Assessment of sorbent impregnated PUF disks (SIPs) for long-term sampling of legacy POPs†
Jasmin K.
Schuster
a,
Rosalinda
Gioia
a,
Tom
Harner
b,
Sum Chi
Lee
b,
Knut
Breivik
cd and
Kevin C.
Jones
a
aCentre for Chemicals Management, Lancaster Environment Centre, Lancaster University, Lancaster, LA1 4YQ, UK
bEnvironment Canada, Science & Technology Branch, Toronto, M3H 5T4, Canada
cNorwegian Institute for Air Research, P.O. Box 100, NO-2027, Kjeller, Norway
dUniversity of Oslo, Department of Chemistry, P.O. Box 1033, NO-0315, Oslo, Norway
First published on 9th November 2011
Abstract
Two field studies were conducted for one year using sorbent-impregnated polyurethane foam (SIP) disks for PCB and PBDE air sampling. SIP disks were introduced by Shoeib et al. (2008) as an alternative passive air sampling medium to the polyurethane foam (PUF) disk and have the advantage of a higher holding capacity for organic chemicals. The first study on SIP disks confirmed their application for measuring volatile perfluorinated compounds (PFCs) and their ability to maintain time-integrated (linear) air sampling. In this study, the suitability of the SIP disks for long-term sampling of polychlorinated biphenyls (PCBs), polybrominated diphenyl ethers (PBDEs) and hexachlorobenzene (HCB) was assessed. SIP disks were deployed at a rural site in the UK and harvested after periods ranging from 35–350 days. Atmospheric POP concentrations were monitored with a high-volume air sampler during the deployment period. Linear uptake was observed for all monitored PCBs and PBDEs over the full exposure time. Air-sampler equilibrium was observed for HCB after 6 months. In a second field study, SIP disks were deployed for one year at 10 sites on a latitudinal transect in the UK and Norway, at which air sampling has been undertaken previously with different passive air sampling media since 1994. The estimated concentrations and spatial distributions derived from the SIP disks were largely in agreement with previously reported data.
Environmental impactLong-term air monitoring studies are important to assess the trends and changes in air concentrations of legacy pollutants. Passive air samplers (PAS) offer a cost-effective and simple alternative to active air samplers. The monitoring of air concentrations at background sites with PAS is hampered by low concentrations and low sampling volumes. A PAS medium with a long linear uptake phase and a high capacity for volatile POPs allows a longer deployment time and therefore increases the probability that sample concentrations exceed the limit of detection. The sorbent-impregnated polyurethane foam disks were found to be applicable for the time-integrated sampling of polychlorinated biphenyls and polybrominated diphenyl ethers for up to one year and for hexachlorobenzene for up to 6 months. |
Introduction
The defining characteristics of persistent organic pollutants (POPs) are their toxicity, tendency to bioaccumulate, persistence and semi-volatility.1 Many monitoring and modelling projects are conducted to improve understanding of the transport mechanisms and fate of POPs. The knowledge gained from this research may also contribute to a better assessment and regulation of newly emerging compounds with similar characteristics. Legacy POPs like PCBs provide a case for a compound group for which production officially stopped and whose fate in the environment can be related to other pollutants with similar properties.Most air monitoring projects for POPs use active air samplers (AAS). Examples include the Integrated Atmospheric Deposition Network (IADN) in the Great Lakes region,2 the Toxic Organic Micro-Pollutants program (TOMPS) in the UK3 and the European Monitoring and Evaluation Programme (EMEP).4 High-volume AAS need to be connected to a power source and require regular maintenance which can make their application in remote background areas expensive and impractical. Passive air samplers (PAS) offer a cost effective alternative that does not depend on access to electric facilities and therefore can be deployed in remote areas. PAS are already used for monitoring atmospheric POP concentrations in several long-term projects like the Global Atmospheric Passive Sampling (GAPS) network5,6 and the deployment of semi-permeable membrane devices in the UK and Norway since 1994.7–11 Drawbacks of the PAS compared to the AAS are that the air volume sampled in a given time is much lower and information on uptake rates is needed. While for most AAS the sampled air volume can be monitored and regulated with a pump, PAS rely on advection and diffusion to supply air to the sampling medium and calibration studies must be performed.
