Dietary fatty acids linking postprandial metabolic response and chronic diseases
Almudena
Ortega†
,
Lourdes M.
Varela†
,
Beatriz
Bermudez†
,
Sergio
Lopez†
,
Rocio
Abia
and
Francisco J. G.
Muriana
*
Laboratory of Cellular and Molecular Nutrition, Instituto de la Grasa (CSIC), 41012, Seville, Spain. E-mail: muriana@ig.csic.es; Fax: +95 4616790; Tel: +95 4611550
First published on 21st October 2011
Abstract
Chronic diseases are by far one of the main causes of mortality in the world. One of the current global recommendations to counteract disability and premature death resulting from chronic diseases is to decrease the consumption of energy-dense high-fat diets, particularly those rich in saturated fatty acids (SFA). The most effective replacement for SFA in terms of risk factor outcomes for chronic disease are polyunsaturated fatty acids (PUFA) and monounsaturated fatty acids (MUFA). The biochemical basis for healthy benefits of such a dietary pattern has been widely evaluated under fasting conditions. However, the increasing amount of data available from multiple studies suggest that the postprandial state, i.e., “the period that comprises and follows a meal”, plays an important, yet underappreciated, role in the genesis of numerous pathological conditions. In this review, the potential of MUFA, PUFA, and SFA to postprandially affect selected metabolic abnormalities related to chronic diseases is discussed.
 Almudena Ortega Almudena Ortega | Almudena Ortega, Rocío Abia, Lourdes M. Varela (down in the photo, from left to right), Sergio López, Beatriz Bermúdez, and Francisco J. G. Muriana (up in the photo, from left to right) belong to the Laboratory of Cellular and Molecular Nutrition at the Instituto de la Grasa of The Spanish National Research Council (CSIC). Their research interests focus on cellular and molecular effects of dietary fats in the postprandial state, and their current research involves topics related with the role of postprandial triglyceride-rich lipoproteins on plaque development and stability. |
1. Introduction
The consumption of monounsaturated fatty acids (MUFA) and polyunsaturated fatty acids (PUFA) in place of saturated fatty acids (SFA) is considered one of the major worldwide nutrition policies for the prevention and control of chronic diseases. This recommended community-wide action arises because we fully recognise the essential role of dietary fatty acids in inducing substantial negative and positive effects on health throughout life. Most importantly, MUFA, PUFA, and SFA adjustment may not only influence present health, but may determine whether an individual will develop obesity, diabetes, hypertension, dyslipidemia, cardiovascular disease, stroke or some types of cancer much later in life.1First, the general dietary goal is to restrict SFA consumption to less than 10% of the total daily calories and less than 7% for high-risk people. Then, the consumption of the other fatty acids is adjusted: PUFA, which is also limited to less than 10% of the total daily calories (ranging from 6% up to 10%), and MUFA, which may supply daily calories that are at least equivalent to those provided by SFA and PUFA together (Fig. 1). The daily total fat intake will range from 15% up to 35% of total daily calories, depending on age, gender, activity, adequate body weight, and the type of dietary fats (e.g. even up to 40% of total daily calories is allowed if virgin olive oil is the main source of MUFA in the diet).2 The monitoring of metabolic parameters, routinely in the fasting state, is often used to ensure successful long-term outcomes. However, transient but repetitive changes in the postprandial lipid metabolism occur every time we eat a fatty meal. In the postprandial state, dietary fatty acids are largely incorporated into nascent triglyceride-rich lipoproteins, which are released from the small intestine into the blood and thereafter cleared by extracellular lipolytic3 and non-lipolytic4 pathways in the tissues. MUFA, PUFA, and SFA have dissimilar postprandial effects on the risk factors for chronic diseases,2,5–8 suggesting that short-term outcomes in response to dietary fatty acid adjustment could be useful to finely tune fat consumption, even for preventing diet-related chronic diseases. In this review, we summarise the evidence in support of the influence of the metabolic control of dietary fatty acids on the risk and pathogenesis of chronic diseases in the context of the postprandial state.
