Bimolecular integrin–ligand interactions quantified using peptide-functionalized dextran-coated microparticles
Jessie E. P.
Sun
ab,
Justin
Vranic
ab,
Russell J.
Composto
ab,
Craig
Streu
c,
Paul C.
Billings
d,
Joel S.
Bennett
e,
John W.
Weisel
f and
Rustem I.
Litvinov
*f
aDepartment of Materials Science and Engineering, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
bNano/Bio Interface Center at the University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
cDepartment of Chemistry and Biochemistry, St. Mary's College of Maryland, St. Mary's City, Maryland 20686, USA
dDepartment of Biochemistry and Biophysics, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA
eHematology-Oncology Division, Department of Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA
fDepartment of Cell and Developmental Biology, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania 19104, USA. E-mail: litvinov@mail.med.upenn.edu; Fax: +1 (215) 898-9871; Tel: +1 (215) 573-4126
First published on 28th November 2011
Abstract
Integrins play a key role in cell–cell and cell–matrix interactions. Artificial surfaces grafted with integrin ligands, mimicking natural interfaces, have been used to study integrin-mediated cell adhesion. Here we report the use of a new chemical engineering technology in combination with single-molecule nanomechanical measurements to quantify peptide binding to integrins. We prepared latex beads with covalently-attached dextran. The beads were then functionalized with the bioactive peptides, cyclic RGDFK (cRGD) and the fibrinogen γC-dodecapeptide (H12), corresponding to the active sites for fibrinogen binding to the platelet integrin αIIbβ3. Using optical tweezers-based force spectroscopy to measure non-specific protein–protein interactions, we found the dextran-coated beads nonreactive towards fibrinogen, thus providing an inert platform for biospecific modifications. Using periodate oxidation followed by reductive amination, we functionalized the bead-attached dextran with either cRGD or H12 and used the peptide-grafted beads to measure single-molecule interactions with the purified αIIbβ3. Bimolecular force spectroscopy revealed that the peptide-functionalized beads were highly and specifically reactive with the immobilized αIIbβ3. Further, the cRGD- and H12-functionalized beads displayed a remarkable interaction profile with a bimodal force distribution up to 90 pN. The cRGD–αIIbβ3 interactions had greater binding strength than that of H12–αIIbβ3, indicating that they are more stable and resistant mechanically, consistent with the platelet reactivity of RGD-containing ligands. Thus, the results reported here describe the mechanistic characteristics of αIIbβ3–ligand interactions, confirming the utility of peptide-functionalized latex beads for the quantitative analysis of molecular recognition.
Insight, innovation, integrationThis work is aimed at mechanistic insights into the adhesive interactions of platelets mediated by the integrin αIIbβ3 that are essential for hemostasis at sites of vascular injury and are responsible for arterial thrombosis. Here, we measured at the single-molecule level the binding strength of the purified αIIbβ3 with peptides mimicking natural protein ligands, utilizing a combination of newly developed technologies of surface chemistry and molecular mechanics. First, microscopic latex beads were grafted with covalently attached dextrans and subsequently functionalized with bioactive peptides. The bimolecular interactions of a surface-bound αIIbβ3 and a peptide were then quantified using optical trap-based force spectroscopy. The results provide valuable nanomechanical characteristics of αIIbβ3–ligand interactions that underlie platelet function. |
Introduction
Protein interaction with peptides is essential for a wide variety of cellular processes.1,2 There are a number of methodological approaches to study protein–peptide interactions, such as grafting peptides on functionalized artificial surfaces, that have become versatile tools in studies of molecular recognition, including single-molecule techniques. Despite the wide range of available functionalized surfaces, the behavior of artificially-grafted biomaterials is difficult to predict because non-specific background reactivity can dominate and confound the interpretation of specific molecular functionality. For example, the detachment strength of erythroleukemia cells from amine-terminated surfaces is the same whether or not the integrin ligand Arg-Gly-Asp (RGD) is tethered to the surface.3 Thus, to discern differences in cell adhesion due to small variations in adhesive peptide structures, for example RGDSPKvs. RGDS, surface treatments that minimize nonspecific interactions are essential.3,4 To address this problem, we have utilized self-assembled monolayers of various alkyl silanes,5 as well as biomimetic dextran-functionalized surfaces.6–10Dextran is a flexible charge-neutral polysaccharide that is poorly adhesive for proteins11 and cells.12 However, it is possible to functionalize dextran with biomolecules, rendering the polymer selectively reactive.13 The biological inertness of dextran, paired with its ability to be functionalized, makes it a promising material for biomedical applications, ranging from tissue engineering and new implants14 to target-specific drug delivery systems15,16 and diagnostic chips.17
In this work, we covalently grafted dextran to the hydrophobic surface of spherical latex beads, functionalized the bound dextran with small bioreactive peptides (Fig. 1), and applied the methodology to biophysical studies of the interaction of the major platelet integrin αIIbβ3 with its natural ligand fibrinogen. We functionalized dextran-grafted beads with two peptides, cyclic RGDFK (cRGD) and the fibrinogen γC-dodecapeptide (H12), each of which is a mimetic for the sites on the fibrinogen molecule that interact with αIIbβ3.18–20Peptide binding to αIIbβ3 was measured using optical tweezers-based force spectroscopy, a method developed in our laboratory. Our results not only validate the combination of dextran–peptide coating and optical trap-based nanomechanical methodology, but provide insights into the interactions between the αIIbβ3 and its ligands that underlie platelet aggregation and platelet thrombus formation.