While a high-volume air sampler can process several hundred m3 air per day, the average sampling rate for a polyurethane foam (PUF) disk deployed in the “flying saucer” device (FSD) was reported as 3–5 m3/day.12,13 For PAS deployed in remote areas with low POP concentrations long sampling periods are therefore important to ensure that the amount of pollutant sequestered in the PAS is detectable and quantifiable. In order to allow the most accurate estimation of the atmospheric concentration from the PAS loads, it is preferable that the sampling period is restricted within the linear uptake phase of the PAS. Among the established PAS sampling media for PCBs are PUF disks,5,6XAD resins,14 semi-permeable membrane devices (SPMDs),7–11polymer-coated glass (POGs)15 and tristearin-coated fibreglass sheets.16 The equilibrium times for PCBs for these vary with rather short equilibrium periods for tristearin-coated fibreglass sheets and POGs (<60 h and <100 h respectively) to medium times for PUF disks (<6 weeks for lighter PCBs) and long equilibrium time periods for XAD and SPMDs (>1 year).
In recent years Shoeib et al.17 developed a mixed sampling medium by impregnating a PUF disk with XAD powder. The XAD powder has a large and highly sorptive surface area and therefore increases the overall sorptive capacity of the sampler. The sorbent impregnated PUF disks (SIPs) combine the advantages of an extended linear uptake period and a simple and cost-effective way for deployment. The SIPs were originally created to monitor perfluorinated compounds (PFCs) which could not be sampled using PUF disks due to their rapid equilibration. An indoor calibration study with PUF disks and SIPs for PFCs proved that the linear uptake period was significantly extended by the XAD powder.17
To evaluate the usability of SIPs as long-term PAS for POPs like PCBs, PBDEs and HCB a year-long field study parallel to an AAS was set up. Corresponding to this SIPs were deployed at remote sampling sites across the UK and Norway. Previous deployment of SPMDs at these sites for several years provides good baseline information on air concentrations of the target compounds.7–11
Methods
Preparation of SIPs
PUF disks (Tisch Environmental, Cleves, USA) were Soxhlet pre-extracted with acetone (12 h) and petroleum ether (18 h) successively. Powdered Amberlite XAD-4 was pre-extracted with methanol (6 h), dichloromethane (DCM) (12 h) and hexane (6 h) consecutively. PUF disks were coated in XAD-4 following the procedure described by Shoeib et al.17SIPs were dried under vacuum and stored in metal tins at −17 °C until deployment and after collection until extraction.Field study parallel to AAS
SIPs were deployed in FSDs (SI-Figure 1†). The FSDs consisted of two stainless steel bowls with diameters of 260 mm (top) and 190 mm (bottom). The SIPs were deployed at the Lancaster University field station Hazelrigg which is part of the TOMPs monitoring program.3 The SIPs were collected in duplicate or triplicate after 4, 7, 14, 21, 28, 35, 42, 112, 182, 266 and 350 days deployment. Field blanks (FB) were collected at the beginning and the end of the field study at the sampling site. FBs were only exposed for the duration of assembling and disassembling of the FSD with the SIP disk.A TOMPs high-volume AAS collected samples parallel to the passive samplers from May 2009 to April 2010. Gaseous and particle phase were collected together using PUF plugs (50 × 75 mm, Klaus Ziemer GmbH Langerwehe, Germany) and glass fibre filters (GF/A, Whatman plc, Maidstone, UK). The AAS sampled consecutively for two week periods. During sample processing, the individual two week samples were bulked and the reported values for TOMPs represent the average air concentration of three months (since the TOMPs program is designed to assess long-term trends over many years). Meteorological data for the sampling period is given in the Supplementary Information.† Further details of the sample preparation, collection and analysis for the TOMPs project are presented in Schuster et al.3
Field study parallel to SPMDs
SIPs were deployed at 10 sites in the UK and Norway from July 2008 to June 2009 (SI-Figure 1†). Two SIPs were deployed in FSDs at each sampling site. FBs and travel blanks (TB) were collected for the whole project. FBs were collected at each site as described above. TBs were defined as SIPs that were not exposed during the sampling deployment and collection process. TBs were collected along the visits to the separate sampling sites and depending on the collection route a TB covered 2–3 sites. Detailed information for the sampling sites is reported in Meijer et al.9 The number of FBs and TBs amounted to 60% of the sample number.Sample extraction and analysis
SIPs were Soxhlet extracted in pre-cleaned cellulose thimbles for 16 h in DCM. Samples were further cleaned using an alumina-silica column, sulphuric acid digestion and gel permeation chromatography. Samples were transferred to 25 μL dodecane containing PCB 30, [13C12] PCB 141, [13C12] PCB 208, PBDE 69 and PBDE 181 as internal standards. Details for the extraction and cleaning methods of the atmospheric samples are described by Chaemfa et al.18 Samples were analyzed by gas chromatography-mass spectrometry (GC-MS) with an EI+ source operating in selected ion mode (SIM) for PCBs, PBDEs, and organochlorine pesticides (OCs). Details of the instruments, temperature program and monitored ions are given elsewhere.19–21 The following compounds were monitored and reported in this publication: tri-PCBs (3CBs) 18, 22 and 28/31; tetra-PCBs (4CBs) 41/64, 44, 49, 52, 60/56, 70 and 74; penta-PCBs (5CBs) 87, 90/101, 95, 99, 105, 110, 118 and 123; hexa-PCBs (6CBs) 138, 141, 149, 151 and 153/132; hepta-PCBs (7CBs) 174, 180, 183, and 187; octa-PCBs (8CBs) 199 and 203; PBDEs 47, 49, 99, 100, 153 and hexachlorobenzene (HCB).Quality control and quality assurance
Surrogate standard was added to the samples prior to extraction to monitor possible losses during the sample processing. Recoveries for PCBs and HCB were controlled using a surrogate standard containing [13C12]-PCB 28, 52, 101, 138, 153 and 180 with average recoveries in the range 85–102%. Recoveries for PBDEs were assessed using a surrogate standard containing PBDE 51, 128 and 190; average recoveries ranged from 79–108%. Samples were not recovery-corrected. Solvent blanks were processed with the samples (approximately every 7 samples) to monitor laboratory contamination. The limit of detection (LOD) was defined as the average of the blanks (FBs and TBs) plus three times their standard deviation or the lowest GC calibration standards, selecting the higher value for each individual congener. LODs ranged from 30–990 pg μL−1 for PCBs and 50–1130 pg μL−1 for PBDEs.Two or three SIPs were deployed in parallel to monitor sampler specific deviations. The average variability between the POP loads for the duplicates and triplicates was 11 ± 9%.
Data analysis
SIP samples parallel to the AAS were collected for a total of 350 days. Data for the PAS samples for <35 days was found to be inconsistent and is not reported here. Further work is planned to explain these observations. The data discussed here was from samples deployed for t = 35, 42, 112, 182, 266 and 350 days.Sampling rates (Rs) for PAS deployed in FSDs were reported as 3–5 m3/day. In this study R was assumed as 3 m3/day to calculate the atmospheric concentrations cair12,13 for HCB (during the linear uptake period) and the majority of PCBs (3–7 chlorine substituents).12,13 To calculate the air concentrations for HCB for the sampling periods after equilibrium, the air volume reached at the end of the observed linear uptake phase was applied (∼546 m3 for 182 days until equilibrium). For 8CBs and PBDEs sampling rates were corrected for the particle associated fraction. Klanova et al.22 found the particle-phase sampling rate to be 10% of the gaseous-phase sampling rate. The air-particle distribution was determined as discussed by Finizio et al.,23 applying site specific parameters for atmospheric particle concentration24 and meteorological factors.
The concentrations of the individual chemicals cAir,i at the field sites in the UK and Norway were estimated from R, the SIP load of the individual chemicals mSIP,i and the deployment time t.
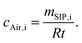 | (3) |
For the UK-Norway transect study, the predicted air concentrations for 2009 were estimated from the 1998–2008 data from the previous PAS studies using SPMDs. The assumption of a first order decline was used to predict the concentration of 2009.11 Details for the method applied to estimate these background concentrations can be found in the Supplementary Information.† Statistical methods and analysis tools applied for data analysis were linear regression (Microsoft Excel, R-project), t-test: paired two sample for means (Microsoft Excel), multiple linear regression and ANOVA (R-project).
Brief comments on the theory of PAS uptake kinetics
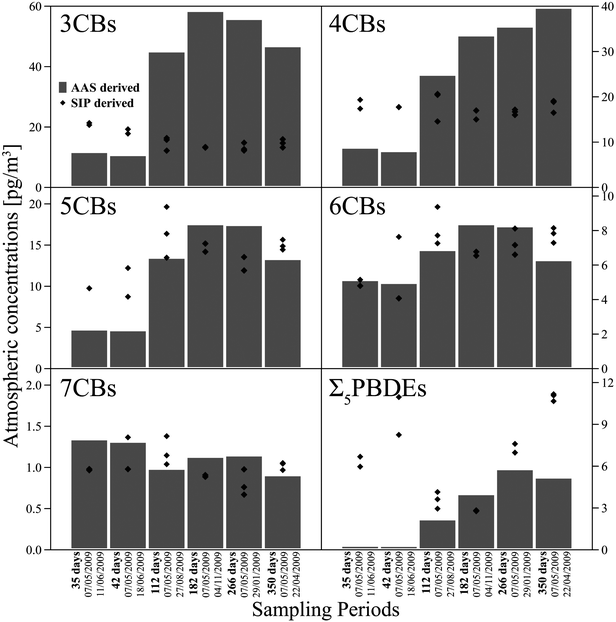
The uptake kinetics for PAS for semi-volatile compounds can be described as a diffusive mass transfer process with three steps defining the overall mass transfer coefficient: (i) The supply by advection of the ambient air into the sampling chamber, (ii) the transfer by diffusion of the target compounds through the air-side boundary layer at the sampler-air interface and (iii) the diffusion through the sampler-side boundary layer and sorption to the sampling media.25 The flux of the target compound from the atmosphere into the sampling media and therefore the sampling rates (R) are controlled by the rate-limiting step of the sampling process. It is assumed that the commonly used PAS chambers, like the Stevenson Screen box and the flying saucer device (FSD), are well ventilated and therefore the transfer of the ambient air into the sampler housing can be discounted as the rate limiting step. If a dependence of the SR to the air-PAS partition coefficient is observed, it indicates that process is sampler- and not air-side restricted. (The mass transfer coefficients depend in varying degrees on PAS dimensions as well as physical chemical properties of the target compounds.) Previous studies for the PAS uptake process for PCBs reported it to be both sampler-side (for SPMDs by Ockenden et al.26) and air-side restricted (for SPMDs and PUFs by Shoeib and Harner12). Wind speed effects as reported by Tuduri et al.13 indicate an air-side restricted uptake process. The mass transfer process between atmosphere and PAS for the target compounds is split by the uptake process into the sampler (defined by the atmospheric concentration cAir and the uptake rate constant kU) and the elimination process from the sampler (defined by the concentration cPAS of the compound sequestered in the PAS and the elimination rate constant kE):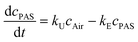 | (1) |
At the initial stage of the sampling process it is assumed that cPAS is very low and the sampler represents a sink for the compound (for kUcAir ≫ kEcPAS). During this linear phase of the sampling process the concentration changes in the PAS are primarily controlled by uptake process. The impact of the elimination process can be neglected at this stage. The sampling profile is linear and cPAS is directly proportional to cAir and deployment time t. With increasing cPAS over time, the revolatilisation of the compound from the sampling medium gains more influence and the impact becomes clear in the sampling profile. The uptake profile shifts from linear to a curvilinear shape and eventually approaches equilibrium (for kUcAir ≈ kEcPAS).
Results and discussion
Field study parallel to AAS
Meteorological data for the Hazelrigg field site was provided daily for the deployment period (May 2009–April 2010). During this period the temperature at the sampling site ranged from −3.2 to 23.4 °C and wind speeds reached a maximum of 13.9 m s−1. The variation of the average temperature and wind speed between the monitored periods of 35–350 days was low, ranging between 9.8–14 °C and 4.1–4.9 m s−1 respectively. The average precipitation for the sampling intervals was 2.2–2.6 mm/day. The POP concentrations monitored with AAS during the sampling period ranged from 19–176 pg m−3, 0.2–13 pg m−3 and 0.1–74 pg m−3 for Σ34PCB, Σ5PBDE and HCB respectively. An overview of the temperature and wind speed is given in SI-Figure 2 and for POP concentrations (as determined with AAS) in Fig. 1 and SI-Figures 3.† Atmospheric POP concentrations at the field site Hazelrigg have been monitored under the TOMPs program since 1992 and have shown a steady decline.3 The high variation in atmospheric data during the field study was therefore not expected. One possible explanation for the highly elevated POP levels during the autumn and winter months 2009 might be excavation and road work starting late summer (continuous for several months within a radius of 1 km from the field station). The soil disturbance may have caused a higher revolatilisation of POPs from surface soil and an increased distribution of POPs associated with particles. Those would be particularly captured by the AAS which samples gaseous and particle phase. | ||
| Fig. 1 Results from the Hazelrigg field site showing amount of chemical accumulated in the SIP disks (mSIP,i) with time for HCB, Σ34PCB and Σ5PBDE. Also shown are air concentrations determined using AAS for consecutive 2-week sampling periods and bulked samples under the TOMPS program. | ||
During the field study, linear uptake was apparent for all PCB and PBDE congeners for the samples collected after 35–350 days deployment. Only HCB was shown to approach equilibrium during this period. The linear uptake phase for HCB ended after ∼6 months. Fig. 1 shows the POP loads per SIP for HCB, Σ34PCB and Σ5PBDE (the plots for the homologue PCB groups are presented in SI-Figure 3†). The mass of POPs sequestered on the SIPs ranged from 5.2–62.0 ng for Σ34PCB, 0.15–3.2 ng for Σ5PBDE and 8.7–52.1 ng for HCB. Value ranges for the homologue PCBs are given in Table 1, for individual PCBs and PBDEs in SI-Table 2.
| POPs | POPs per | Atmospheric concentration | |
|---|---|---|---|
| SIP [ng] | SIP [pg m−3] | AAS [pg m−3] | |
| 3CBs | 1.8–20.0 | 12.2–21.3 | 10.8–58.6 |
| 4CBs | 1.5–12.1 | 14.6–20.6 | 8.1–39.8 |
| 5CBs | 1.0–16.4 | 8.7–19.6 | 4.7–17.6 |
| 6CBs | 0.70–12.3 | 4.1–9.4 | 5.0–8.4 |
| 7CBs | 0.10–1.1 | 0.7–1.4 | 0.9–1.3 |
| 8CBs | 0.002–0.081 | 0.01–0.08 | 0.03–0.1 |
| Σ34PCB | 5.2–62.0 | 47.4–67.2 | 30.1–119 |
| Σ5PBDE | 0.15–3.2 | 0.9–3.5 | 0.3–5.8 |
| HCB | 8.7–52.1 | 30.2–129 | 0.9–33.0 |
As noted above, a media with a high capacity for the target compounds and an optimal linear uptake period is required for sampling in remote areas with low concentrations. Chaemfa et al.18 reported linear uptake phases for PCBs in PUF disks of ∼4 weeks for 3CBs and 4CBs, >6 weeks for 5CBs, 6CBs and 7CBs in an outdoor study. The linear uptake phase for HCB was reported as >8 weeks. Somewhat longer linear uptake periods were reported for the 3CBs in PUF disks and SPMDs (50–130 days and 140–310 days, respectively) during indoor calibrations. The longer equilibration times may be due to slower uptake kinetics indoor due to lower air velocities.12 Sampling rates for passive samplers are wind-speed dependent. For air-side controlled sampling processes the thickness of the boundary layer is a restricting factor during the uptake process. While the boundary layer thickness is mainly defined by the passive sampling media configurations, Tuduri et al.13 reported a decrease of the boundary layer thickness with increasing wind speeds (and therefore an increase in the sampling rates but possible decrease of the linear uptake phase). In this study for the SIP disks it was ascertained that (except for HCB) none of the lighter congeners was approaching equilibrium during the 350 day sampling period.
The daily uptake for PCBs and PBDEs ranged from 0.09–18.4 pg/day for individual congeners. The data was analysed applying multiple linear regression and ANOVA for dependence of the congener specific properties molar mass M, octanol-air partition coefficient Koa (surrogate for the SIP-air partition coefficient) and their abundance f in the samples (cAir,i/cAir,Σ34PCB). The statistical parameters indicate that the uptake rate on the sampler is mainly defined by the abundance and the partitioning coefficients of the target compound.
Atmospheric concentrations calculated for the SIPs ranged from 47.4–67.2 pg m−3, 0.9–3.5 pg m−3, 52–127 pg m−3 for Σ34PCB, Σ5PBDE and HCB respectively (HCB concentrations were only reported for the samples during the linear uptake phase). HCB concentrations derived from SIPs far exceeded those from the AAS data. This might be explained with high breakthrough of the HCB in the AAS sampler for the 2 weeks sampling period. Fig. 2 shows the average atmospheric POP concentrations for the separate deployment periods derived from the TOMPs data and the SIP loads for the homologue groups 3CB, 4CB, 5CB, 6CB, 7CB and Σ5PBDE. In general higher differences between the air concentrations derived with AAS and SIP disks were observed for the samples collected until day 42. Average concentrations monitored for the deployment periods of 112 up to 350 days showed better agreement between results of AAS and SIP disks. For the lighter PCB congeners higher concentrations were observed with the AAS. AAS derived concentrations were (72 ± 5)% higher for the 3CBs and (46 ± 15)% for the 4CBs. The concentration values of the higher homologue groups were not considered significantly different based on the paired t-test (average differences of (20 ± 13)% for 5CBs, (18 ± 10)% for 6CBs, (21 ± 12)% for 7CBs, (66 ± 38)% for 8CBs). Values from SIPs were higher for Σ5PBDE, but not significantly different for the congeners BDE 47, 99 and 100 (average difference of (32 ± 23)%, (38 ± 18)% and (21 ± 13)% respectively). The SIP derived concentrations for PBDEs and 8CBs would have been significantly lower than the AAS derived concentrations if the sampling rates would not have been corrected for the particle-phase associated fraction. The concentrations for 3–7CBs were calculated both for a constant sampling rate of 3 m3/day and the weighted sampling rate considering the possible partitioning to the particle phase. Better agreement to the AAS derived air concentrations was found for applying the constant sampling rate of 3 m3/day to calculate the SIP data. The 3–7CBs partition less strongly to the particle phase and the application of weighted sampling rates is therefore not necessary.
 | ||
| Fig. 2 Results from the Hazelrigg field site showing air concentrations based on SIP disks and AAS for 3CBs, 4CBs, 5CBs, 6CBs, 7CBs and Σ5PBDEs. | ||
However, the data for most PCBs suggests that the actual sampling rates might be lower than the commonly reported 3–5 m3/day. SI-Table 1† gives an overview of Rs under different sampling conditions reported in literature. PCB Rs for sheltered PUF disks in an outdoor environment were reported as ranging from 2.5–12 m3/day.12,13,18,27,28 For SPMDs the reported values range from 3.5–13 m3/day.12,26 For SIPs Genualdi et al. reported site specific Rs of 2.3–12.5 m3/day (monitored with depuration compounds on co-deployed PUF disks).29 For the XAD samplers introduced by Wania et al.14 PCB sampling rates are still being investigated.30,31 The sampling rates reported for the XAD sampler for OCs ranged from 0.52–2.1 m3/day. Sampling rates for HCB were reported by Chaemfa et al.18 as 7.7 m3/day. The values suggested for PBDERs range from 1.2–12.5 m3/day for PUF disks in FSDs. The reported Rs for PCBs and PBDEs show no specific trend between SIPs, PUFs and SPMDs and no advantages between the different media can be established based on the sampling rates. SI-Table 2† presents a rough estimate of the average sampling rates in this study. The values have to be considered as approximate estimates, due to the high variability in atmospheric concentrations during the long-term field deployment. These would cause high uncertainties in sampling rate values derived from this deployment period.
A clear advantage of the SIP disks to sampling media such as PUF disks and SPMD is the long linear uptake period that was observed in this study. This is especially important if the lighter PCB congeners are among the target compounds. The SIP derived air concentrations show a good agreement with average concentrations monitored by AAS. A further advantage of SIP disks is the relatively simple and economic deployment. Under consideration of these arguments, SIPs can be recommended for long-term sampling of PCBs and PBDEs.
Field deployment parallel to SPMDs
From summer 2008–2009 SIPs were deployed in FSDs along 4 sampling sites in the UK and 6 in Norway (SI-Figure 1†). PCBs, PBDEs and HCB were monitored in all samplers, but unrealistically high values for PBDEs at the Norwegian sites suggest a possible contamination (either during transportation or storage) and so the data are not included here. The chosen sites are established passive sampling sites since 19947–11 and atmospheric PCB data for these sites was available (passive air monitoring with SPMDs, which were deployed in Stevenson screen boxes for two years consecutive years from 1994–2008 and analysed for the here discussed PCB congeners for the samples of 1998–2008). Average PCB concentrations for the sampling period 2008–2009 were estimated by extrapolating the reported data (assuming first order kinetics for decline11). Details on the estimations can be found in SI-Text 1. From the field study results (Fig. 1) HCB reaches equilibrium in the sampler after 6 months and the equilibrium concentrations for the one year deployment were calculated from the volume sampled during the 6 months period. HCB concentrations estimated from SIP disks ranged from 1.3–2.8 pg m−3 and were significantly lower than the predicted values (0.2–8.5 pg m−3).The atmospheric PCB concentrations monitored with the SIPs and the estimated concentrations is given in the SI-Table 3† for the homologue groups. For Σ34PCB concentrations ranged from 3.7–33.0 pg m−3 for SIP monitored values and 1.7–13.0 pg m−3 for estimated values from previous sampling campaigns (see SI-Text 1 and SI-Table 3 for details†). Correlations between estimated and monitored concentrations were significant for all homologue PCB groups except 8CBs. It was observed that the values monitored in the SIPs were on average higher by (28 ± 18)% for 4CBs, (30 ± 15)% for 5CBs, (18 ± 17)% for 6CBs and (31 ± 27)% for 7CBs. As noted above, it was proposed by Shoeib and Harner12 that 3CBs might reach equilibrium in SPMDs after only 50 days and 4CBs after 210 days. This would lead to an underestimation of the concentrations monitored with SPMDs for a 2-year deployment period. This was mostly corrected in the estimation of the SPMD derived concentrations by applying site specific sampling rates and effective sampling volumes (which already consider the equilibrium approach of the SPMD). Differences between the data sets of the lighter congeners still indicate an underestimation for the SPMD derived/extrapolated concentrations for 2009. The paired t-test for all sampling sites finds the estimated and monitored values as not significantly different for all homologue groups except 3CBs and 8CBs. Fig. 3 shows the concentrations for 4CBs, 5CBs, 6CBs and 7CBs.
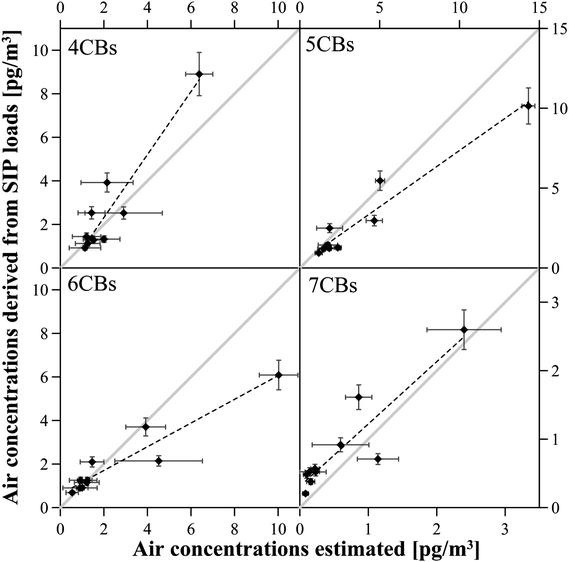 | ||
| Fig. 3 A direct comparison of air concentrations extrapolated from SPMD data (1998–2008) and derived from SIP disks deployed in the UK and Norway shows better agreement for the higher chlorinated PCBs. | ||
Conclusions
The current study has demonstrated the applicability of SIP disks over long deployment periods of up to a year for investigations of semivolatile compounds such as PCBs, PBDEs and HCB. With the exception of HCB that reached saturation in ∼6 months, all of the target compounds experienced linear-phase sampling for the entire deployment period. Linear phase sampling is required for generating time-weighted air concentrations. SIP disks are a good alternative to the conventional PUF disk sampler, especially in cases where longer deployment times are required and for studies involving more volatile POPs. The use of SIP disks is also advantageous with regard to practical sampling aspects such as ease of deployment and cost-efficiency.In the current study, SIP disk derived air concentrations were determined using a previously reported (default) sampling rate of 3 m3/day. Because of their high sorptive capacity, SIP disk samplers do not lend themselves to the use of depuration compounds (DCs) for generating site-specific linear sampling rates. However, in cases where greater accuracy is required it would be possible to collocate a conventional PUF disk sampler spiked with DCs to assess the sampling rate.
Another consideration for using SIP disks and other passive samplers for trace analysis of POPs in air is to ensure that appropriate measures are taken to minimize blank contamination and to take a generous quantity of laboratory and field blanks. This is particularly relevant for some of the emerging POPs that experience relatively high concentrations indoors or that may be present in commercial products in the lab environment.
Acknowledgements
We would like to thank Dr Douglas A. Lane from Environment Canada and Dr Yani Najman from the Lancaster Environment Centre for providing the means to prepare the XAD powder and the UK Department of Environment, Food and Rural Affairs and the Research Council of Norway for financial support.References
- United Nations Environment Programme UNEP, Geneva, Switzerland, 24–27 March 2003.
- M. Venier and R. A. Hites, Environ. Sci. Technol., 2010, 44, 8050–8055 CrossRef CAS.
- J. K. Schuster, R. Gioia, A. J. Sweetman and K. C. Jones, Environ. Sci. Technol., 2010, 44, 8068–8074 CrossRef CAS.
- EMEP (European Monitoring and Evaluation Programme), EMEP, http://tarantula.nilu.no/projects/ccc/emepdata.html, Accessed 01/12/2009, 2009.
- T. Harner, K. Pozo, T. Gouin, A. M. Macdonald, H. Hung, J. Cainey and A. Peters, Environ. Pollut., 2006, 144, 445–452 CrossRef CAS.
- K. Pozo, T. Harner, F. Wania, D. C. G. Muir, K. C. Jones and L. A. Barrie, Environ. Sci. Technol., 2006, 40, 4867–4873 CrossRef CAS.
- R. Gioia, E. Steinnes, G. O. Thomas, S. N. Mejier and K. C. Jones, J. Environ. Monit., 2006, 8, 700–710 RSC.
- F. M. Jaward, S. N. Meijer, E. Steinnes, G. O. Thomas and K. C. Jones, Environ. Sci. Technol., 2004, 38, 2523–2530 CrossRef CAS.
- S. N. Meijer, W. A. Ockenden, E. Steinnes, B. P. Corrigan and K. C. Jones, Environ. Sci. Technol., 2003, 37, 454–461 CrossRef CAS.
- W. A. Ockenden, A. J. Sweetman, H. F. Prest, E. Steinnes and K. C. Jones, Environ. Sci. Technol., 1998, 32, 2795–2803 CrossRef CAS.
- J. K. Schuster, R. Gioia, K. Breivik, E. Steinnes, M. Scheringer and K. C. Jones, Environ. Sci. Technol., 2010, 44, 6760–6766 CrossRef CAS.
- M. Shoeib and T. Harner, Environ. Sci. Technol., 2002, 36, 4142–4151 CrossRef CAS.
- L. Tuduri, T. Harner and H. Hung, Environ. Pollut., 2006, 144, 377–383 CrossRef CAS.
- F. Wania, L. Shen, Y. D. Lei, C. Teixeira and D. C. G. Muir, Environ. Sci. Technol., 2003, 37, 1352–1359 CrossRef CAS.
- T. Harner, N. J. Farrar, M. Shoeib, K. C. Jones and F. A. P. C. Gobas, Environ. Sci. Technol., 2003, 37, 2486–2493 CrossRef CAS.
- J. F. Muller, D. W. Hawker, D. W. Connell, P. Komp and M. S. McLachlan, Atmos. Environ., 2000, 34, 3525–3534 CrossRef CAS.
- M. Shoeib, T. Harner, S. C. Lee, D. Lane and J. P. Zhu, Anal. Chem., 2008, 80, 675–682 CrossRef CAS.
- C. Chaemfa, J. L. Barber, T. Gocht, T. Harner, I. Holoubek, J. Klanova and K. C. Jones, Environ. Pollut., 2008, 156, 1290–1297 CrossRef CAS.
- G. O. Thomas, A. J. Sweetman, C. A. Parker, H. Kreibich and K. C. Jones, Chemosphere, 1998, 36, 2447–2459 CrossRef CAS.
- W. A. Ockenden, B. P. Corrigan, M. Howsam and K. C. Jones, Environ. Sci. Technol., 2001, 35, 4536–4543 CrossRef CAS.
- T. Gouin, G. O. Thomas, I. Cousins, J. Barber, D. Mackay and K. C. Jones, Environ. Sci. Technol., 2002, 36, 1426–1434 CrossRef CAS.
- J. Klánová, P. Èupr, J.i. Kohoutek and T. Harner, Environ. Sci. Technol., 2007, 42, 550–555 CrossRef.
- A. Finizio, D. Mackay, T. Bidleman and T. Harner, Atmos. Environ., 1997, 31, 2289–2296 CrossRef CAS.
- R. G. M. Lee and K. C. Jones, Environ. Sci. Technol., 1999, 33, 3596–3604 CrossRef CAS.
- M. E. Bartkow, K. Booij, K. E. Kennedy, J. F. Muller and D. W. Hawker, Chemosphere, 2005, 60, 170–176 CrossRef CAS.
- W. A. Ockenden, H. F. Prest, G. O. Thomas, A. Sweetman and K. C. Jones, Environ. Sci. Technol., 1998, 32, 1538–1543 CrossRef CAS.
- C. Chaemfa, J. L. Barber, K. S. Kim, T. Harner and K. C. Jones, Atmos. Environ., 2009, 43, 3843–3849 CrossRef CAS.
- S. Hazrati and S. Harrad, Chemosphere, 2007, 67, 448–455 CrossRef CAS.
- S. Genualdi, S. C. Lee, M. Shoeib, A. Gawor, L. Ahrens and T. Harner, Environ. Sci. Technol., 2010, 44, 5534–5539 CrossRef CAS.
- X. Zhang and F. Wania, in SETAC Europe 20th Annual Meeting, Seville, Editon edn, 2010 Search PubMed.
- X. Zhang and F. Wania, in SETAC Europe 21st Annual Meeting, Milan, Editon edn, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SI-Figure 1–3, SI-Table 1–3, SI-Text 1. See DOI: 10.1039/c1em10697j |
| This journal is © The Royal Society of Chemistry 2012 |