal ions of interest in microsamples containing complicated matrices.
3.4. Continuous determination of extracellular trace metals in a rat brain
To further evaluate the applicability of the proposed online MD/in-loop PTFE SPE/ICP-MS system, the MD probe perfused with saline solution (flow rate: 1.0 μL min−1) was implanted in the exposed brain to continuously collect microdialysate sample solutions, which were immediately online mixed with maleic buffer solution to ensure the appropriate pH (pH 8) for the following extraction experiment. Because the concentrations of transition and other heavy metals in the ECF are usually very low [(sub)nanograms per millilitre], the microdialysate had to be collected over a fixed time interval to provide sufficient sample volumes that met the detection requirements of the ICP-MS system. As illustrated in Fig. 1, to simplify the hyphenated system, a 20 μL PTFE tubing as a sample/extraction loop was used to collect and extract analyte ions from the mixed microdialysate/buffer solution, each at a flow rate of 1 μL min−1 over 14 min; thus, the temporal resolution for this system was 14 min. Using this methodology, we readily measured the basal levels of Ni (0.36 ± 0.16 μg L−1), Zn (3.09 ± 0.31 μg L−1), and Mn (1.31 ± 0.06 μg L−1) in the extracellular fluid of the rat brain in 14 min MD fractions, as illustrated in Fig. 5. These values agree well with those reported in the literature.4 In the brain, the blood–brain and blood–cerebrospinal fluid (CSF) barriers to protein-bound and non-protein-bound Mn are relatively permeable.17 Therefore, to evaluate the potential of this system for the use in dynamic monitoring, MnSO4 (1000 mg kg−1 body weight) was injected intraperitoneally (n = 3) and then determined the continuous change in the level of Mn ions in the brain microdialysates. Fig. 5 reveals an apparent increase in the level of Mn in the extracellular fluid of the brain 14 min after injection. Thus, the developed system not only has practicability for the continuous, in vivo, and on-site monitoring of the levels of trace metal ions in rat brains but also provides a novel in-loop SPE method for separating trace metal ions from volume-limited saline samples.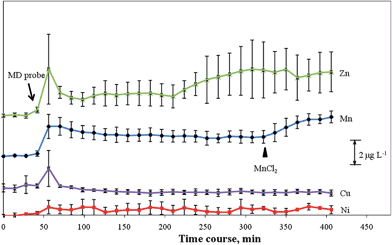 | ||
| Fig. 5 Time course of the concentrations of trace metals in the brain ECF of a living rat following insertion of a microprobe into the brain. Error bars represent standard deviations (n = 3). ▲: Intraperitoneal dose of MnSO4 (1000 mg kg−1 body weight). | ||
4. Conclusions
In summary, the selective polymer–ion interaction was used to online separate metal ions from highly saline, volume-limited microdialysate samples. By taking advantage of the separation principles operating in the in-loop PTFE SPE system, an online MD/in-loop PTFE SPE/ICP-MS system that is capable of performing in vivo analyses of multiple trace elements in the ECF of living animals was developed. This novel analytical system has potential to be used to explore the pathological and toxicological processes involved in neurological disorders caused by variations in the concentrations of Zn, Cu, Ni, and Mn ions after physiological stimulation or acute exposure to metal-containing substances.Acknowledgements
The authors would like to record their gratitude to Prof. Mo-hsiung Yang for his supervision and advice. We are also grateful for the financial support provided by the National Science Council of the Republic of China (Taiwan).References
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS.
- C. J. Frederickson, J. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef CAS.
- S. L. Sensi, P. Paoletti, A. I. Bush and I. Sekler, Nat. Rev. Neurosci., 2009, 10, 780–791 CrossRef CAS.
- C. K. Su, Y. C. Sun, S. F. Tzeng, C. S. Yang, C. Y. Wang and M. H. Yang, Mass Spectrom. Rev., 2010, 29, 392–424 CAS.
- J. A. Bourne, Clin. Exp. Pharmacol. Physiol., 2003, 30, 16–24 CrossRef CAS.
- C. J. Watson, B. J. Venton and R. T. Kennedy, Anal. Chem., 2006, 78, 1391–1399 CrossRef.
- J. Wang and E. H. Hansen, TrAC, Trends Anal. Chem., 2003, 22, 836–846 CrossRef CAS.
- J. L. Todolí and J. M. Mermet, TrAC, Trends Anal. Chem., 2005, 24, 107–116 CrossRef.
- Z. L. Fang, M. Sperling and B. Welz, J. Anal. At. Spectrom., 1991, 6, 301–306 RSC.
- X. P. Yan, R. Kerrich and M. J. Hendry, Anal. Chem., 1998, 70, 4736–4742 CrossRef CAS.
- X. P. Yan, M. J. Hendry and R. Kerrich, Anal. Chem., 2000, 72, 1879–1884 CrossRef CAS.
- X. P. Yan and Y. Jiang, TrAC, Trends Anal. Chem., 2001, 20, 552–562 CrossRef CAS.
- D. C. Pereira, D. L. A. de Faria and V. R. L. Constantino, J. Braz. Chem. Soc., 2006, 17, 1651–1657 CrossRef CAS.
- G. A. Parks, Chem. Rev., 1965, 65, 177–198 CrossRef CAS.
- K. E. Geckeler and K. Volchek, Environ. Sci. Technol., 1996, 30, 725–734 CrossRef CAS.
- A. J. Paulson, Anal. Chem., 1986, 58, 183–187 CrossRef CAS.
- A. Takeda, S. Ishiwatari and S. Okada, J. Neurosci. Res., 1999, 56, 93–98 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ja10168d |
| This journal is © The Royal Society of Chemistry 2012 |