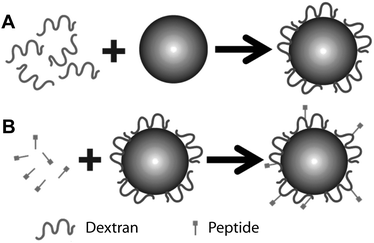 | ||
| Fig. 1 Schema for producing dextran-coated latex beads functionalized with small peptides. (A) Step 1: covalent coupling of oxidized dextran to an amine-modified latex bead. (B) Step 2: coupling of a bead coated with re-oxidized dextran with peptide. | ||
Experimental section
Materials
Dextran (MW = 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, MW/Mn = 1.52) prepared from Leuconostoc spp. was supplied by Fluka Chemie AG (Switzerland). Tetramethyl-rhodamine isothiocyanate-labeled dextran (TRITC-dextran), sodium periodate (NaIO4), sodium borohydride (NaBH4), ethanolamine, concanavalin A-agarose, n-octyl-β-D-glucoside, and prostaglandin E1 (PGE1) were purchased from Sigma-Aldrich Co. (St. Louis, MO). Halt™ protease inhibitor cocktail was from Pierce Biotechnology, Inc. (Rockford, IL). Human fibrinogen was purchased from HYPHEN BioMed (France). NH2-functionalized latex beads were purchased from Bangs Laboratories, Inc. (Fisher, IL). The peptidescyclo(Arg-Gly-Asp-D-Phe-Lys) (cRGD) and cyclo(Arg-Ala-Asp-D-Phe-Lys) (cRAD) were purchased from Peptides International (Louisville, KY). The fibrinogen γC dodecapeptide His-His-Leu-Gly-Gly-Ala-Lys-Gln-Ala-Gly-Asp-Val (H12) was purchased from Bachem Americas, Inc. (Torrance, CA). All solutions were made using Milli-Q water and were sterilized by filtration.
000 Da, MW/Mn = 1.52) prepared from Leuconostoc spp. was supplied by Fluka Chemie AG (Switzerland). Tetramethyl-rhodamine isothiocyanate-labeled dextran (TRITC-dextran), sodium periodate (NaIO4), sodium borohydride (NaBH4), ethanolamine, concanavalin A-agarose, n-octyl-β-D-glucoside, and prostaglandin E1 (PGE1) were purchased from Sigma-Aldrich Co. (St. Louis, MO). Halt™ protease inhibitor cocktail was from Pierce Biotechnology, Inc. (Rockford, IL). Human fibrinogen was purchased from HYPHEN BioMed (France). NH2-functionalized latex beads were purchased from Bangs Laboratories, Inc. (Fisher, IL). The peptidescyclo(Arg-Gly-Asp-D-Phe-Lys) (cRGD) and cyclo(Arg-Ala-Asp-D-Phe-Lys) (cRAD) were purchased from Peptides International (Louisville, KY). The fibrinogen γC dodecapeptide His-His-Leu-Gly-Gly-Ala-Lys-Gln-Ala-Gly-Asp-Val (H12) was purchased from Bachem Americas, Inc. (Torrance, CA). All solutions were made using Milli-Q water and were sterilized by filtration.
Covalent coupling of dextran to amine-modified latex beads (Fig. 1A and 2A)
Dextran with a molecular mass of 110 kDa (0.6 g) was dissolved in 25 mL of deionized water. In addition to lower dispersity, high molecular weight dextran has the advantage of tethering peptide with its native structure away from the surface latex, thereby facilitating binding interactions. NaIO4 (1.5 g) was then added and the dextran solution was allowed to oxidize in the dark for 16 hours at 25 °C with constant mixing. A 20 μL suspension of NH2-functionalized 2 μm latex beads was washed three times by sedimentation and re-suspension in 1 mL of 20 mM HEPES/150 mM NaCl, pH 7.4, after which the beads were re-suspended in 1 mL of the oxidized dextran solution. The beads were then allowed to react with the oxidized dextran in the dark for 24 hours at 25 °C with constant mixing. Following dextran attachment, the beads were sedimented by centrifugation and re-suspended in 1 mL of 0.1 M NaBH4 solution. After 12 hour incubation at 25 °C with constant mixing, they were washed with 20 mM HEPES/150 mM NaCl, pH 7.4, and stored in this solution at 4 °C.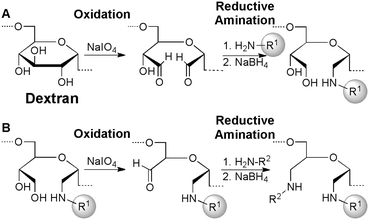 | ||
| Fig. 2 Dextran-mediated immobilization of peptides is accomplished by first functionalizing latex beads displaying free amines (H2N–R1) with oxidized dextran (A), followed by an additional oxidation and reductive amination sequence with the desired peptide (H2N–R2) to yield peptides covalently bound to latex beads (B). (A) Reaction of 1,2-diols in dextran with NaIO4 results in oxidative cleavage of the sugar subunits. Subsequent reductive amination with latex beads displaying free amines (H2N–R1) yields sugar-coated beads. (B) An additional NaIO4-mediated oxidative cleavage of 1,2-diols within the immobilized dextran yields a bead-tethered aldehyde, which is ideal for linking the latex beads to amines on the desired peptide (H2N–R2) using another reductive amination. | ||
Ligand peptide coupling to the dextran-coated bead (Fig. 1B and 2B)
After storage, the dextran-coated beads were re-oxidized using 0.01 M NaIO4 and washed three times in 0.2 M Na2HPO4. They were then re-suspended in 1 mL of 0.2 M Na2HPO4 containing 20 μM ligand peptide and incubated in the dark for 4 hours at 4 °C with constant mixing. After covalent coupling of the ligand peptide, the beads were re-suspended in 1 mL of 0.1 M ethanolamine in 0.2 M Na2HPO4 and incubated again in the dark for 16 hours at 4 °C with constant mixing. Following ethanolamine quenching, the beads were washed three times using 20 mM HEPES/150 mM NaCl, pH 7.4, and stored in suspension at 4 °C. If stored for more than 2–3 days, 0.02% sodium azide was added to prevent bacterial growth.Optical tweezers-based force spectroscopy
