Two dimensional graphene–SnS2 hybrids with superior rate capability for lithium ion storage†
Bin
Luo
a,
Yan
Fang
a,
Bin
Wang
a,
Jisheng
Zhou
b,
Huaihe
Song
b and
Linjie
Zhi
*a
aNational Center for Nanoscience and Technology, Zhongguancun, Beiyitiao No.11, 100190, Beijing, P.R. China. E-mail: zhilj@nanoctr.cn; Fax: + 86 10 82545578; Tel: + 86 10 82545578
bState key laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, 100029, Beijing, P.R. China
First published on 23rd November 2011
Abstract
A novel porous nanoarchitecture composed of 2D graphene–SnS2 (G–SnS2) units is developed via a two-step approach in this work. The special structure endows the high-rate transportation of electrolyte ions and electrons throughout the electrode matrix, resulting in remarkable electrochemical performance when it was used as anode in lithium ion batteries.
Broader contextLithium ion batteries (LIBs) have great potential for applications in electric and hybrid vehicles due to their high energy density, high voltage, and long lifespan. However, their rate performance, one of the most important factors for the above applications, urgently needs to be improved. Nanostructured tin-based materials have attracted enormous research interest as high-capacity anode materials for next-generation LIBs. In this study, we report for the first time a facile approach towards two dimensional (2D) graphene–SnS2 hybrids by transforming tin oxide nanoparticles into 2D tin disulfide nanoplates directly on/between graphene nanosheets. The unique structure endows the high-rate transportation of lithium ions and electrons throughout the electrode matrix, resulting in remarkable electrochemical performance when it is used as anode in LIBs. |
The discovery of graphene has triggered a gold rush for worldwide material scientists to search for its potential applications. One of the most attractive characteristics of graphene is its unique two dimensional (2D) atomic thickness structure which leads to various extraordinary properties.1–6 Among these, the very high theoretical surface area (2600 m2 g−1) and its excellent electron transfer behavior render graphene a highly attractive candidate as an electrode material in energy storage devices, such as lithium ion batteries and supercapacitors.7–10 Particularly, the combination of graphene with a second phase material can further enhance the electrochemical properties of the obtained composites. Many materials, including various 0D metal or metal oxide nanoparticles, 1D carbon nanotubes, and polymers, have been used as the second phase to incorporate with graphene.11–17 However, to completely utilize the 2D nature of graphene, an ideal second phase material should be 2D as well, which will significantly improve the diffusion efficiency of lithium ions and thus advance the rate performance of lithium ion batteries.15,18–20 Nevertheless, known examples of 2D nanomaterial–graphene composites are still rarely reported.21,22 Recently, SnSe2 nanoplate–graphene composites were prepared through a solution approach and applied as anode materials in lithium ion batteries, showing promising storage performance superior to SnSe2 nanoplates or graphene alone.18 However, the complicated synthesis procedure for stable colloidal 2D nanomaterials and their low efficiency hybridization with high quality graphene might limit the practical applications of the composites.23 Very recently, composites of SnO2 nanosheets and graphene were prepared via a hydrothermal process followed with a chemical reduction procedure. This SnO2–graphene hybrid structure exhibits enhanced lithium storage properties with high reversible capacities and good cycling performance.22
SnS2 nanoplates have a layered CdI2-type structure, composed of tin atoms sandwiched between two layers of hexagonally disposed close packed sulfur atoms. Recent investigation has revealed that SnS2 nanoplates showed remarkably enhanced electrochemical properties as an electrode material in lithium ion batteries compared to their bulk counterparts. This is mainly due to their unique morphology, consisting of a finite lateral sized and well-defined layered structure.24–28 However, the main drawback of this system has stemmed from the large volume changes and accompanying decrease in capacity that occurs during electrochemical cycling.29 One method of lessening the volume expansion in tin-based electrodes is to distribute the tin-based materials evenly throughout another phase matrix.30–33
Herein, we report for the first time a facile approach towards 2D graphene–SnS2 hybrids by transforming tin oxide nanoparticles into 2D tin disulfide nanoplates directly on/between graphene nanosheetsvia a solution approach followed with a simple chemical vapor deposition (CVD) process (Scheme 1). As a result, a novel porous nanoarchitecture composed of 2D graphene–SnS2 (G–SnS2) units was obtained. Remarkably, high reversible capacity (ca. 650 mA h g−1) and excellent high-rate capability (ca. 230 mA h g−1 at the rate of 6400 mA g−1) were achieved for G–SnS2 when it was used as the anode in lithium ion batteries.