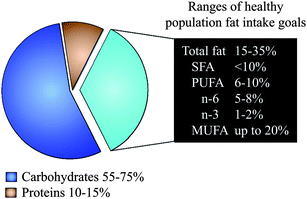 | ||
| Fig. 1 Ranges of population fat intake goals. SFA: saturated fatty acids, PUFA: polyunsaturated fatty acids, MUFA: monounsaturated fatty acids. | ||
2. Dietary fatty acid identities
A fatty acid is a carboxylic acid that often has a long unbranched aliphatic chain. Fatty acids are divided into SFA and unsaturated fatty acids based on structural and chemical properties (Fig. 2). SFA do not contain any double bonds or other functional groups along the chain, which is fully saturated with hydrogen atoms. Palmitic acid [16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 (PA)] is composed of 16 carbon atoms and is the principal SFA in the diet. SFA is found chiefly in animal products, including meats and dairy foods, but is also found in some plant sources, including coconut, cottonseed, and palm kernel oils. MUFA are unsaturated fatty acids that contain one pair of carbon atoms linked by a cis double bond. The major dietary MUFA is oleic acid [18:1n-9 (OA)], which has 18 carbon atoms with the double bond occurring 9 carbon atoms away from the methyl end of the fatty acid molecule. OA is the primary component of olive oil, but also can be found in hazelnut, canola, and peanut oils. A carbon chain that contains two or more cis double bonds with the first double bond located between the third and fourth or sixth and seventh carbon atom from the methyl end of the fatty acid molecule characterises the families of n-3 or n-6 PUFA. These families cannot be synthesised by the human body (double bonds can be introduced into all positions of the fatty acid chain except for the n-3 and n-6 positions) and must be obtained from the diet either as alpha-linolenic acid [18:3n-3 (αLN)] and linoleic acid [18:2n-6 (LA)], or their long-chain PUFA derivatives.9 Of these fatty acids, eicosapentaenoic acid [20:5n-3 (EPA)], docosahexaenoic acid [22:6n-3 (DHA)], dihomo-gamma linolenic acid [20:3n-6 (DGLA)], and arachidonic acid [20:4n-6 (AA)] are the most metabolically significant. While conversion of LA to DGLA and AA is typically very efficient, conversion of αLN to EPA and DHA is much less so.10 This fact has particular importance in people with compromised αLN availability or conversion enzyme activity. Therefore, not only αLN and LA but also EPA and DHA should be considered as essential fatty acids. LA and αLN can be found in vegetable oils, LA in safflower, sunflower, soybean, maize, and cottonseed oils, and αLN in flaxseed, blackcurrant, walnut, rapeseed, and soybean oils. EPA and DHA are abundant in cold-water fatty fish, including herring, sardines, mackerel, salmon, tuna, and shellfish. The competition between EPA and DHA with DGLA and AA and the opposing effects of their oxidative metabolites suggest the importance of an optimum ratio for the consumption of n-3 and n-6 PUFA.11 It is widely accepted that the ratio of n-3 to n-6 consumption in a typical Western diet is imbalanced and extends into the range of 1:10 to 1:25, which is far from the currently established ideal consumption ratio of between 1:1 and 1:4.12
:0 (PA)] is composed of 16 carbon atoms and is the principal SFA in the diet. SFA is found chiefly in animal products, including meats and dairy foods, but is also found in some plant sources, including coconut, cottonseed, and palm kernel oils. MUFA are unsaturated fatty acids that contain one pair of carbon atoms linked by a cis double bond. The major dietary MUFA is oleic acid [18:1n-9 (OA)], which has 18 carbon atoms with the double bond occurring 9 carbon atoms away from the methyl end of the fatty acid molecule. OA is the primary component of olive oil, but also can be found in hazelnut, canola, and peanut oils. A carbon chain that contains two or more cis double bonds with the first double bond located between the third and fourth or sixth and seventh carbon atom from the methyl end of the fatty acid molecule characterises the families of n-3 or n-6 PUFA. These families cannot be synthesised by the human body (double bonds can be introduced into all positions of the fatty acid chain except for the n-3 and n-6 positions) and must be obtained from the diet either as alpha-linolenic acid [18:3n-3 (αLN)] and linoleic acid [18:2n-6 (LA)], or their long-chain PUFA derivatives.9 Of these fatty acids, eicosapentaenoic acid [20:5n-3 (EPA)], docosahexaenoic acid [22:6n-3 (DHA)], dihomo-gamma linolenic acid [20:3n-6 (DGLA)], and arachidonic acid [20:4n-6 (AA)] are the most metabolically significant. While conversion of LA to DGLA and AA is typically very efficient, conversion of αLN to EPA and DHA is much less so.10 This fact has particular importance in people with compromised αLN