To test the binding properties of ligand-coupled dextran-coated beads, we used optical tweezers-based forced spectroscopy as previously described.21–23 Briefly, a dextran-coated 2 μm-latex bead, either coupled or uncoupled to peptide, was trapped by the laser light and brought to a distance of 2 to 3 μm from an αIIbβ3- or fibrinogen-coupled 5 μm-spherical silica pedestal pre-coated with a thin layer of polyacrylamide activated with glutaraldehyde. After oscillating the bead at 10 Hz with a 0.8 μm peak-to-peak amplitude (2000 pN s−1 loading rate), the bead was brought into intermittent contact with the pedestal by micromanipulation using a keyboard-controlled piezoelectric stage. Data collection was initiated at the first contact between the bead and the pedestal. Rupture forces following repeated contacts between the pedestal and the bead were collected for periods of about 1 min and displayed as normalized force histograms for each experimental condition. Because only a small percentage of contact/detachment cycles resulted in effective receptor–ligand binding/unbinding, data from 10 experiments, representing 103 to 104 individual measurements, were combined. Optical artifacts observed with or without trapped latex beads produced signals that appeared as forces below 10 pN. Accordingly, rupture forces in this range were not considered when the data were analyzed. Individual forces measured during each contact–detachment cycle were collected into 5 pN-wide bins. The number of events in each bin was plotted against the average force for that bin after normalizing for the total number of interaction cycles. The percentage of events in a particular force range (bin) represents the probability density of rupture events at that tension.Purification of αIIbβ3 from outdated human platelets
αIIbβ3 was purified from outdated platelets as previously described.24 Briefly, outdated platelets, obtained from the Blood Bank of the Hospital of the University of Pennsylvania, were washed in HEN buffer (10 mM HEPES/150 mM NaCl/3 mM EDTA/1 μM PGE1, pH 6.5), re-suspended in HEN buffer containing Halt™ protease inhibitor cocktail, flash frozen in liquid nitrogen, and stored at −80 °C. The frozen platelets were then slowly thawed at room temperature and disrupted by nitrogen cavitation at 1300 psi for 10 min followed by extrusion into a centrifugation tube on ice. The resulting turbid suspension was centrifuged for 15 min at 4 °C and 7000 × g to remove cellular debris and centrifuged again at 30000 × g to obtain a membrane-containing pellet. The membrane-containing pellet was then dissolved in a minimum volume of 10 mM HEPES buffer, pH 6.8, containing 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, and 1% Triton X-100. After centrifuging the dissolved pellet at 10000 × g to remove undissolved material, the supernatant was decanted into a Falcon tube containing concanavalin A-agarose resin and agitated lightly with a mixer for 2 hours at 4 °C. The resin was then gently sedimented at 600 rpm for 2 min and the supernatant removed. After washing the concanavalin A-resin twice in the re-suspension buffer, αIIbβ3 was eluted using 200 mM methyl-α-D-mannopyranose in 10 mM HEPES buffer, pH 6.8, containing 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, and 80 mM n-octyl-β-D-glucoside. The αIIbβ3 content of elution fractions was determined by 4–12% SDS-PAGE and fractions containing the greatest amounts of αIIbβ3 were combined and concentrated to 1 mg mL−1 using Amicon® Ultra 30k Centrifugal filters. Protein concentration was determined using the BCA technique. The polypeptide composition, purity, and monomeric state of αIIbβ3 and fibrinogen were verified by SDS-polyacrylamide gel electrophoresis and by transmission electron microscopy using rotary shadowing with tungsten.25 Functionality of the αIIbβ3 preparations was confirmed by the ability of the integrin to bind fibrinogen in solution and its susceptibility to activating effects of dithiothreitol and manganese ions.22
Dynamic light scattering (DLS)
DLS was used to compare changes in bead diameter after washing in different methods of dextran attachment to the latex bead surfaces. Briefly, 200 μL of each bead suspension was vortexed and thoroughly sonicated for a minute before being added to 1 mL of 20 mM HEPES buffer, pH 7.4. Samples were then vortexed and sonicated and added to a disposable PMMA cuvette. All DLS measurements were conducted on a Malvern Zetasizer Nano S at 21 °C.Fourier transform infrared spectroscopy (FTIR)
FTIR spectroscopy was used to identify chemical changes that occur in dextran during oxidation and covalent coupling. All measurements were made on cleaned silicon wafers. Bead suspensions were pipetted onto individual pieces of silicon and placed in a dessicator for 24 hours. Following this initial drying, pieces of the silicon were placed in a drying oven at 160 °C for 72 hours. FTIR spectra were recorded on a Perkin-Elmer infrared spectrometer (Spectrum RX 1 FTIR) at a resolution of 8 cm−1, averaging 256 scans.Results
Covalent grafting of dextran to latex beads
We used fluorescently-labeled dextran and fluorescence microscopy after multiple washes to verify that dextran was covalently-coupled to the NH2-functionalized latex beads. Compared to beads coated with TRITC-dextran by physisorption, multiply-washed beads containing covalently-coupled TRITC-dextran exhibited substantially greater fluorescence under identical exposure time, gain and offset of the photomultiplier (Fig. 3), suggesting that physisorbed dextran was removed during the washing cycles, whereas covalently-coupled dextran remained stably attached to the bead surface. This suggestion was confirmed using dynamic light scattering (DLS) to compare the average diameter of beads containing covalently-coupled versus physisorbed dextran as a function of the number of washes (Fig. 4). The average diameter of beads coated with dextran by physisorption gradually decreased from ∼2.2 μm to 1.9 μm between the first and fifth washing, whereas the diameter of beads coated covalently with dextran remained relatively constant between 1.90 μm and 1.95 μm. In addition, the average diameter of beads coated covalently with dextran was consistently larger than that of the uncoated beads, the difference attributed to a layer of dextran. The estimated thickness of the engrafted dextran layer was ∼45 nm, which is consistent with studies by Elender et al.26 and Piehler et al.27 However, the swelling of dextran depends on the coupling chemistry, molecular weight, and measuring technique8 and the thickness we measured is ∼5 times less than previous measurements for photoactivated dextran on planar silicon surfaces, possibly due to a higher grafting density in the previous studies.8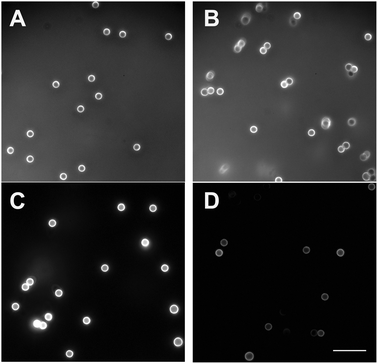 | ||
