Oxidation-hydroxymethylation-reduction: a one-pot three-step biocatalytic synthesis of optically active α-aryl vicinal diols
Saravanakumar
Shanmuganathan
a,
Dessy
Natalia
a,
Lasse
Greiner
*ab and
Pablo
Domínguez de María
*a
aInstitut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringerweg 1, 52074, Aachen, Germany. E-mail: dominguez@itmc.rwth-aachen.de; Fax: +49 241 8022177; Tel: +49 241 8020468
bDECHEMA e.V. Karl-Winnacker-Institut, Theodor-Heuss-Allee 25, D-60486, Frankfurt am Main, Germany. E-mail: greiner@dechema.de; Fax: +49 697564388; Tel: +49 697564337
First published on 8th December 2011
Abstract
A one-pot multi-step approach comprising enzymatic oxidation-hydroxymethylation-reduction enables the synthesis of optically active α-aryl vicinal diols with high yields and enantioselectivities. Formaldehyde required for the hydroxymethylation step is enzymatically produced in situ using less hazardous methanol as substrate.
The production of enantiopure building blocks for manufacturing pharmaceuticals or biologically-active compounds usually involves elaborate multi-step synthetic protocols. Typically, isolation of intermediates and tedious protection-deprotection steps are needed, often leading to low yields and high E-factors. One powerful approach towards sustainable organic synthesis lies in the development of one-pot multi-step syntheses, e.g.enzyme cascades inspired by the in vivobiosynthesis. For the successful implementation of one-pot catalytic multi-step reaction systems, stringent conditions need to be orchestrated for optimal results. Reaction rates need balancing; compatibility of catalysts has to be achieved; and productivity has to be optimized.1–3 A few multi-step catalytic processes have been reported, comprising excellent examples of enzymatic and metal-enzyme combinations.4–10 Furthermore, recent elegant approaches combine organo- and biocatalysis as well.11–12
In this communication, we disclose a one-pot multi-enzymatic concept utilizing oxidoreductases and benzaldehyde lyase (BAL), a thiamine-diphosphate-dependent (ThDP) lyase, for the production of optically active α-aryl vicinal diols, which are valuable building blocks for fine chemicals and pharmaceuticals.13 The use of multi-step biocatalytic reactions for the production of these important building blocks has been testimonial, with only few examples involving lyases and oxidoreductases (in two-pot reactions),14 or chemo-catalysis.15,16 Herein, the core step – a C–C bond formation – is enabled by the capability of BAL to catalyze the hydroxymethylation of aromatic aldehydes with formaldehyde to afford α-aryl-2-oxo-1-hydroxy ketones.17–20 Alternatively, this step could also be conducted viacarbene-based organocatalysis,21–23 mimicking the function of ThDP-lyases.24–26 Yet, in itself, the efficient organocatalytic approach requires either stoichiometric amounts of bases or elevated temperatures, thus combining this with enzymatic steps in one-pot could prove difficult.
In modern sustainable synthesis the required efficient and selective protocols need to be aligned with minimal waste production. Hazardous chemicals must be avoided or at least efficiently converted to safer chemicals. To combine these requirements with the supply of formaldehyde for the BAL-catalyzed hydroxymethylation17,18 two strategies were devised: A) Co-utilization of formaldehyde for cofactor regeneration in the subsequent alcohol dehydrogenase (ADH) catalyzed enantioselective ketone reduction (Scheme 1, route A); or B) Enzymatic in situ formation of formaldehyde through oxidation of the less hazardous methanol (Scheme 1, route B).
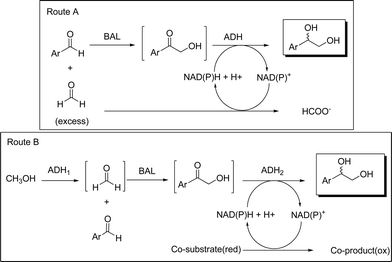 | ||
| Scheme 1 Conceptual outline for the multi-step biocatalytic reactions. | ||
As first step, the BAL-catalyzed carboligation of benzaldehyde and formaldehyde was carefully assessed to minimize the excess of formaldehyde needed to allow quantitative conversion in 1, as the competitive formation of benzoin potentially reduces overall selectivity. 2-Methyltetrahydrofuran (2-MeTHF) was employed as (bio-based) co-solvent, since it provides an excellent operational window for BAL, while avoiding significant formation of wastewater and further environmental issues associated with other co-solvents (e.g.DMSO, MTBE), leading to a diminished E factor in the process.27 Thus, a 3-fold excess of formaldehyde assured quantitative conversion in α-aryl-2-oxo-1-hydroxy ketone 1 (Scheme 2).