availability or conversion enzyme activity. Therefore, not only αLN and LA but also EPA and DHA should be considered as essential fatty acids. LA and αLN can be found in vegetable oils, LA in safflower, sunflower, soybean, maize, and cottonseed oils, and αLN in flaxseed, blackcurrant, walnut, rapeseed, and soybean oils. EPA and DHA are abundant in cold-water fatty fish, including herring, sardines, mackerel, salmon, tuna, and shellfish. The competition between EPA and DHA with DGLA and AA and the opposing effects of their oxidative metabolites suggest the importance of an optimum ratio for the consumption of n-3 and n-6 PUFA.11 It is widely accepted that the ratio of n-3 to n-6 consumption in a typical Western diet is imbalanced and extends into the range of 1:10 to 1:25, which is far from the currently established ideal consumption ratio of between 1:1 and 1:4.12
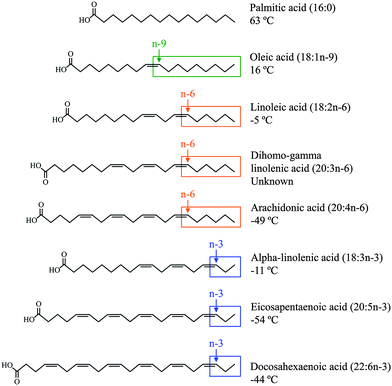 | ||
| Fig. 2 The structure and melting point of the most significant dietary fatty acids. | ||
Requirements in MUFA, n-3 and n-6 PUFA are satisfied by the diet. MUFA can be synthesised from acetyl-CoA within mammalian tissues. However, it is unclear whether the entire MUFA requirement can be met by de novo metabolic machinery. MUFA, and specifically OA, represent one of the core components of the Mediterranean diet (mainly due to the liberal use of virgin olive oil), which represents a prototypical dietary model associated with a long life expectancy and a low occurrence of chronic diseases.2
3. The role of dietary fatty acids in the risk factors for chronic diseases
There is increasing evidence to suggest that postprandial hypertriglyceridemia, which also severely affects insulin secretion and action,6 contributes to the pathogenesis of type 2 diabetes, atherosclerosis, coronary heart disease (CHD), and stroke.13–15 Worldwide, these chronic diseases led to approximately 30% of all deaths, and the proportion is predicted to substantially increase in the next few years. The most important behavioural risk factor for type 2 diabetes, CHD, and stroke is unhealthy diet,16,17 mainly due to a high-intake of SFA. However, changes in the quality of dietary fatty acids and their effects on postprandial TRL and insulin excursions have received little attention as simple and cost-effective strategies for the prevention and management of these chronic diseases.3.1. Blood lipid abnormalities
The small intestine can secrete two different triglyceride-rich lipoproteins: chylomicrons (CM) and very low-density lipoproteins (VLDL),18,19 both referred herein to as TRL (Fig. 3). CM are formed after eatingfatty foods, whereas VLDL are the major lipoproteins secreted during fasting. Studies involving lipid infusion in animals and lipid interaction in an in vitro model of human intestinal cells have shown that triglycerides and phospholipids induce the formation of CM and VLDL, respectively.20 The function of the postprandial TRL is to collect the absorbed dietary lipids and fat-soluble components, stabilising them for transport in the aqueous plasma environment. CM and VLDL also carry one molecule of apolipoprotein (apo)-B48 and apoB100 per particle, respectively. However, exaggerated postprandial hypertriglyceridemia, which is characterised by increased production and decreased clearance of postprandial TRL, is emerging as an important risk factor for chronic diseases.13–15 The fatty acid composition of dietary triglycerides influences the size and number of postprandial TRL,21 which may be better predictors of atherosclerosis than conventional triglyceride measurements.13 A recent proton NMR spectroscopic study of non-fractionated plasma revealed that compared to an SFA-based meal, the consumption of a MUFA-rich meal leads to the formation of fewer large TRL particles.21 There is no consensus on the effects of MUFA, PUFA, and SFA on the intensity of the postprandial triglyceride peak. Some studies indicate that SFA-rich meals elicit either a lower, higher or comparable postprandial peak value for triglycerides than meals rich in MUFA and PUFA.22–26 However, it is consistent that postprandial TRL enriched in MUFA and PUFA are more efficiently cleared compared to SFA-containing postprandial TRL. This efficiency probably depends on several factors, including the extent of triglyceride intraluminal lipolysis, the ability of triglyceride-depleted particles to accept cholesterol from donors, and the rate of receptor-mediated uptake in hepatic and extrahepatic tissues.21,27,28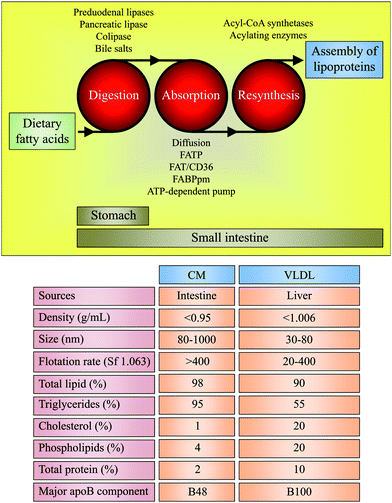 | ||