| Fig. 3 Fluorescent microscopy of NH2-modified latex beads coated with TRITC-labeled dextran either covalently (A, C) or by physisorption (B, D) before (A, B) and after (C, D) washing. The background fluorescence before washing is due to TRITC-dextran present in the solution. Physisorption of TRITC-dextran to bead surfaces occurred during a 24 hour incubation at 25 °C with untreated beads. Covalent binding of TRITC-dextran was made as described in Methods and shown in Fig. 2A. Each type of coated bead was washed five times by centrifugation using 20 mM HEPES, pH 7.4, containing 150 mM NaCl. Before and after washing, the beads were viewed in a fluorescent microscope (Olympus IX70) using the excitation wavelengths of 542–567 nm and the emission wavelengths of 602–662 (red filter) under identical exposure time, gain and offset of the photomultiplier. Magnification bar = 10 μm. | ||
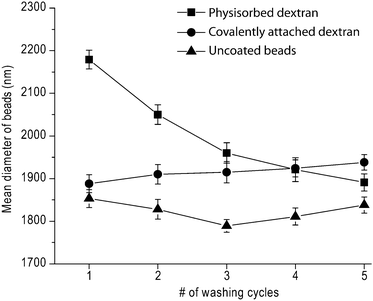 | ||
| Fig. 4 The average diameter of dextran-coated and uncoated latex beads as a function of the number of washing cycles. Beads size was determined by dynamic light scattering (DLS) as described in the “Experimental section”. | ||
FTIR spectroscopy was then used to confirm the formation of free aldehyde groups following dextran oxidation and their disappearance after coupling with primary amines, as well as the formation of secondary amines characteristic of a Schiff base. The FTIR spectrum of beads containing covalently-coupled dextran was compared to that of oxidized but unbound dextran and of covalently-bound, re-oxidized dextran-coated beads. As shown in Fig. 5A, an aldehyde peak was present at ∼1700 cm−1 in the oxidized unbound dextran sample. This peak disappeared following covalent coupling and peaks at ∼1500 cm−1 and 3100 cm−1 appeared, corresponding to the appearance of secondary amines. An aldehyde peak re-emerged at ∼1700 cm−1 for the covalently-bound re-oxidized dextran-coated beads (Fig. 5B). Thus, these studies, as well as the DLS, indicate that dextran can be covalently-coupled to amine-modified latex beads and that the engrafted dextran is stable following the reoxidation chemistry necessary for subsequent peptide attachment.
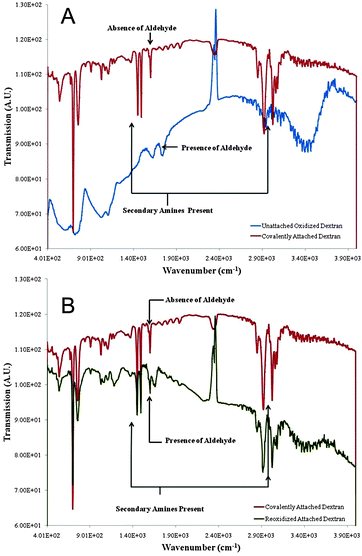 | ||
| Fig. 5 Fourier transform infrared spectroscopy (FTIR) of dextran after the oxidation and covalent binding to NH2-modified latex beads. (A) Covalently-attached dextran (upper curve) vs. unattached oxidized dextran (lower curve). Disappearance of the aldehyde group indicates covalent coupling. (B) Covalently-attached dextran (upper curve) vs. re-oxidized attached dextran (lower curve) demonstrating re-appearance of aldehyde groups in the re-oxidized sample. | ||
Non-specific interaction of dextran-coated latex beads with proteins
An objective of coating latex beads with dextran is to minimize non-specific interaction of the beads with biomolecules. To characterize the background reactivity of the dextran-coated latex beads, we used optical tweezers-based force spectroscopy. In these experiments, we compared the background reactivity of the latex beads, either covalently- or non-covalently-coated with dextran, with beads covalently-coupled with bovine serum albumin (BSA), a protein used previously as the standard for non-specific background.23 Each type of bead was brought into contact with silica pedestals coated with fibrinogen, a highly adhesive blood plasma protein.28 The resulting rupture force histograms (Fig. 6) and the derived quantitative parameters (Table 1) indicate that beads containing covalently-bound dextran had a substantially lower cumulative binding probability (0.9 ± 0.2% vs. 2.2 ± 0.7%, p < 0.01) and lower maximum rupture force (∼20 pN vs. ∼40 pN) than BSA-coated beads. Physisorbed dextran did not protect the beads from a wide range of non-specific interactions, with a cumulative binding probability of 8.2 ± 1.6% and a binding strength reaching ∼60 pN. Thus, latex beads coated covalently with dextran exhibit significantly lower non-specific interaction with fibrinogen compared to beads coated with BSA.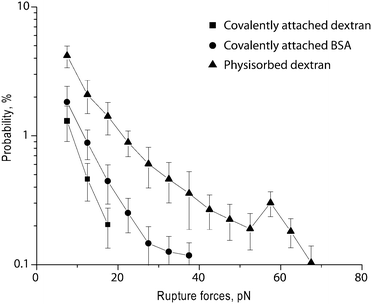 | ||
| Fig. 6 Non-specific reactivity of beads coated covalently with dextran, coated with dextran by physisorption, and coated covalently with bovine serum albumin (BSA). Data shown are rupture force histograms of the coated beads and immobilized human fibrinogen and represent averages from individual pedestal–bead pairs ±1 standard deviation. | ||
| Interacting surface coatings | Cumulative binding probability, % | Maximum force, pN | Most probable forcea, pN |
|---|---|---|---|
| a The most probable force is derived from the peak position of the Gaussian-like portion of the rupture force distribution. b p < 0.05. | |||
| Covalently attached dextran + fibrinogen | 0.9 ± 0.2 | 20 | No peak |
| BSA + fibrinogen | 2.2 ± 0.7 | 40 | No peak |
| Physisorbed dextran + fibrinogen | 8.2 ± 1.6 | ∼70 | No peak |