 | ||
| Scheme 1 Illustration of the formation of the G–SnS2 involving two steps: a) tin oxide nanoparticles were firstly decorated on graphene nanosheets and b) converted into metal disulfide nanoplates via a solid-gas reaction. | ||
As illustrated in Scheme 1, the overall synthetic procedure of G–SnS2 involves two steps. Graphene oxide (GO) was firstly decorated with tin oxide nanoparticles by heating ethylene glycol solutions containing SnCl2 at atmospheric pressure. The obtained graphene-supported SnO2 nanoparticles (G–SnO2) were annealed and then heated under a gas mixture of H2S and Ar at 300 °C for a certain amount of time. Detailed synthetic procedures are provided in experimental section of the ESI.†
Field emission scanning electron microscopy (FE-SEM) characterization of a typical G–SnS2 shows that a disordered 3D network structure composed of 2D hexagonal SnS2 nanoplates and flexible graphene nanosheets is obtained (Fig. 1a,b). SnS2 nanoplates are grown in the graphene nanosheets, forming 2D SnS2–graphene units, and distributed uniformly in the whole network. Additionally, the enlarged view of the hybrid material reveals that the size of the SnS2 nanoplates is about 200 nm (Fig. 1b). The XRD pattern supports the assignment of a 2T-type layered structure (JCPDS card #: 23-0677) to the SnS2 nanoplates (Fig. 1c). The thickness of the SnS2 nanoplates is estimated as ca. 6.9 nm, using the Scherrer equation according to the (001) peak.34 The presence of both SnS2 and graphene is further confirmed by Raman spectroscopy (Fig. 1d). The Raman spectrum of G–SnS2 exhibits an intense peak at about 311 cm−1, which is attributed to the A1g mode of SnS2 according to the group theory analysis conducted by previous studies.35,36 The peak at about 1585 cm−1 (G band), corresponding to an E2g mode of graphite, was related to the vibration of the sp2-bonded carbon atoms in a two dimensional hexagonal lattice, while the peak at about 1325 cm−1 (D band) was related to the defects and disorder in the hexagonal graphitic layers.37 The intensity ratio of the D band to the G band (ID/IG) was calculated as 1.19, 1.01, and 0.87 for G–SnS2, G–SnO2 and GO, respectively. Compared with GO, the increased ID/IG for both G–SnO2 and G–SnS2 can be attributed to the presence of unrepaired defects after the removal of partial oxygen moieties38 and the widely separated graphene layers promoted by the intercalation of SnO2 or SnS2 nanoobjects. FT-IR measurements (Figure S2†) confirmed that GO has been reduced during the solution step, which is consistent with the XRD results (Figure S3†).
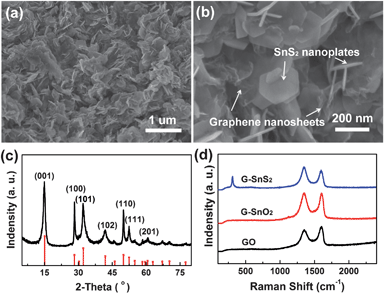 | ||
| Fig. 1 (a, b) SEM images of G–SnS2 at different magnifications. (c) XRD pattern of G–SnS2 (the bottom of the image indicates the JCPDS data (JCPDS: 23-0677) for SnS2). (d) Raman spectra of GO, G–SnO2, and G–SnS2 in the wavelength range of 100–2400 cm−1. | ||
The morphology and the microstructure of the as-prepared G–SnS2 are further investigated by field emission transmission electron microscopy (FE-TEM) measurements as shown in Fig. 2. Cross-sectional TEM and HRTEM images (Fig. 2a, b) confirm the corrugated nature of the graphene nanosheets and relatively rigid structure of SnS2 nanoplates. A side view of the SnS2 nanoplate reveals a thickness of ca. 7 nm (Fig. 2b) which is complying with the estimated thickness from XRD characterization. The HRTEM image (Fig. 2b) exhibits parallel fringes with a spacing of 5.9 Å and 2.8 Å, which can be assigned to the (001) and (101) plane of SnS2, respectively. Meanwhile, the corrugated parallel fringes with a basal spacing of about 3.4 Å can be assigned to the (002) plane of several-layer graphene. Specifically, a typical SnS2 nanoplate lying flat on a grid has lattice fringes with interplanar distances of 3.2 Å and 1.8 Å (Fig. 2c). These distances are consistent with the respective (100) and (-1-10) planes of the hexagonal 2T-SnS2. The fast Fourier transformation (FFT) image (Fig. 2c, inset) of the SnS2 nanoplate shows the characteristic spots for a hexagonal structure with the inner six spots and the outer six spots indicating reflections of the {100} and the {110} planes, respectively. Based on these analyses and the results from viewing different crystallographic orientations, it is reasonable to conclude that the SnS2 nanoplates are single crystalline and have a 2D layered structure with hexagonal symmetry. Element mapping of G–SnS2 hybrid on carbon, tin, and sulfur further demonstrates the homogeneous and highly efficient combination of graphene and 2D SnS2 nanoplates (Fig. 2d), in agreement with energy-dispersive X-ray (EDX) analysis (Supporting Information, Figure S2†).