| Fig. 3 Digestion of triglycerides containing dietary fatty acids, absorption of fatty acids (and sn-2-monoglycerides), resynthesis to triglycerides for intestinal lipoprotein assembly, and physical properties and composition of intestinal lipoproteins: chylomicrons (CM) and very low-density lipoproteins (VLDL). FATP: fatty acid-transport proteins, FAT/CD36: fatty acid translocase/cluster of differentiation 36, FABPpm: plasma membrane-associated fatty acid-binding protein, Sf: Svedberg flotation, apoB: apolipoprotein-B. Values are approximate. | ||
Dietary fatty acids might modulate the clearance of triglycerides in postprandial TRL using a lipoprotein lipase (LPL)–mediated lipolytic complex, which is tethered to the capillary endothelium viaheparin sulphate proteoglycans.29,30 A recent study indicates that dietary fatty acids can modulate the LPL inhibitor angiopoietin-like protein 4 (Angptl4).31 In a transgenic mouse model of Angptl4 deletion, it was found that Angptl4 protects against the severe proinflammatory effects in mesenteric lymph nodes and resident macrophages of PA-rich meals and that this effect is specific for PA because OA and LA did not cause an inflammatory response. Interestingly, OA and LA were much more potent inducers of Angptl4 expression compared to PA, suggesting that dietary SFA may disturb the homeostatic negative feedback mechanism of Angptl4 on LPL. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein-1 (GPIHBP1) has the ability to stabilise LPL and partially prevents LPL inhibition by Angptl4.32GPIHBP1, which is expressed on the luminal side of the endothelium, has been also suggested to bind postprandial TRL and to facilitate their processing.33 The tissue pattern of GPIHBP1 gene expression is similar to that of LPL. GPIHBP1 and Angptl4 genes are transcriptionally regulated by peroxisome proliferator-activated receptors (PPAR). Fatty acids can be intracellularly generated from postprandial TRL by enzymatic and non-enzymatic mechanisms,34 and they are the best-known bonafide PPAR activators.35 Therefore, it can be hypothesised that distinctive effects of MUFA, PUFA, and SFA on PPAR activity could play a role on GPIHBP1 and Angptl4-mediated TRL lipolysis in the postprandial state.
Delayed clearance of postprandial TRL has been reported to increase lipid exchange between postprandial TRL and cholesterol-rich lipoproteins mediated by cholesterol ester transfer protein.36 This reciprocal exchange leads to the formation of cholesterol-enriched postprandial TRL, whereas cholesterol-rich lipoproteins are enriched in triglycerides.37 Moreover, MUFA-rich meals, when compared to SFA and PUFA-rich meals, promote the largest area-under-the-curve for cholesterol in postprandial TRL,38 suggesting a high lipid exchange turnover with cholesterol-rich lipoproteins. These observations further support the importance of dietary fatty acids in TRL and cholesterol-rich lipoprotein remodelling in the postprandial state.
Defective clearance of postprandial TRL likely influences the lipid accumulation into the arterial wall,39 which is considered an initial step in atherogenesis. Lipid deposition begins with the entry of TRL and low-density lipoproteins (LDL) and the initiation of a proinflammatory cascade that attracts monocytes into the subendothelial space. Postprandial TRL and oxidised or modified LDL can be taken up by infiltrated macrophages, eventually becoming foam cells. However, monocytes circulate together with TRL in the postprandial bloodstream and may start accumulating lipids even prior to their migration to tissues and differentiation to macrophages.4In vivo, postprandial TRL are removed by circulating monocytesvia the apoB48 receptor (apoB48R). Monocytes from healthy volunteers displayed an early time-dependent lipid accumulation in response to a high-fat meal that was paralleled by increased apoB48R mRNA levels and activation. These effects were coincident with an increase of plasma TRL carrying apoB48, with a decrease of plasma free fatty acid levels, and with no change of plasma fractions containing VLDL and cholesterol-rich lipoproteins. During the late postprandial phase, the acceleration in the rate of plasma triglyceride clearance was hypothesised to be a harbinger of apoB48R gene transcription, lipid accumulation, and activation decline in monocytes. Interestingly, when compared to postprandial TRL rich in SFA and PUFA, postprandial TRL rich in MUFA diminish the apoB48R transcriptional activity, lipid accumulation, and activation in human primary and THP-1 monocytes (unpublished data). These findings suggest that postprandial TRL are involved in the process of atherosclerosis prior to their entry into the subendothelial space and highlight the role of dietary fatty acids in modulating pro-atherogenic events in the postprandial state.