| cRGD + αIIbβ3 | 18.3 ± 5.3 | 90 | 57 ± 5b |
| cRAD + αIIbβ3 | 2.1 ± 0.6 | 50 | No peak |
| cRGD + αIIbβ3 + RGDS | 10.1 ± 2.7 | ∼60 | No peak |
| H12 + αIIbβ3 | 37.7 ± 11.3 | 80 | 49 ± 4b |
| H12 + αIIbβ3 + RGDS | 12.8 ± 4.2 | ∼80 | 46 ± 7 |
Interaction of peptide-coupled dextran-coated latex beads with αIIbβ3
Next, we coupled peptides corresponding to sites in the fibrinogen molecule that interact with αIIbβ3 to the dextran-coated latex beads and measured the interaction of these beads with purified and immobilized αIIbβ3 using optical tweezers-based force spectroscopy. The peptides studied were a cyclic Arg-Gly-Asp (RGD)-containing peptide (cRGD) and the γC-dodecapeptide (H12) corresponding to the extreme C-terminal 12 residues of the human fibrinogen γ chain. To confirm the specificity of the interaction of the peptide-coupled beads with αIIbβ3, rupture force measurements were made in the absence and presence of the integrin antagonist RGDS, as well as with dextran-coated beads that had either not been coupled to peptides or had been coupled to cRAD, known to not interact with αIIbβ3.The results of these studies are shown in Fig. 7 and 8 and Table 1. Whereas beads coated with dextran alone barely interacted with αIIbβ3 (binding probability 0.9 ± 0.2% and maximum rupture force ∼20 pN), latex beads coupled to cRGD displayed a bimodal interaction profile with a cumulative binding probability of 18.3 ± 5.3% and a maximum rupture force of ∼90 pN (Fig. 7). Addition of 1 mM RGDS resulted in a significant decrease in both the probability of binding (10.1 ± 2.7%, p < 0.05) and the maximum rupture force (∼60 pN). It is noteworthy that the force histograms for cRGD in the absence and presence of RGDS diverge at forces >30 pN, implying that rupture forces greater than 30 pN resulted from specific cRGD–αIIbβ3 bonds. Beads coated with cRAD were significantly less reactive with maximal rupture forces barely reaching 50 pN and a binding probability of 2.1 ± 0.6%.
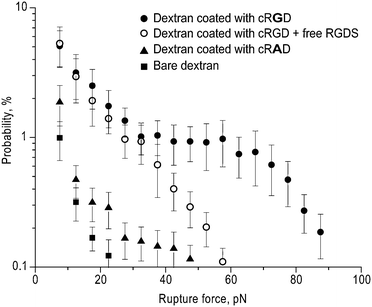 | ||
| Fig. 7 Rupture force histograms of the interaction of cRGD-coupled dextran-coated latex beads with immobilized αIIbβ3 purified from human platelets. Specific cRGD binding to αIIbβ3 was determined by performing the measurements in the presence of 1 mM RGDS or using cRAD-coupled dextran-coated latex beads. Each point represents averages from individual pedestal–bead pairs ±1 standard deviation. | ||
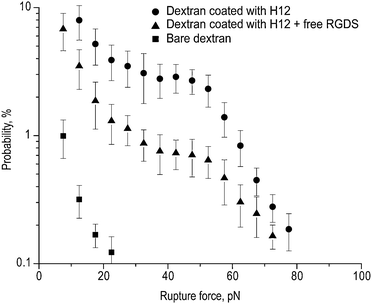 | ||
| Fig. 8 Rupture force histograms of the interaction of H12 peptide-bound dextran-coated latex beads with immobilized purified αIIbβ3. Specific H12 peptide binding to αIIbβ3 was determined by performing the measurements in the presence of 1 mM RGDS. Each point represents averages from individual pedestal–bead pairs ±1 standard deviation. | ||
We then measured the interaction of dextran-coated beads coupled to the H12 peptide with αIIbβ3 (Fig. 8). Compared to beads coupled to cRGD, there was a significantly greater cumulative probability of interacting with αIIbβ3 (37.7 ± 11.3%), but with a maximum rupture force that was comparable at ∼80 pN. The effect of 1 mM RGDS on this interaction was also different. Whereas RGDS significantly decreased the binding probability to 12.8 ± 4.2% (p < 0.01), similar to its effect on the interaction of cRGD-coupled beads with αIIbβ3, it did so without affecting the maximum rupture force. Thus, these results indicate that although both cRGD and H12 interact with αIIbβ3, their interaction is not equivalent.
Discussion
We have studied adhesive molecular interactions mediated by the platelet integrin αIIbβ3 using novel chemical and biophysical approaches. First, we have developed methodology to functionalize microscopic spherical surfaces which can be used in various applications related to receptor–ligand interaction studies, such as biomembrane force probe, magnetic or optical tweezers, atomic force microscopy, and particle–cell binding reactions. Next, we applied a highly sensitive biophysical technique, optical tweezers, which is capable of measuring adhesive forces between two spherical microparticles that are repeatedly touched and separated.To synthesize peptide-coated latex beads, we utilized a two-step double oxidation procedure based on the ability of oxidized dextran to form a Schiff base with primary amine groups to couple biologically active peptides to latex beads (Fig. 1 and 2). The successful covalent coupling of dextran to latex beads is supported by three independent studies. First, fluorescent dextran covalently-bound to the beads did not detach after repeated washing cycles. By contrast, the chemically-untreated physisorbed TRITC-dextran was desorbed during washing, resulting in beads that were not visible by fluorescence microscopy (Fig. 3). Second, beads coupled with oxidized dextran did not change their size upon multiple washings, whereas beads bearing physisorbed dextran, although initially larger in diameter, became smaller with each washing cycle, behavior manifestly different from beads to which oxidized dextran was covalently attached (Fig. 4). It is noteworthy that the average diameter of the dextran-coated beads was consistently greater than that of the untreated control beads without dextran, further evidence for the presence of a stable, hydrated grafted polymer. Third, FTIR spectra provided direct evidence for chemically-induced structural changes in dextran, corresponding to successive steps of oxidation and reductive amination that resulted in the formation of covalent bonds between the polymer and the beads (Fig. 5).