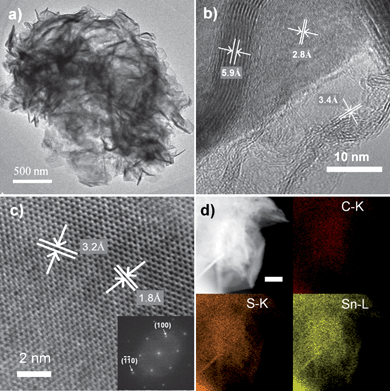 | ||
| Fig. 2 (a) TEM, (b–c) HRTEM, and (d) Element mapping images of the G–SnS2. The inset in (c) is the fast Fourier transformation (FFT) image of the SnS2 nanoplate. Scale bar in (d) is 200 nm. | ||
In order to elucidate the formation mechanism of the 2D G–SnS2 hybrids, time-dependent experiments were carried out. Figure S3† illustrates the XRD patterns of GO, unannealed G–SnO2, annealed G–SnO2, G–SnO2–SnS2 (samples collected after heat treatment in H2S/Ar for 1 h), and G–SnS2, respectively. The major broad diffraction peaks in Figure S3d† can be indexed to the tetragonal rutile SnO2 phase with small crystal size. And there are no distinct diffraction peaks for graphene nanosheets due to its low content. After one hour of heat treatment in H2S/Ar atmosphere, G–SnO2–SnS2 shows the existence of both SnO2 and SnS2 phases and no other phases such as SnO, SnS, and Sn2S3, are observed (Figure S3b†), indicating the direct transformation from SnO2 nanoparticles to SnS2 nanoplates. TEM measurements (Figure S5a, b†) also observed SnO2 nanoparticles attaching on the surface of SnS2 nanoplates during the heat treatment in H2S/Ar.
On the basis of these observations, it is reasonable to speculate that the G–SnS2 hybrid is fabricated via such a two-step process. Firstly, SnO2 nanoparticles are formed on the surface of graphene nanosheets through the solution-based reactions, during which Sn2+ ions are hydrolyzed to form SnO2 nanoparticles at room temperature, while the graphene oxide nanosheets are synchronously reduced to graphene nanosheets. These in situ formed SnO2 nanoparticles are able to effectively separate the stacking of graphene nanosheets. Then, in the CVD procedure, SnO2 nanoparticles react with H2S and are converted into 2D SnS2 nanoplates. The following solid-gas reaction (1) is suggested to be involved for the whole conversion process, during which graphene nanosheets serve as both a platform for the nucleation of the sulfide phase and a good buffer preventing the aggregation of the SnS2 nanoplates.
| SnO2(s) + 2H2S(g) → SnS2(s) + 2H2O(g) | (1) |
In addition, G–SnO2 has much sharper diffraction peak after annealing at 500 °C for 2 h (Figure S3c–d†), demonstrating an increase of crystalline grain size of SnO2 nanoparticles, which is consistent with the TEM results (Figure S4a–d†). Figure S4a and b† demonstrate that SnO2 nanoparticles decorated uniformly on graphene have an average size of less than 5 nm with low crystallinity, while the average size is increased to ∼10 nm after annealing (Figure S4c, d†).