3.2. Blood insulin abnormalities
The damage to beta-cell function and the promotion of insulin resistance are known to contribute to the development of type 2 diabetes, which is a metabolic disorder that progresses over the course of months to years. Lifestyle modification is the cornerstone of both treatment and attempts to prevent type 2 diabetes. Compensatory hyperinsulinemia due to enhanced beta-cell function is considered to be an obligate accompanying feature in insulin resistance syndromes.40 Euglycemic clamps or frequently sampled intravenous glucose tolerance tests are the reference methods to determine beta-cell sensitivity to glucose and the sensitivity of body tissues to insulin.41 However, these tests are far from physiological because insulin secretion or activity is only measured in the steady-state. Empirical and model-based indices based on the oral glucose tolerance test (OGTT) provide a reasonable approximation of postprandial beta-cell function and whole-body insulin sensitivity.41,42 An important caveat of the OGTT is that the events associated with the ingestion of a pure glucose solution are not wholly equivalent to the numerous metabolic events associated with eating a mixed high-fat meal when both carbohydrates and fatty acids are ingested.It has been hypothesised that insulin resistance syndromes might be a postprandial phenomenon linked to the acute metabolism of dietary fatty acids.43 Exaggerated postprandial hypertriglyceridemia is indeed an inherent feature of diabetic dyslipidemia and is frequently found even in diabetic patients with normal fasting triglycerides.44 There is strong evidence that SFA selectively desensitises the response of peripheral tissues to insulin, whereas MUFA may counteract this effect.45 Such phenomena would be consistent with studies linking SFA-rich meals to dysfunctions in insulin secretion and the frequency of type 2 diabetes.6,46 It is probable that MUFA, PUFA, and SFA could compete at the level of the beta-cell (Fig. 4). The islet tissue, which expresses LPL, could access triglycerides from postprandial TRL as a source of free fatty acids, in which case, the type and concentration of the fatty acid in the immediate vicinity of the beta-cells is likely to be dependent on the nature of the dietary fatty acids.6 The input of fatty acids into the beta-cell can be mediated by cell surface G protein-coupled receptors (i.e., GPR40)47 and fatty acid-translocase FAT/CD36.48 ApoE-dependent and independent recognition sites could also cooperate with LPL to selectively remove postprandial TRL and to immediately generate intracellular fatty acidsvia catabolic pathways.49–53 Free fatty acid deprivation in islet tissue has been reported to impede glucose-stimulated insulin secretion, a process rapidly reversed by replacement with exogenous free fatty acids.54In vitro, OA elicits half of the insulinotropic potency of PA or stearic acid (18:0).55 When mixed high-fat meals with different proportions of dietary fatty acids are administered to healthy subjects, they become less insulin resistant postprandially as the proportion of MUFA to SFA in dietary fatty acids increase.56 In addition, when compared to SFA-rich meals, the MUFA and PUFA-rich meals induce a lower early postprandial insulin response.56,57 These findings are consistent with the notion that in comparison with SFA, MUFA might moderate the compensatory hyperactivity of beta-cells in the postprandial state,6 although whether this maintenance of glucose tolerance during feeding periods could prevent or delay the development of overt type 2 diabetes remains to be elucidated.
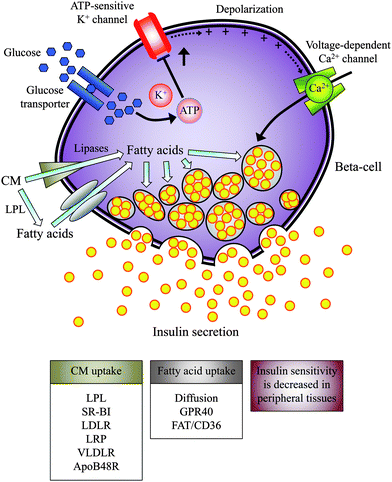 | ||
| Fig. 4 The potential impact of dietary fatty acids on beta-cell function in the postprandial state. In conjugation or not with LPL, postprandial TRL could be taken up in islets by different receptors and then hydrolyzed inside the beta-cell. Fatty acids from local LPL activity could enter the beta-cellvia passive diffusion and different receptors. Elevated beta-cell fatty acid levels could modulate insulin secretion, which largely would depend on the type of fatty acid in the diet. The postprandial state is characterized by a decrease in insulin sensitivity. LPL: lipoprotein lipase, TRL: triglyceride-rich lipoproteins, CM: chylomicrons, SR-BI: scavenger receptor class B member 1, LDLR: low-density lipoprotein receptor, LRP: LDL receptor-related protein, VLDLR, very low-density lipoprotein receptor, apoB48R: apolipoprotein-B48 receptor, GPR40: G protein-coupled receptor 40, FAT/CD36: fatty acid translocase/cluster of differentiation 36. | ||
Fasting hypertriglyceridemia results from either overproduction of triglycerides by the liver, impaired lipolysis, or a combination of both. In hypertriglyceridemic patients, the overproduction of triglycerides is disproportionately greater than the increase in apoB100 production, resulting in the formation of large triglyceride-rich VLDL particles.58 Obesity and insulin resistance result in increased hepatic supply of fatty acids and overproduction of triglycerides.59Insulin inhibits VLDL production in an effort to reduce the postprandial triglyceride response to a high-fat meal. A recent randomised and within-subject crossover study in volunteers who were newly diagnosed with type IIb or IV hyperlipoproteinemia revealed that postprandial beta-cell function and insulin sensitivity are improved with MUFA when compared to SFA,60 therefore extending the relationship between MUFA-rich meals and the benefits on postprandial glucose homeostasis observed in subjects with normal fasting triglyceride levels56 to a population of subjects with high fasting triglyceride levels.60 Furthermore, with regard to resistance to insulin-mediated glucose disposal, SFA was found to stimulate additional insulin secretion to maintain postprandial glucose homeostasis, suggesting a mechanism of lipid-induced deterioration of insulin sensitivity coupled with compensatory insulin secretion that is distinctively modulated by dietary fatty acids. Whether lipoprotein receptor-mediated signalling pathways in skeletal muscle and other insulin target tissues, such as adipose tissue and liver, are sensitive to the types of fatty acids in postprandial TRL, which largely depend on the nature of dietary fatty acids embodied in the meal, and whether they are connected to insulin signalling pathways, should be established.