Previous studies29 investigated the effect of molecular weight on dextran grafting to surfaces. For dextran prepared from Leuconostoc ssp., molecular weights ranging from 1 kDa to 100 kDa (close to that used in this study) were covalently grafted to surfaces and characterized by a range of methods including atomic force microscopy to determine roughness and coverage. The ∼100 kDa dextran provided the lowest contact angle and thickest film. Moreover, the highest molecular weight dextran best mimicked the endothelial surface in measurements of bubble adhesion measurement. Furthermore, a detailed study of the oxidation kinetics of 110 kDa dextran was presented.6
To test the adhesive properties of dextran-coated latex beads, they were brought into intermittent contact with a surface coated with the plasma protein fibrinogen. In vivo, fibrinogen is enzymatically converted to a fibrin polymer, a scaffold for blood clots and thrombi, and therefore is involved in multiple adhesive interactions in the vasculature and other tissues.28 The non-specific adhesive properties of covalently attached dextran were compared with physisorbed dextran and BSA, a relatively non-reactive blood plasma protein that is widely used as an inert blocker at bio-interfaces. The force histograms presented in Fig. 6 and their quantitative parameters in Table 1 clearly show that beads with physisorbed dextran were “stickier” to a fibrinogen-coated surface. On the other hand, beads with covalently attached dextran displayed remarkably low non-specific adhesiveness towards a fibrinogen-coated surface that was even less than that of BSA. The physisorbed dextran shows stronger adhesion to fibrinogen compared with covalently attached dextran because the bare polystyrene particles are attractive for proteins and the physisorbed (thinner) layer does not shield the core particle as well as the grafted dextran brush. Thus, the protocol for covalent coupling of oxidized dextran to NH2-modified latex beads enabled us to synthesize micron-size particles with extremely low protein adhesion, a prerequisite for functionalizing them for use in bioassays.
Dextran-coated beads were further modified to introduce bioactive peptides with high binding selectivity. One peptide, RGD, is a binding motif for at least 8 of 24 integrins of different structure and cellular origin30 and X-ray crystallography has begun to reveal the structural basis for these interactions.31–33Dextran coating containing immobilized RGD strongly promotes attachment and spreading of cells compared to dextran surfaces without RGD.34,35 Here, we asked whether cRGD, covalently-bound to dextran-coated latex beads, is able to interact specifically with the purified αIIbβ3, which is the most abundant platelet receptor for the RGD-containing protein ligands fibrinogen, von Willebrand factor, fibronectin, and vitronectin, and is required for platelet aggregation at the sites of vascular injury.18,20 In addition to RGD, αIIbβ3 interacts with residues located at the extreme C-terminus of the fibrinogen γ chain and represented by the H12 dodecapeptide used in this study. It is noteworthy that 10–15% of the fibrinogen γ chain occurs as an alternatively spliced variant (γ′), in which the C-terminal 400–411 motif is eliminated by adding new amino acids from 408 to 427.36 It is possible that in the absence of the γ chain C-terminal dodecapeptide, the RGD sequences in the Aα chain may play a role as the major integrin-binding site. Understanding the molecular basis for interaction of αIIbβ3 with RGD and the H12 sequence remains an unresolved issue whose kinetics and thermodynamics can be studied at the single-molecule level.
As a highly precise tool to assess the molecular mechanisms of the αIIbβ3–peptide interactions, we used an original optical tweezers-based model system developed in our laboratory to study individual receptor–ligand interactions.21,23,37 This system permits the measurement of discrete rupture forces up to about 100 pN, which are in the range of forces known to break non-covalent bimolecular interactions observed in biology.38
An important feature of the laser tweezers system is that it was specifically designed to ensure that the majority of observed rupture events are due to single bimolecular attachments. Evidence that this is indeed the case has been previously reported in detail.23 Briefly, this evidence includes the observation that rupture events always occur in one step, whereas the rupture of multiple attachments should occur in sequential multiple steps. Here, we found that >80% of the ruptures occurred in one step and the remainder occurred in two or more steps manifested as jagged signals (not shown). Only single-step interactions were included in our analysis. Further, the distribution of rupture forces of multiple identical attachments should appear as a series of peaks that are multiples of a single value of force and have probabilities inversely proportional to the number of bonds. As seen in Fig. 7 and 8, for interactions with rupture forces greater than 40 pN, we observed only a single well-defined peak in the force histograms. Lastly, to ensure that the majority of observed rupture events are due to single αIIbβ3–peptide bonds, surface-coating conditions are tuned to keep protein density low. Thus, the frequency of binding events comprised <20% of total interface contacts, the exception being H12–αIIbβ3 interactions, which had a cumulative binding probability of ∼40% due to increased surface density (Table 1).