Notably, the annealing treatment of G–SnO2 before the CVD process and the flow rate of the H2S/Ar gas mixture significantly affect the morphologies of the obtained SnS2 nanoplates. Fig. 3 illustrates the XRD patterns and SEM images of three typical G–SnS2 samples with different lateral sizes of SnS2 plates. In order to facilitate the later statement, they are marked as G–SnS2-S, G–SnS2-M and G–SnS2-L with the increasing diameters, respectively. It is observed that a high temperature (e.g. 500 °C) pre-annealing of G–SnO2 leads to a tendency of forming SnS2 plates with small diameters (<300 nm, Fig. 3b, c), while the same CVD processing on a non pre-annealed G–SnO2 tends to produce SnS2 plates as large as 500 nm in diameter (Fig. 3d). More interestingly, the diameter of the SnS2 nanoplates is tunable by controlling the flow rate of gas mixture as well. For example, when the flow rate of H2S/Ar is 400 sccm, the estimated average diameter of plates obtained is ca. 100 nm, as shown in Fig. 3b, which is much smaller than those (ca. 200 nm) obtained at a lower flow rate (e.g. 200 sccm) (Fig. 3c). Additionally, according to a calculation based on the (001) diffraction peak of the samples,34 the average thickness of the SnS2 plates is increasing along with the increase of their diameters, which is in agreement with the results obtained by TEM characterization. The size distribution of SnS2 nanoplates of these three samples obtained for different experimental conditions is listed in Table S1.† In view of the possible formation mechanism of the hybrids discussed above, one of the major factors influencing the size of SnS2 plates might be the crystal structure of the SnO2 nanoparticles. The formation of large-sized SnS2 plates from unannealed SnO2 nanoparticles can be attributed to its low crystallinity and high surface energy of the small SnO2 nanoparticles which are favorable for the growth of disulfide phase. In addition, high flow rate of gas mixture might lead to fast nucleation of disulfide phase, resulting in small-sized SnS2 nanoplates.
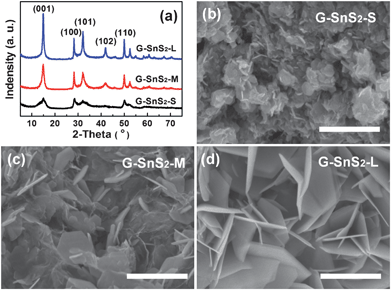 | ||
| Fig. 3 (a) XRD patterns and (b–d) SEM images of three typical G–SnS2 samples with different lateral sizes of SnS2 plates prepared in different conditions. Scale bars in (b–d) are 500 nm. | ||
Based on the unique nanoarchitecture, one promising application of the G–SnS2 hybrids can be envisioned in the context of lithium ion storage. As one example, G–SnS2 hybrid with small SnS2 nanoplate (G–SnS2-S) was selected as the anode in lithium ion batteries. For comparison, bare SnS2 (B-SnS2) was prepared via a similar route (see the Experimental section) and investigated under the same electrochemical conditions. The cyclic voltammetry (CV) behavior (Figure S6 in the Supporting information†) of G–SnS2 is generally consistent with that of pure SnS2, indicating similar electrochemical reaction pathway of the two electrode materials.27,39 However, galvanostatic discharge–charge experiments on these two samples led to totally different electrochemical performances. As shown in Fig. 4, a first reversible capacity of as high as 687 mA h g−1 is observed on G–SnS2-S at 50 mA g−1 (Fig. 4b), which is much higher than that of B-SnS2 (504 mA h g−1). Surprisingly, the reversible capacity of G–SnS2-S is still stable at ca. 650 mA h g−1 even after 30 cycles, while the reversible capacities of B-SnS2 gradually reduce to 277 mA h g−1. The G–SnS2-S manifests exceptional cycling performance compared to not only B-SnS2 but the other SnS2-based anode materials reported previously.24–26,33,40,41Thermogravimetric analysis (TGA) of G–SnS2-S reveals that the weight fraction of graphene in the composite is ca. 5% (see the Supporting Information†). The excellent cycling durability of G–SnS2-S demonstrates that such a small fraction of graphene in the composite fulfils efficiently its role as both buffer matrix and conducting pathways.
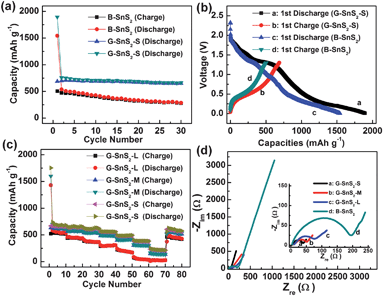 | ||
| Fig. 4 (a) Cycle performance and (b) the first discharge and charge curves of G–SnS2-S and B-SnS2 at a rate of 50 mA g−1. (c) Rate performances of G–SnS2-S, G–SnS2-M, and G–SnS2-L electrodes. (d) Nyquist plots of the G–SnS2-S, G–SnS2-M, G–SnS2-L and B-SnS2 electrodes obtained by applying a sine wave with an amplitude of 5.0 mV in the frequency range from 100 kHz to 10 MHz. | ||
More exciting results come from the rate performance of G–SnS2-S. As demonstrated in Fig. 4c, the reversible capacity of G–SnS2-S is stable at ca. 668 mA h g−1 after 10 cycles at a rate of 100 mA g−1. Upon increasing the discharge–charge rates to 200, 400, 800, 1600, 3200 and 6400 mA g−1, its reversible capacities are maintained at ca. 650, 630, 560, 480, 375, and 230 mA h g−1, respectively. The retention of the reversible capacity of the G–SnS2-S electrodes at 6400 mA g−1 is nearly 93% after 70 cycles. This is in contrast to the traditional carbonaceous materials, which show a continuous and progressive capacity decay along with the cycling processes.42 However, when the diameter of SnS2 nanoplate becomes larger and larger, the corresponding composites (G–SnS2-M and G–SnS2-L) display inferior rate performance, as shown in Fig. 4c. The growing of the SnS2 nanoplates results in an increase of the surface area of the composite (Figure S8†), but in the mean time, leads to a separation between the nanoplates and the graphene layers (Fig. 3b–d) since the growth direction is not exactly along the graphene surface as demonstrated by SEM characterization. On the contrary, small SnS2 nanoplates are much easier to combine with graphene efficiently, as shown in Fig. 3b. Accordingly, it is reasonable to conclude that the 2D nature and the efficient combination of graphene and SnS2 nanoplates are the critical points for the excellent cycling and rate performance of the hybrids; and this is actually the most attractive advantage of the herein developed in situ strategy, which is different from the traditional two-step solution approach.18
To understand deeper the reasons for the excellent rate capability of G–SnS2 and the size effect of SnS2 plates, electrochemical impedance spectroscopy (EIS) measurements were carried out for G–SnS2-S, G–SnS2-M, G–SnS2-L, and B-SnS2 electrodes after the 10th cycle at a rate of 100 mA g−1. Nyquist plots obtained from these four electrodes consist of one depressed semicircle at high/medium frequency and an inclined line at low frequency (Fig. 4d). In general, the semicircle is attributed to the summation of the contact, the solid-electrolyte interphase resistance, and the charge-transfer resistance, while the inclined line at a ca. 45° angle to the real axis corresponds to the lithium-diffusion process within the electrodes. Apparently, the semicircle diameters of G–SnS2 electrodes increase with the increasing size of SnS2 nanoplates and B-SnS2 has the largest one. This is clear evidence that the G–SnS2-S possesses the highest electrical conductivity and the fastest charge-transfer reaction for lithium ion insertion and extraction among all the four samples, further confirming the highly efficient combination of graphene and SnS2 nanoplates.
It should be noted that a large irreversible capacity of nearly 1200 mA h g−1 is observed on the G–SnS2-S electrode during the first discharge and charge process. This may be partly attributed to both the existence of defects in graphene and the disorder of the unique 2D structure of SnS2. In the mean time, it is an interesting point as well for future studies to understand the reasons why so many lithium ions can be inserted into this hybrid.
In summary, a new strategy has been developed to prepare a novel 2D G–SnS2 hybrid. Electrode materials composed of 2D G–SnS2 units are obtained via a successful transformation of SnO2 nanoparticles into 2D SnS2 nanoplates directly on/between graphene nanosheets through a solution approach followed with a simple CVD. Such a unique hybrid system provides favorable transport kinetics for both lithium ions and electrons. As a result, high reversible capacity (ca. 650 mA h g−1) and excellent rate capability (ca. 230 mA h g−1 at the rate of 6400 mA g−1) has been achieved for lithium ion batteries using G–SnS2 as the anode. In addition, this is expected to extend the palette of available methods for the growth and assembly of various inorganic–graphene composites, which definitely encourages extensive applications in areas such as supercapacitors, catalysis, and photovoltaic devices.
Financial supported from the National Natural Science Foundation of China (Grant No. 20973044), the Ministry of Science and Technology of China (No. 2009AA03Z328 and No.2009DPA41220), the Chinese Academy of Sciences (No. KJCX2-YW-H21), and the Guangdong-CAS strategic cooperation Program (2009B091300007) is acknowledged.
References
- K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- K. Geim, Science, 2009, 324, 1530–1534 CrossRef.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170–1171 CrossRef CAS.
- X. L. Feng, S. B. Yang, X. C. Wang and K. Mullen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef.
- G. X. Wang, X. P. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- M. H. Liang and L. J. Zhi, J. Mater. Chem., 2009, 19, 5871–5878 RSC.
- D. Chen, L. H. Tang and J. H. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 RSC.
- Z. J. Fan, J. Yan, L. J. Zhi, Q. Zhang, T. Wei, J. Feng, M. L. Zhang, W. Z. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS.
- D. H. Wang, R. Kou, D. Choi, Z. G. Yang, Z. M. Nie, J. Li, L. V. Saraf, D. H. Hu, J. G. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587–1595 CrossRef CAS.
- S. M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72–75 CrossRef CAS.
- S. B. Yang, X. L. Feng, S. Ivanovici and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS.
- S. B. Yang, X. L. Feng, L. Wang, K. Tang, J. Maier and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 4795–4799 CAS.
- E. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
- S. B. Yang, G. L. Cui, S. P. Pang, Q. Cao, U. Kolb, X. L. Feng, J. Maier and K. Mullen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS.
- J. Choi, J. Jin, I. G. Jung, J. M. Kim, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 5241–5243 RSC.
- Y. Q. Zou and Y. Wang, Nanoscale, 2011, 3, 2615–2620 RSC.
- S. Yang, X. Feng and K. Mullen, Adv. Mater., 2011, 23, 3575–3579 CrossRef CAS.
- H. L. Wang, H. S. Casalongue, Y. Y. Liang and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS.
- S. J. Ding, D. Y. Luan, F. Y. C. Boey, J. S. Chen and X. W. Lou, Chem. Commun., 2011, 47, 7155–7157 RSC.
- K. H. Park, K. Jang and S. U. Son, Angew. Chem., Int. Ed., 2006, 45, 4608–4612 CrossRef CAS.
- J. W. Seo, J. T. Jang, S. W. Park, C. J. Kim, B. W. Park and J. W. Cheon, Adv. Mater., 2008, 20, 4269–4273 CrossRef CAS.
- Zhai, N. Du and H. Z. D. Yang, Chem. Commun., 2011, 47, 1270–1272 RSC.
- S. A. Liu, X. M. Yin, L. B. Chen, Q. H. Li and T. H. Wang, Solid State Sci., 2010, 12, 712–718 CrossRef CAS.
- T. J. Kim, C. Kirn, D. Son, M. Choi and B. Park, J. Power Sources, 2007, 167, 529–535 CrossRef CAS.
- T. Momma, N. Shiraishi, A. Yoshizawa, T. Osaka, A. Gedanken, J. J. Zhu and L. Sominski, J. Power Sources, 2001, 97–8, 198–200 CrossRef.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395–1397 CrossRef CAS.
- Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS.
- G. L. Cui, Y. S. Hu, L. J. Zhi, D. Q. Wu, I. Lieberwirth, J. Maier and K. Mullen, Small, 2007, 3, 2066–2069 CrossRef CAS.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336–2340 CrossRef CAS.
- H. S. Kim, Y. H. Chung, S. H. Kang and Y. E. Sung, Electrochim. Acta, 2009, 54, 3606–3610 CrossRef CAS.
- L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef.
- R. Wang, K. B. Tang, Q. Yang and Y. T. Qian, Chem. Phys. Lett., 2002, 357, 371–375 CrossRef.
- J. Smith, P. E. Meek and W. Y. Liang, J. Phys. C: Solid State Phys., 1977, 10, 1321–1333 CrossRef.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- J. F. Shen, Y. Z. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. X. Ye, Chem. Mater., 2009, 21, 3514–3520 CrossRef CAS.
- H. Mukaibo, A. Yoshizawa, T. Momma and T. Osaka, J. Power Sources, 2003, 119, 60–63 CrossRef.
- S. A. Liu, X. M. Yin, Q. Y. Hao, M. Zhang, L. M. Li, L. B. Chen, Q. H. Li, Y. G. Wang and T. H. Wang, Mater. Lett., 2010, 64, 2350–2353 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. H. Liu and J. S. Xue, Science, 1995, 270, 590–593 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ee02800f |
| This journal is © The Royal Society of Chemistry 2012 |