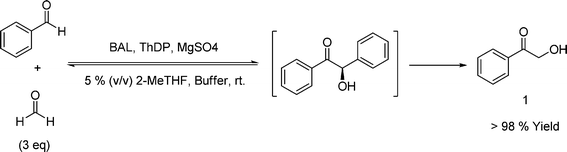 | ||
| Scheme 2 BAL-catalyzed hydroxymethylation. 20 mL phosphate buffer 50 mmol L−1, pH 8.0, 5% v/v 2-MeTHF, 0.25 mmol benzaldehyde, 0.75 mmol formaldehyde, 2.5 mmol L−1MgSO4, 0.15 mmol L−1ThDP, 10 U BAL. | ||
Once the reaction conditions of the enzymatic hydroxymethylation were optimized, the next step was to capitalize the excess of formaldehyde (Scheme 2) as ancillary substrate (cofactor regeneration) for the enantioselective ketone reduction. To this end, glycerol dehydrogenase from Cellulomonas sp. (GDH) was selected for the enantioselective reduction of 1.28 For cofactor regeneration (NADH), formaldehyde dehydrogenase from Pseudomonas putida was chosen.29 After careful optimization of absolute and relative enzyme and cofactor loadings with reaction times, high conversion and excellent enantioselectivity for the desired product diol (R)-2 were achieved (isolated yield 75%, ee > 99%) (Scheme 3).
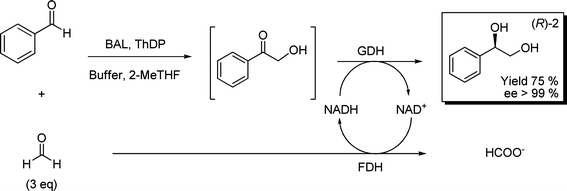 | ||
| Scheme 3 Enzymatic multi-step process for the enantioselective synthesis of (R)-2. 10 mL phosphate buffer 50 mmol L−1, pH 8.0, 5% v/v 2-MeTHF, 0.13 mmol benzaldehyde, 0.4 mmol formaldehyde, 2.5 mmol L−1MgSO4, 0.15 mmol L−1ThDP, 10 U BAL, 354 U GDH, 21 U FDH, NADH/NAD+ 0.5 mmol L−1 each. | ||
Alternatively, to avoid issues related to toxicity inherent to formaldehyde (still one equivalent present after hydroxymethylation and oxidation steps), the enzymatic in situformaldehyde formation and consumption was assessed, thus providing the basis for a “formaldehyde-free” hydroxymethylation. To this end, the FAD-dependent alcohol oxidase from Hansenula sp. proved to be an efficient catalyst for the oxidation of methanol with oxygen.30,31 The coupled product H2O2 was converted by catalase, regenerating oxygen in solution and avoiding detrimental effects to the enzymes. Firstly reactions were separated in time, that is, enzymatic methanol oxidation being conducted for 8 h, followed by BAL and benzaldehyde addition for additional 16 h. 1 was isolated in ∼93% yield along with traces of benzoin. Presumably, enzymatic oxidation steps are slower compared to the C–C ligation reaction. By systematically varying enzyme loadings, yield of 1 was quantitative, and remarkably colored impurities were diminished, compared to route A (with addition of excess of formaldehyde). Finally, since no deactivation of BAL in the presence of methanol or alcohol oxidase was observed, oxidation and hydroxymethylation were combined in one step. Gratifyingly, 1 was again formed in quantitative conversion. Consequently, all three reactions were conducted in a one-pot sequential array: first the in situoxidation of methanol to formaldehyde, followed by concomitant BAL-catalyzed cross condensation with benzaldehyde, and followed by Lb-ADH addition to afford (S)-2 quantitatively (isolated yield) with ee > 99% (Scheme 4). It is important to mention that in this case NADPH-dependent alcohol dehydrogenase from Lactobacillus brevis (LbADH) was used,32 affording the (S)-enantiomer. Thus, by smartly using different enzymes of a toolbox, access to both enantiomers might be achieved.
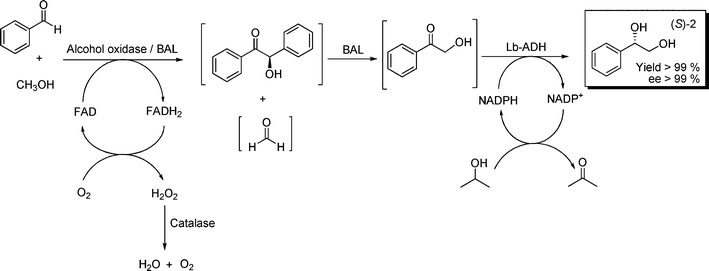 | ||
| Scheme 4 Four enzymes in one-pot reaction: In situoxidation-hydroxymethylation-reduction. 20 mL, phosphate buffer 50 mmol L−1, pH 8.0, 5% v/v 2-MeTHF, 0.12 mmol benzaldehyde, 0.90 mmol methanol, 33 U alcohol oxidase, 14 U catalase, 2.5 mmol L−1MgSO4, 0.15 mmol L−1ThDP, 40 U BAL, 10 U LbADH. 0.5 mmol L−1NADPH, 0.1 mmol L−1 FAD. | ||
Finally, the methodology was extended to more challenging aromatic aldehydes like furfural. Furan ethane diols can be used as building blocks for the preparation of levoglucosenone, a valuable synthon for biologically-sound intermediates.33–35 Traditional synthetic approaches involve Lewis acid-catalyzed glucal rearrangement, typically with poor enantioselectivity.33–35 Since furfural-based diols are unstable, they were chemically dibenzoylated in situ. The first approach following route A (Scheme 1) provided a successful proof-of-principle, with low isolated yields (25%) albeit with excellent (R)-enantioselectivity (Scheme 5).
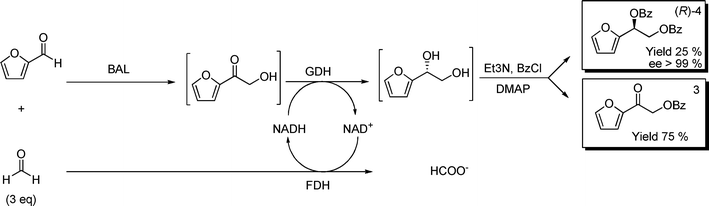 | ||
| Scheme 5 Biocatalytic formation of chiral furane-diolsvia route A. 40 mL phosphate buffer 50 mmol L−1, pH 8.0, 5% v/v 2-MeTHF, 1.6 mmol furaldehyde, 4.8 mmol formaldehyde, 2.5 mmol L−1MgSO4, 0.15 mmol L−1ThDP, 168 U BAL, 1500 U GDH, 95 U FDH, NADH/NAD+ 1 mmol L−1 each. | ||
With the considerable amount of 3 as intermediate despite the higher amount of GDH applied, the bottleneck was supposed to be at low activity of GDH towards furan compounds. In fact, when LbADH was applied, with glucose dehydrogenase (GlucDH) for cofactor regeneration, this resulted in excellent isolated yields (87%) and enantioselectivity (>99% ee, (S)) (Scheme 6). Therefore, the herein reported conceptual approach may accept other enzymes with different enantioselectivity or different bias towards organic molecules. Due to the ample portfolio of oxidoreductases already available,31,36 further successful combinations may be expected.
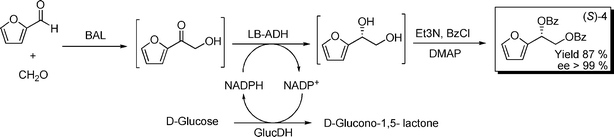 | ||
| Scheme 6 Biocatalytic formation of chiral furan-diols. 40 mL phosphate buffer 50 mM, pH 8.0, 5% v/v 2-MeTHF, 1.6 mmol furaldehyde, 3.6 mmol glucose, 4.4 mmol CaCO3, 4.8 mmol formaldehyde, 2.5 mmol L−1MgSO4, 0.15 mmol L−1ThDP, 168 U BAL, 50 U Lb-ADH, 2800 U GlucDH, 0.5 mmol L−1NADPH. | ||
By means of these strategies a significant reduction of the E factor can be achieved, since the amount of organic solvent (for product extraction), as well as water for the enzymatic process can be reduced to one third, due to the setup of just one final downstream processing. Importantly, these inherent environmental advantages of the herein reported concept still need to be complemented with optimized reaction times, substrate loadings and less enzyme concentrations, eventual biocatalyst recyclability, etc. In this respect, a promising approach for future research may be the incorporation of whole-cells with overexpressed enzymes, as it has been successfully reported for lyases or oxidoreductases in processes with enhanced enzyme stability and higher substrate loadings.37,38 With such implementation at hand, the smart combination of enzymatic cascade processes together with high productivities and efficiencies may be reached.
In summary one-pot multi-step biocatalytic concepts have been reported, comprising oxidation, hydroxymethylation, and a further reduction to afford chiral α-aryl vicinal diols. By in situ production or its subsequent utilization as reducing agent, the use of hazardous formaldehyde is addressed. Another advantage is that the concept allows the use of different oxidoreductases, providing access to both enantiomers (“on-demand”) and furan derivatives. Future sustainable (industrial) processes will adopt more and more such highly integrated multi-catalytic steps with diminished waste formation, together with optimized substrate concentration and enzyme loadings.
Acknowledgements
Financial support from DFG training group 1166 “BioNoCo” (“Biocatalysis in non-conventional Media”) is gratefully acknowledged. Mrs. Hannelore Eschmann and Ms. Julia Wurlitzer (both RWTH Aachen University) are acknowledged for support in analytical measurements (HPLC).References
- E. García-Junceda in Multi-Step Enzyme Catalysis – Biotransformations and Chemoenzymatic Synthesis, ed. S. G. Burton and M. le Roes-Hill, Wiley-VCH, Weinheim, 2008, pp 41–58 Search PubMed.
- T. Fischer and J. Pietruszka, Top. Curr. Chem., 2010, 297, 1–43 Search PubMed.
- H. C. Hailes, P. A. Dalby and J. M. Woodley, J. Chem. Technol. Biotechnol., 2007, 82, 1063–1066 CrossRef CAS.
- X. Tong, B. El-Zahab, X. Zhao, Y. Liu and P. Wang, Biotechnol. Bioeng., 2011, 108, 465–469 CrossRef CAS.
- A. Znabet, E. Ruijter, F. J. J. de Kanter, V. Köhler, M. Helliwell, N. J. Turner and R. V. A. Orru, Angew. Chem., Int. Ed., 2010, 49, 5289–5292 CrossRef CAS.
- F. R. Bisogno, I. Lavandera, W. Kroutil and V. Gotor, J. Org. Chem., 2009, 74, 1730–1732 CrossRef CAS.
- C V. Voss, C. C. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 741–745 CrossRef CAS.
- E. Burda, W. Hummel and H. Gröger, Angew. Chem., Int. Ed., 2008, 47, 9551–9554 CrossRef CAS.
- H. Ankati, D. Zhu, Y. Yang, E. R. Bieh and L. Hua, J. Org. Chem., 2009, 74, 1658–1662 CrossRef CAS.
- S. K. Karmee, C. Roosen, C. Kohlmann, S. Lütz, L. Greiner and W. Leitner, Green Chem., 2009, 11, 1052–1055 RSC.
- K. Baer, M. Krausser, E. Burda, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2009, 48, 9355–9358 CrossRef CAS.
- G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 7944–7947 Search PubMed.
- R. Kadyrov, R. M. Koenigs, C. Brinkmann, D. Voigtlaender and M. Rueping, Angew. Chem., Int. Ed., 2009, 48, 7556–7559 Search PubMed.
- D. Kihumbu, T. Stillger, W. Hummel and A. Liese, Tetrahedron: Asymmetry, 2002, 13, 1069–1072 CrossRef CAS.
- L. C. Bencze, C. Paizs, M. L. Tosa, F. Dan Irimie and J. Retey, ChemCatChem, 2011, 3, 343–346 Search PubMed.
- A. Kamal, M. Sandbhor, K. Ahmed, S. F. Adil and A. Ali Shaik, Tetrahedron: Asymmetry, 2003, 14, 3861–3866 Search PubMed.
- A. S. Demir, P. Ayhan, A. C. Igdir and A. N. Duygu, Tetrahedron, 2004, 60, 6509–6512 CrossRef CAS.
- A. Cosp, C. Dresen, M. Pohl, L. Walter, C. Röhr and M. Müller, Adv. Synth. Catal., 2008, 350, 759–771 CrossRef CAS.
- M. Brovetto, D. Gamenara, P. Saenz Méndez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 Search PubMed.
- P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcántara and P. Domínguez de María, Acc. Chem. Res., 2010, 43, 288–299 CrossRef CAS.
- N. Kuhl and F. Glorius, Chem. Commun., 2011, 47, 573–575 RSC.
- R. Hackstell, I. Jovel, M. Beller, M. Hateley, C. Weckbecker and K. Huthmacher, US 7504542, 2009.
- T. Matsumoto, M. Ohishi and S. Inoue, J. Org. Chem., 1985, 50, 603–606 CrossRef CAS.
- P. Domínguez de María and S. Shanmuganathan, Curr. Org. Chem., 2011, 15, 2083–2097 Search PubMed.
- D. Enders and A. A. Narine, J. Org. Chem., 2008, 73, 7857–7870 CrossRef CAS.
- D. Enders, M. R. M. Hüttl, O. Niemeier, in Organocatalysis, ed., M. T. Reetz, B. List, S. Jaroch, and H. Weinmann, Springer-Verlag, Berlin 2008, pp 45–124 Search PubMed.
- S. Shanmuganathan, D. Natalia, A. van den Wittenboer, C. Kohlmann, L. Greiner and P. Domínguez de María, Green Chem., 2010, 12, 2240–2245 RSC.
- D. Degenring, I. Schröder, C. Wandrey, A. Liese and L. Greiner, Org. Process Res. Dev., 2004, 8, 213–218 Search PubMed.
- M. Ando, T. Yoshimoto, S. Ogushi, K. Rikitake, S. Shibata and D. Tsuru, J. Biochem., 1979, 85, 1165–1172 CAS.
- B. Yoon, H. S. Kim, T. J. Kwon, J. W. Yang, G. S. Kwon, H. S. Lee, J. S. Ahn and T. I. Mheen, J. Microbiol. Biotechnol., 1992, 12, 278–283 Search PubMed.
- F. Hollmann, I. W. C. E. Arends, K. Bühler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC.
- S. Leuchs and L. Greiner, Chem. Biochem. Eng. Q., 2011, 25, 267–281 Search PubMed.
- C. Müller, M. A. Gómez-Zurita, D. Ballinari, S. Colombo, A. Bitto, E. Martegani, C. Airoldi, A. van Neuren, M. Stein, J. Weiser, C. Battistini and F. Peri, ChemMedChem, 2009, 4, 524–528 Search PubMed.
- A. M. Sarotti, R. A. Spanevello and A. G. Suárez, Green Chem., 2007, 9, 1137–1140 RSC.
- J. M. Harris, M. D. Keränen, H. Nguyen, V. G. Young and G. A. O'Doherty, Carbohydr. Res., 2000, 328, 17–36 CrossRef CAS.
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2314 RSC.
- P. Domínguez de María, T. Stillger, M. Pohl, M. Kiesel, A. Liese, H. Gröger and H. Trauthwein, Adv. Synth. Catal., 2008, 350, 165–173 CrossRef CAS.
- A. Jakoblinnert, R. Mladenov, A. Paul, F. Sibilla, U. Schwaneberg, M. B. Ansorge-Schumacher and P. Domínguez de María, Chem. Commun., 2011, 47, 12230–12232 RSC.
| This journal is © The Royal Society of Chemistry 2012 |