Two conclusions are immediately apparent from an analysis of the rupture force histograms for cRGD–αIIbβ3 and H12–αIIbβ3 (Fig. 7 and 8). First, the peptide-functionalized beads are much more reactive than bare dextran-coated beads. Coupling cRGD or H12 resulted in strong interactions with αIIbβ3, producing a broad range of forces up to 90 pN, appearing in two relatively distinctive ranges, one with exponentially decreasing probability and the other as a Gaussian-like peak. Importantly, the bimodal force distributions we observed are similar to those we previously detected for the interaction of αIIbβ3 with fibrinogen, but with lower binding strengths, as might be expected since the coupled peptides only partially correspond to the binding interface of the whole fibrinogen molecule.23 It is noteworthy that the broad force range is an intrinsic property of single-molecule force spectroscopy, originating from the stochastic nature of the interactions, molecular heterogeneity, and extraordinary sensitivity of the optical trap. Second, the interactions we observed are specific, inasmuch as they were sensitive to the presence of RGDS, a competitive inhibitor of fibrinogen binding to αIIbβ3.39 RGDS at 1 mM reduced the probability of interaction between αIIbβ3 and beads coated with cRGD or H12 by 45% and 66%, respectively, comparable to the 45% reduction in the interaction of αIIbβ3 with beads coated with fibrinogen by the same concentration of RGDS.23 The RGDS competitor more effectively suppressed higher forces, indicating that they reflected the most specific portion of the integrin–peptide interactions. Similarly, we found that beads coated with non-reactive peptide cRAD were only slightly stickier than beads coated with dextran but uncoupled to peptide. The latter result also confirms that re-oxidizing and further processing dextran neither physically degrades the latex bead surface nor changes the dextran chemically to make it less inert.
The bimodal rupture force distribution observed for cRGD–αIIbβ3 and H12–αIIbβ3, as we previously suggested,23,37 indicates that the complex of αIIbβ3 bound to ligands likely exists in two states with different mechanical stabilities. Based on the effect of RGDS on αIIbβ3 binding to beads coupled with cRGD, the interaction can be segregated into relatively non-specific interactions with rupture forces <25–30 pN and more specific interactions whose rupture forces exceed this value. We used the latter part of the rupture force distribution, which appeared as a Gaussian-like peak, to determine the binding strength of αIIbβ3 to cRGD, defined as the most probable force or a peak position on the X-axis. Under our experimental conditions, the binding strength was ∼60 pN. This value is smaller than the unbinding force of 93 pN determined by AFM for the single molecular complex of an RGD-containing peptide and αIIbβ3 on the platelet membrane.40 The difference is likely attributable to the substantially higher loading rate used in the AFM studies (12000 pN s−1) compared to ours (2000 pN s−1), given the logarithmic dependence of measured rupture forces on the rate at which load is applied to the molecular complex.41
Comparison of the cRGD–αIIbβ3vs. the H12–αIIbβ3 interactions reveals two major differences, one in the binding probability and the other in the binding strength. The 2-fold difference in the binding probability (18% for cRGD and 38% for H12) may be attributable to the different surface density of the immobilized peptides, despite the fact that the binding procedures for the two peptides were similar. H12 has two potentially reactive amine groups, the α-amine of the N-terminal glycine and the ε-amine of lysine; thus, it is twice as reactive with oxidized dextran as cRGD which has only one reactive amine group. Further, there is a moderate, but statistically significant, difference between the values of binding strength for cRGD and H12, indicating that the cRGD–αIIbβ3 complex is more mechanically stable. Although the results are not fully comparable due to a number of major experimental differences, this conclusion is consistent with the results of AFM experiments using live platelets showing that the zero-force kinetic off-rate (Koff) was much greater for H12 than for an RGD-containing peptide, indicating that the H12–αIIbβ3 complex dissociates faster upon mechanical separation.42
Conclusions
We used a combination of a newly developed method for coating latex beads with peptide-functionalized dextran and optical trap-based single-molecule force spectroscopy to measure the binding strength of canonical αIIbβ3–peptide binding partners. The dextran-coated beads were shown to be inert to fibrinogen, an adhesive protein involved in blood clotting and were even less reactive than beads coated with BSA, a standard inert blocker protein. The inertness of dextran and its ability to undergo specific biochemical modifications hold great potential for such future biomedical applications. We successfully functionalized the surface of dextran with biologically active fibrinogen-related peptides, cRGD and γC dodecapeptide, and showed that peptide-functionalized dextran-coated latex beads exhibited high binding reactivity and molecular specificity towards αIIbβ3. Finally, using optical trap-based single-molecule force spectroscopy we revealed remarkable interaction profiles for each of the peptides with a bimodal force distribution. The cRGD–αIIbβ3 interactions were more stable and resistant mechanically than that of the H12–αIIbβ3 complexes, consistent with the remarkable platelet reactivity with RGD-containing ligand proteins. The results provide quantitative mechanistic insights into αIIbβ3–ligand interactions that underlie platelet function.Acknowledgements
This research was supported by the NSF/NanoBio Interface Center under grant DMR08-32802 (RJC, JWW) as well as grants NSF/DMR09-07493 (RJC), NIH HL060230 (RJC), HL030954/HL090774 (JWW), and HL40387/HL81012 (JSB). We thank the Microscopy Core (Director—Andrea Stout) of the Department of Cell and Developmental Biology, Perelman School of Medicine, University of Pennsylvania, for assistance with fluorescent microscopy.References
- E. Petsalaki and R. B. Russell, Curr. Opin. Biotechnol., 2008, 19, 344–350 CrossRef CAS.
- P. Vanhee, J. Reumers, F. Stricher, L. Baeten, L. Serrano, J. Schymkowitz and F. Rousseau, Nucleic Acids Res., 2009, 38, D545–D551 CrossRef.
- M. H. Lee, D. A. Brass, R. Morris, R. J. Composto and P. Ducheyne, Biomaterials, 2005, 26, 1721–1730 CrossRef CAS.
- M. H. Lee, C. S. Adams, D. Boettiger, W. F. Degrado, I. M. Shapiro, R. J. Composto and P. Ducheyne, J. Biomed. Mater. Res., Part A, 2007, 81, 150–160 CrossRef.
- M. H. Lee, D. Boettiger, P. Ducheyne and R. J. Composto, Silanes and Other Coupling Agents, 2007, 4, 1–16 Search PubMed.