4. Conclusions
Dietary fatty acids are nutritional signals that play a relevant role in modulating hypertriglyceridemia and compensatory hyperinsulinemia to decreased insulin sensitivity in the postprandial state, which are major risk factors for chronic diseases such as atherosclerosis and type 2 diabetes. Contrary to SFA, MUFA leads to improved acute lipid tolerance and insulin action. PUFA does not appear to confer additional benefit over MUFA. Therefore, this review is consistent with dietary policies that specifically promote the consumption of MUFA in place of SFA and that highlight the need to study the mechanisms and pathways that might account for transient but repetitive daily dietary fatty acid-induced postprandial metabolic abnormalities. Such phenomena would imply a greater propensity towards chronic diseases, as a metabolic abnormality becomes more exaggerated and prolonged in the postprandial state. This largely subclinical and underappreciated condition further links the metabolism of SFA with atherogenic and diabetogenic disorders and suggests a new role for MUFA, particularly OA, as a critical player in preventing and controlling chronic diseases.Acknowledgements
This manuscript was supported by MICINN grants AGL2008-02811 and AGL2011-29008, and Marie Curie grant PERG07-GA-2010-268413.5. References
- WHO Technical Report Series 916, 2003, Geneva, Switzerland Search PubMed.
- J. Lopez-Miranda, F. Perez-Jimenez, E. Ros, R. De Caterina, L. Badimon, I. Covas, E. Escrich, J. M. Ordovas, F. Soriguer, R. Abia, C. Alarcon, M. Battino, D. Corella, J. Chamorro-Quiros, J. Delgado-Lista, D. Giugliano, K. Esposito, R. Estruch, J. M. Fernandez-Real, J. J. Gaforio, C. La Vecchia, D. Lairon, F. Lopez-Segura, P. Mata, J. A. Menendez, F. J. G. Muriana, J. Osada, D. B. Panagiotakos, J. A. Paniagua, P. Perez-Martinez, M. A. Peinado, M. Pineda-Priego, H. Poulsen, J. L. Quiles, M. C. Ramirez-Tortosa, J. Ruano, L. Serra-Majem, R. Sola, M. Solanas, V. Solfrizzi, R. de la Torre-Fornell, A. Trichopoulou, M. Uceda, J. M. Villalba-Montoro, J. R. Villar-Ortiz, F. Visioli and N. Yiannakouris, Nutr., Metab. Cardiovasc. Dis., 2010, 20, 284–294 CrossRef CAS.
- G. M. Dallinga-Thie, R. Franssen, H. L. Mooij, M. E. Visser, H. C. Hassing, F. Peelman, J. J. Kastelein, M. Peterfy and M. Nieuwdorp, Atherosclerosis, 2010, 211, 1–8 CrossRef CAS.
- L. M. Varela, A. Ortega, B. Bermudez, S. Lopez, Y. M. Pacheco, J. Villar, R. Abia and F. J. G. Muriana, Am. J. Clin. Nutr., 2011, 93, 918–925 CrossRef CAS.
- B. Bermudez, S. Lopez, A. Ortega, L. M. Varela, Y. M. Pacheco, R. Abia and F. J. G. Muriana, Curr. Pharm. Des., 2011, 17, 831–843 CrossRef CAS.
- S. Lopez, B. Bermudez, R. Abia and F. J. G. Muriana, Curr. Opin. Lipidol., 2010, 21, 15–20 CrossRef CAS.
- A. N. Margioris, Curr. Opin. Clin. Nutr. Metab. Care, 2009, 12, 129–137 CrossRef CAS.
- Y. Jimenez-Gomez, J. Lopez-Miranda, L. M. Blanco-Colio, C. Marin, P. Perez-Martinez, J. Ruano, J. A. Paniagua, F. Rodriguez, J. Egido and F. Perez-Jimenez, Atherosclerosis, 2009, 204, 70–76 CrossRef.
- J. Bezard, J. P. Blond, A. Bernard and P. Clouet, Reprod. Nutr. Dev., 1994, 34, 539–568 CrossRef CAS.
- J. T. Brenna, N. Salem Jr., A. J. Sinclair and S. C. Cunnane, Prostaglandins Leukot. Essent. Fatty Acids, 2009, 80, 85–91 CrossRef CAS.
- R. Wall, R. P. Ross, G. F. Fitzgerald and C. Stanton, Nutr. Rev., 2010, 68, 280–289 CrossRef.
- A. P. Simopoulos, Exp. Biol. Med., 2008, 233, 674–688 CrossRef CAS.
- B. G. Nordestgaard, M. Benn, P. Schnohr and A. Tybjaerg-Hansen, JAMA, J. Am. Med. Assoc., 2007, 298, 299–308 CrossRef CAS.
- W. B. Kannel and R. S. Vasan, Curr. Opin. Cardiol., 2009, 24, 345–350 CrossRef.
- B. G. Nordestgaard and J. J. Freiberg, Curr. Vasc. Pharmacol., 2011, 9, 281–286 CrossRef CAS.
- K. Lock, R. D. Smith, A. D. Dangour, M. Keogh-Brown, G. Pigatto, C. Hawkes, R. M. Fisberg and Z. Chalabi, Lancet, 2010, 376, 1699–1709 CrossRef.
- M. Cecchini, F. Sassi, J. A. Lauer, Y. Y. Lee, V. Guajardo-Barron and D. Chisholm, Lancet, 2010, 376, 1775–1784 CrossRef.
- D. D. Black, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 293, 519–524 CrossRef.
- C. M. Mansbach and F. Gorelick, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 293, 645–650 CrossRef.
- H. Mu and C. E. Hoy, Prog. Lipid Res., 2004, 43, 105–133 CrossRef CAS.
- P. Perez-Martinez, J. M. Ordovas, A. Garcia-Rios, J. Delgado-Lista, N. Delgado-Casado, C. Cruz-Teno, A. Camargo, E. M. Yubero-Serrano, F. Rodriguez, F. Perez-Jimenez and J. Lopez-Miranda, Nutr., Metab. Cardiovasc. Dis., 2011, 21, 39–45 CrossRef CAS.
- C. J. Masson and R. P. Mensink, J. Nutr., 2011, 141, 816–821 CrossRef CAS.
- T. Tholstrup, B. Sandstrom, A. Bysted and G. Holmer, Am. J. Clin. Nutr., 2001, 73, 198–208 CAS.
- N. Mekki, M. Charbonnier, P. Borel, J. Leonardi, C. Juhel, H. Portugal and D. Lairon, J. Nutr., 2002, 132, 3642–3649 CAS.
- K. G. Jackson, E. J. Wolstencroft, P. A. Bateman, P. Yaqoob and C. M. Williams, Br. J. Nutr., 2005, 93, 693–700 CrossRef CAS.
- Y. M. Pacheco, B. Bermudez, S. Lopez, R. Abia, J. Villar and F. J. G. Muriana, Am. J. Clin. Nutr., 2006, 84, 342–349 CAS.
- R. Abia, Y. M. Pacheco, J. S. Perona, E. Montero, F. J. G. Muriana and V. Ruiz-Gutierrez, J. Nutr., 2001, 131, 59–65 CAS.
- R. Abia, Y. M. Pacheco, E. Montero, V. Ruiz-Gutierrez and F. J. G. Muriana, Life Sci., 2003, 72, 1643–1654 CrossRef CAS.
- D. Lairon, Atheroscler. Suppl., 2008, 9, 45–48 CrossRef CAS.
- A. Bergouignan, I. Momken, D. A. Schoeller, C. Simon and S. Blanc, Prog. Lipid Res., 2009, 48, 128–147 CrossRef CAS.
- L. Lichtenstein, F. Mattijssen, N. J. de Wit, A. Georgiadi, G. J. Hooiveld, R. van der Meer, Y. He, L. Qi, A. Koster, J. T. Tamsma, N. S. Tan, M. Muller and S. Kersten, Cell Metab., 2010, 12, 580–592 CrossRef CAS.
- W. K. Sonnenburg, D. Yu, E. C. Lee, W. Xiong, G. Gololobov, B. Key, J. Gay, N. Wilganowski, Y. Hu and S. Zhao, J. Lipid Res., 2009, 50, 2421–2429 CrossRef CAS.
- M. M. Weinstein, L. Yin, Y. Tu, X. Wang, X. Wu, L. W. Castellani, R. L. Walzem, A. J. Lusis, L. G. Fong, A. P. Beigneux and S. G. Young, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 20–23 CrossRef CAS.
- O. Ziouzenkova, S. Perrey, L. Asatryan, J. Hwang, K. L. MacNaul, D. E. Moller, D. J. Rader, A. Sevanian, R. Zechner, G. Hoefler and J. Plutzky, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2730–2735 CrossRef CAS.
- A. K. Hihi, L. Michalik and W. Wahli, Cell. Mol. Life Sci., 2002, 59, 790–798 CrossRef CAS.
- B. H. Chung, B. H. Cho, P. Liang, S. Doran, L. Osterlund, R. A. Oster, B. Darnell and F. Franklin, Am. J. Clin. Nutr., 2004, 80, 1145–1158 CAS.
- S. Patel, R. Puranik, S. Nakhla, P. Lundman, R. Stocker, X. S. Wang, G. Lambert, K. A. Rye, P. J. Barter, S. J. Nicholls and D. S. Celermajer, Atherosclerosis, 2009, 204, 424–428 CrossRef CAS.
- S. Lopez, B. Bermudez, Y. M. Pacheco, G. Lopez-Lluch, W. Moreda, J. Villar, R. Abia and F. J. G. Muriana, J. Nutr., 2007, 137, 1999–2005 CAS.
- S. Sudheendran, C. C. Chang and R. J. Deckelbaum, Prostaglandins Leukot. Essent. Fatty Acids, 2010, 82, 205–209 CrossRef CAS.
- G. M. Reaven, Panminerva Med., 2005, 47, 201–210 CAS.
- C. Cobelli, G. M. Toffolo, C. Dalla Man, M. Campioni, P. Denti, A. Caumo, P. Butler and R. Rizza, Am. J. Physiol.: Endocrinol. Metab., 2007, 293, 1–15 CrossRef.
- E. Bartoli, G. P. Fra and G. P. Carnevale Schianca, Eur. J. Intern. Med., 2011, 22, 8–12 CrossRef CAS.
- M. T. Pedrini, A. Niederwanger, M. Kranebitter, C. Tautermann, C. Ciardi, T. Tatarczyk and J. R. Patsch, Diabetologia, 2006, 49, 1612–1618 CrossRef CAS.
- N. Tentolouris, C. Arapostathi, D. Perrea, D. Kyriaki, C. Revenas and N. Katsilambros, Diabetes Care, 2008, 31, 2276–2278 CrossRef CAS.
- J. H. Moon, J. Y. Lee, S. B. Kang, J. S. Park, B. W. Lee, E. S. Kang, C. W. Ahn, H. C. Lee and B. S. Cha, Lipids, 2010, 45, 1109–1116 CrossRef CAS.
- A. Misra, N. Singhal and L. Khurana, J. Am. Coll. Nutr., 2010, 29, 289–301 Search PubMed.
- M. A. Kebede, T. Alquier, M. G. Latour and V. Poitout, Diabetes, Obes. Metab., 2009, 11, 10–20 CrossRef CAS.
- T. Wallin, Z. Ma, H. Ogata, I. H. Jørgensen, M. Iezzi, H. Wang, C. B. Wollheim and A. Bjorklund, Biochim. Biophys. Acta, 2010, 1801, 191–197 CAS.
- M. L. Brown, M. P. Ramprasad, P. K. Umeda, A. Tanaka, Y. Kobayashi, T. Watanabe, H. Shimoyamada, W. L. Kuo, R. Li, R. Song, W. A. Bradley and S. H. Gianturco, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7488–7493 CrossRef CAS.
- R. Out, J. K. Kruijt, P. C. Rensen, R. B. Hildebrand, P. de Vos, M. Van Eck and T. J. Van Berkel, J. Biol. Chem., 2003, 278, 18368–18375 CrossRef.
- M. E. Roehrich, V. Mooser, V. Lenain, J. Herz, J. Nimpf, S. Azhar, M. Bideau, A. Capponi, P. Nicod, J. A. Haefliger and G. Waeber, J. Biol. Chem., 2004, 279, 18401–18406 CrossRef.
- A. von Eckardstein and R. A. Sibler, Curr. Opin. Lipidol., 2011, 22, 26–32 CrossRef CAS.
- A. Lass, R. Zimmermann, M. Oberer and R. Zechner, Prog. Lipid Res., 2011, 50, 14–127 CrossRef CAS.
- K. Pinnick, M. Neville, A. Clark and B. Fielding, J. Cell. Biochem., 2010, 109, 683–692 CAS.
- C. E. Wrede, R. Buettner, H. Wobser, I. Ottinger and L. C. Bollheimer, Horm. Metab. Res., 2007, 39, 482–488 CrossRef CAS.
- S. Lopez, B. Bermudez, Y. M. Pacheco, J. Villar, R. Abia and F. J. G. Muriana, Am. J. Clin. Nutr., 2008, 88, 638–644 CAS.
- M. D. Robertson, K. G. Jackson, B. A. Fielding, C. M. Williams and K. N. Frayn, Br. J. Nutr., 2002, 88, 635–640 CrossRef CAS.
- M. J. Caslake and C. J. Packard, Curr. Opin. Lipidol., 2004, 15, 387–392 CrossRef CAS.
- D. C. Chan, H. P. Barrett and G. F. Watts, Am. J. Cardiovasc. Drugs, 2004, 4, 227–246 CrossRef CAS.
- S. Lopez, B. Bermudez, A. Ortega, L. M. Varela, Y. M. Pacheco, J. Villar, R. Abia and F. J. G. Muriana, Am. J. Clin. Nutr., 2011, 93, 494–499 CrossRef CAS.
Footnote |
| † To be considered equal first authors |
| This journal is © The Royal Society of Chemistry 2012 |