- D. Miksa, E. R. Irish, D. Chen, R. J. Composto and D. M. Eckmann, Biomacromolecules, 2006, 7, 557–564 CrossRef CAS.
- M. H. Lee, D. Boettiger and R. J. Composto, Biomacromolecules, 2008, 9, 2315–2321 CrossRef CAS.
- M. C. Ferrer, S. Yang, D. M. Eckmann and R. J. Composto, Langmuir, 2010, 26, 14126–14134 CrossRef.
- I. Y. Tsai, N. Tomczyk, J. I. Eckmann, R. J. Composto and D. M. Eckmann, Colloids Surf., B, 2011, 84, 241–252 CrossRef CAS.
- I. Y. Tsai, C. C. Kuo, N. Tomczyk, S. J. Stachelek, R. J. Composto and D. M. Eckmann, Soft Matter, 2011, 7, 3599–3606 RSC.
- M. S. Wang, L. B. Palmer, J. D. Schwartz and A. Razatos, Langmuir, 2004, 20, 7753–7759 CrossRef CAS.
- S. P. Massia, J. Stark and D. S. Letbetter, Biomaterials, 2000, 21, 2253–2261 CrossRef CAS.
- S. P. Massia, M. M. Holecko and G. R. Ehteshami, J. Biomed. Mater. Res., Part A, 2004, 68, 177–186 Search PubMed.
- D. Machy, P. Carteron and J. Jozefonvicz, J. Biomater. Sci., Polym. Ed., 2002, 13, 963–975 CrossRef CAS.
- D. Letourneur, C. Parisel, S. Prigent-Richard and M. Cansell, J. Controlled Release, 2000, 65, 83–91 CrossRef CAS.
- T. G. Van Thienen, K. Raemdonck, J. Demeester and S. C. De Smedt, Langmuir, 2007, 23, 9794–9801 CrossRef CAS.
- C. Jonsson, M. Aronsson, G. Rundstrom, C. Pettersson, I. Mendel-Hartvig, J. Bakker, E. Martinsson, B. Liedberg, B. MacCraith, O. Ohman and J. Melin, Lab Chip, 2008, 8, 1191–1197 RSC.
- J. S. Bennett, Ann. N. Y. Acad. Sci., 2001, 936, 340–354 CrossRef CAS.
- J. S. Bennett, J. Clin. Invest., 2005, 115, 3363–3369 CrossRef CAS.
- B. S. Coller and S. J. Shattil, Blood, 2008, 112, 3011–3025 CrossRef CAS.
- R. I. Litvinov, H. Shuman, J. S. Bennett and J. W. Weisel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7426–7431 CrossRef CAS.
- R. I. Litvinov, C. Nagaswami, G. Vilaire, H. Shuman, J. S. Bennett and J. W. Weisel, Blood, 2004, 104, 3979–3985 CrossRef CAS.
- R. I. Litvinov, J. S. Bennett, J. W. Weisel and H. Shuman, Biophys. J., 2005, 89, 2824–2834 CrossRef CAS.
- M. C. Hillman, N. I. Zolotarjova, D. R. Patrick, D. D. McCabe, K. Shen, J. I. Corman, J. T. Billheimer, D. A. Seiffert, G. F. Hollis and R. Wynn, Protein Expression Purif., 2002, 25, 494–502 CrossRef CAS.
- J. W. Weisel, C. Nagaswami, G. Vilaire and J. S. Bennett, J. Biol. Chem., 1992, 267, 16637–16643 CAS.
- G. Elender, M. Kuhner and E. Sackmann, Biosens. Bioelectron., 1996, 11, 565–577 CrossRef CAS.
- J. Piehler, A. Brecht, K. Hehl and G. Gauglitz, Colloids Surf., B, 1999, 13, 325–336 CrossRef CAS.
- J. W. Weisel, Adv. Protein Chem., 2005, 70, 247–299 CrossRef CAS.
- M. Ombelli, D. M. Eckman and R. J. Composto, Mater. Res. Soc. Symp. Proc., 2003, 734, B10.17.11–B10.17.16 Search PubMed.
- E. Ruoslahti, Annu. Rev. Cell Dev. Biol., 1996, 12, 697–715 CrossRef CAS.
- J. P. Xiong, T. Stehle, R. Zhang, A. Joachimiak, M. Frech, S. L. Goodman and M. A. Arnaout, Science, 2002, 296, 151–155 CrossRef CAS.
- T. Xiao, J. Takagi, B. S. Coller, J. H. Wang and T. A. Springer, Nature, 2004, 432, 59–67 CrossRef CAS.
- T. A. Springer, J. Zhu and T. Xiao, J. Cell Biol., 2008, 182, 791–800 CrossRef CAS.
- M. Dubs, J. Weissler, R. Linke, A. Pfuch, D. Imhof and M. Schnabelrauch, Materialwiss. Werkstofftech., 2009, 40, 853–860 CrossRef CAS.
- S. P. Massia and J. Stark, J. Biomed. Mater. Res., 2001, 56, 390–399 CrossRef CAS.
- S. Uitte de Willige, K. F. Standeven, H. Philippou and R. A. Ariens, Blood, 2009, 114, 3994–4001 CrossRef CAS.
- R. I. Litvinov, V. Barsegov, A. J. Schissler, A. R. Fisher, J. S. Bennett, J. W. Weisel and H. Shuman, Biophys. J., 2011, 100, 165–173 CrossRef CAS.
- J. W. Weisel, H. Shuman and R. I. Litvinov, Curr. Opin. Struct. Biol., 2003, 13, 227–235 CrossRef CAS.
- J. S. Bennett, S. J. Shattil, J. W. Power and T. K. Gartner, J. Biol. Chem., 1988, 263, 12948–12953 CAS.
- I. Lee and R. E. Marchant, Surf. Sci., 2001, 491, 433–443 CrossRef CAS.
- E. Evans and K. Ritchie, Biophys. J., 1997, 72, 1541–1555 CrossRef CAS.
- I. Lee and R. E. Marchant, Ultramicroscopy, 2003, 97, 341–352 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |