Additive-free synthesis of unique TiO2 mesocrystals with enhanced lithium-ion intercalation properties†
Zhensheng
Hong
a,
Mingdeng
Wei
*a,
Tongbin
Lan
a,
Lilong
Jiang
a and
Guozhong
Cao
*b
aInstitute of New Energy Technology and Nano-Materials, Fuzhou University, Fuzhou, Fujian 350002, China. E-mail: wei-mingdeng@fzu.edu.cn
bDepartment of Materials Science and Engineering, University of Washington, 302M Roberts Hall, Seattle, WA 98195, USA. E-mail: gzcao@u.washington.edu
First published on 1st December 2011
Abstract
Unique nanorod-like mesocrystals constructed from ultrathin rutileTiO2 nanowires were successfully fabricated for the first time using a low-temperature additive-free synthetic route, and the mesocrystal formation requirements and mechanism in the absence of polymer additives were discussed. The ultrathin nanowires were highly crystalline and their diameters were found to be ca. 3–5 nm. The rutile TiO2 mesocrystals were formed through homoepitaxial aggregation of the ultrathin nanowiresvia face-to-face oriented attachment, accompanied and promoted by simultaneous phase transformation from the precursor hydrogen titanate to rutile TiO2. The rutile TiO2 mesocrystals thus synthesized were subjected to detailed structural characterization by means of scanning and transmission electron microscopy (SEM/TEM) including high-resolution TEM (HRTEM) and selected area electron diffraction (SAED), X-ray diffraction (XRD) and Raman spectroscopy. The rutile TiO2 mesocrystals were tested for lithium-ion intercalation and demonstrated large reversible charge–discharge capacity and excellent cyclic stability, which could be attributed to the intrinsic characteristics of the mesostructured TiO2 constructed from ultrathin nanowires offering large specific surface area for intercalation reaction and easy mass and charge transport, as well as sufficient void space accommodating volume change.
Broader contextMesocrystals are a class of new material that may simultaneously provide good crystallinity and large specific surface area. The mesocrystals have been therefore recognized as a promising material that may have wide applications to electric devices such as dye-sensitized solar cells and lithium-ion batteries. It is critically important to find a simple route for the fabrication of high-quality mesocrystals. This paper reports an experimental study of a new synthesis strategy for the formation of unique rutile TiO2 mesocrystals constructed from ultrathin nanowires in the absence of an additive. The rutile TiO2 mesocrystals were used for the first time as the electrode in LIBs and exhibited a large reversible lithium-ion charge–discharge capacity and excellent cyclic stability. Thus, such TiO2 mesocrystals are a promising anode material for high-power lithium-ion batteries. |
1. Introduction
Superstructures are the subject of intensive research owing to their unique and tunable chemical and physical properties and new collective functions they offer.1–4 Mesocrystals are a new class of solid material constructed of ordered superstructures with controlled architectures.5–7 They can be regarded as assemblies of crystallographically oriented nanocrystals, and have attracted a great deal of research interest since they were first published in 2005.5 Mesocrystals are porous quasi-single crystals consisting of ordered assemblies of aligned nanoparticles which form an entirely new class of superstructured materials. Mesocrystals are similar to zeolites in that they both are porous single crystals; however, they are vastly different. Zeolites possess pores defined by a crystal lattice,8,9 while mesocrystals form a porous structure due to either non-uniform nanocrystals or non-close packing of nanocrystals. In comparison with zeolites, mesocrystals permit significant versatility in chemical compositions and would allow the introduction of a variety of functional properties. Mesocrystals offer the advantages inherited from single crystals and the huge specific surface area, and are considered as promising substitutes for single-crystalline or polycrystalline materials in many applications such as catalysis, sensing and energy storage and conversion.5–7 Some mesocrystals including calcium carbonate,10–13 organic molecules14 and metal oxides6 have been synthesized, characterized and reported. However, the formation mechanism of mesocrystals remains a topic of study, synthesis approaches are materials dependent, the expensive organic additives seemingly have to be used during the synthesis, and the large-scale applications of mesocrystals are limited.7 In addition, the removal of polymer additives after synthesis may present a challenge, as it may alter the surface chemistry unfavorably for certain applications.TiO2 is one of the most important inorganic crystals and has found many technical and industrial applications; yet many new applications are to be fully explored. For example, it is considered as one of the best candidates for photocatalysis and is promising for applications in energy storage and conversion.15–17 A great deal of attention has been paid to nanostructured TiO2 for lithium-ion insertion, because it has many potential advantages including its safety, good cyclic stability and high rate performance.18–21 The performance of TiO2 depends largely on its crystalline phase, crystallinity, surface state, and microstructures.22,23TiO2 mesocrystals with desired crystal phases and tunable architectures would offer the potential of significantly enhanced or revolutionized chemical and physical properties for a wide range of applications, and thus become a focus of intensive research,18–20,24–27 since anatase TiO2 mesocrystals were first prepared by a topotactic conversion from NH4TiOF3 mesocrystals.28,29 However, the synthesis of rutile TiO2 mesocrystals has been very limited.30,31 They were synthesized by using organic additives30 or by the microwave assisted hydrothermal method at relatively high temperatures.31 It would be highly desirable to develop an additive-free synthesis of TiO2 mesocrystals under mild conditions.
This paper reports an experimental study of a new synthesis strategy for the formation of unique rutile TiO2 mesocrystals constructed from ultrathin nanowires in the absence of a polymer additive and at a low temperature. The assembly process of the rutile TiO2 mesocrystals with a single-crystal-like structure through homoepitaxial aggregation of hydrogen titanate nanowires as the primary building blocks is proposed, promoted and accompanied by simultaneous phase transition from hydrogen titanate to rutile titania. Possible requirements for the mesocrystal formation in the absence of polymer additives are also proposed. The rutile TiO2 mesocrystals were tested and exhibited a large reversible lithium-ion charge–discharge capacity and excellent cyclic stability for Li-ion battery applications.
2. Experimental
The synthesis and formation of rutile TiO2 mesocrystals consisted of two sequential steps: first titanate nanowires were synthesized by means of hydrothermal growth in highly basic aqueous solution and acid-washed, and then titanate nanowires dispersed in acidic aqueous solution were allowed to aggregate at mild temperatures. Synthesis of titanate nanowires is similar to the process reported in the literature.32 Typically, 1 g of TiO2 (anatase) was dispersed in a 50 mL of 15 M aqueous KOH solution. After stirring for 10 min, the resulting suspension was transferred into a Teflon-lined stainless steel autoclave with a capacity of 75 mL. The autoclave was kept at 170 °C for 72 h and then naturally cooled to room temperature. The resulting precipitate was washed with 0.1 M HNO3 solution repeatedly until pH 1–2 was reached. The final product was then collected by centrifugation and dried at 70 °C for 12 h in air. Synthesis of rutile TiO2 mesocrystals started with dispersing 150 mg of precursor titanate nanowires in 50 mL of 2 M HNO3 solution under stirring at 50 °C for up to 7 days, and the final product was obtained by centrifugation and washed thoroughly with distilled water. Scanning electron microscopy (SEM, S4800 instrument) and transmission electron microscopy (TEM, FEI F20 S-TIN instrument) were applied for the structural characterization of the resulting nanowires and mesocrystals. X-Ray diffraction (XRD) patterns were recorded on a PANalytical X'Pert spectrometer using the Co Kα radiation (λ = 1.78897 Å), and the data were changed to Cu Kα data. The Raman spectra were recorded in a Renishaw inVia Raman microscope with a 785 nm laser.For the electrochemical measurement of Li-ion intercalation, rutile TiO2 mesocrystals were admixed with polyvinylidene fluoride (PVDF) binder and acetylene black carbon additive in a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10, following a standard method as widely used in the literature.33 The mixture was spread and pressed on copper foil circular flakes as working electrodes (WE), and dried at 120 °C in a vacuum for 12 h. Lithium foils were used as the counter electrodes. The electrolyte was 1 M LiPF6 in a 1/1/1 (volume ratio) mixture of ethylene carbonate (EC), ethylene methyl carbonate (EMC) and dimethyl carbonate (DMC). The separator was a UP 3093 (Japan) micro-porous polypropylene membrane. The cells were assembled in a glove box filled with highly pure argon gas (O2 and H2O levels <1 ppm), and charge/discharge tests were performed in the voltage range of 1 to 3 V (Li+/Li) at different current densities on a Land automatic batteries tester (Land CT 2001A, Wuhan, China). The cyclic voltammetry (CV) measurements were performed on an IM6 at a scan rate of 1 mV s−1.
:20:10, following a standard method as widely used in the literature.33 The mixture was spread and pressed on copper foil circular flakes as working electrodes (WE), and dried at 120 °C in a vacuum for 12 h. Lithium foils were used as the counter electrodes. The electrolyte was 1 M LiPF6 in a 1/1/1 (volume ratio) mixture of ethylene carbonate (EC), ethylene methyl carbonate (EMC) and dimethyl carbonate (DMC). The separator was a UP 3093 (Japan) micro-porous polypropylene membrane. The cells were assembled in a glove box filled with highly pure argon gas (O2 and H2O levels <1 ppm), and charge/discharge tests were performed in the voltage range of 1 to 3 V (Li+/Li) at different current densities on a Land automatic batteries tester (Land CT 2001A, Wuhan, China). The cyclic voltammetry (CV) measurements were performed on an IM6 at a scan rate of 1 mV s−1.
3. Results and discussion
Rutile TiO2 mesocrystals were synthesized by a two-step process: firstly, titanate nanowires were fabricated by means of hydrothermal growth by dissolving anatase TiO2 in KOH aqueous solution and the resulting potassium titanate nanowires were acid-washed thoroughly to form hydrogen titanate nanowires, and secondly, such homogeneously dispersed hydrogen titanate nanowires in HNO3 aqueous solution form aggregated bundles through homoepitaxial aggregation at 50 °C at ambient pressure. Further aging was necessary for the completion of phase transition from hydrogen titanate to rutile titania. Detailed experimental evidence and possible explanation are given below.The analytical results for the titanate nanowires are depicted in Fig. S1†; it is revealed that the precursor of titanate nanowires can be indexed to the same structure of layered titanate of K2Ti6O13 (JCPDS 40-0403), the similar results were reported in the literature.32 It is likely that potassium titanate nanowires were first synthesized during the hydrothermal growth and then converted to hydrogen titanate during the subsequent acid-wash.
Fig. 1a shows the XRD pattern of the rutile TiO2 mesocrystals synthesized at 50 °C for 7 days. All the diffraction peaks can be well indexed to pure rutile TiO2 (JCPDS 76-0318). The average crystallite sizes were calculated to be ca. 12 nm using the Scherer equation based on the (110) diffraction peak, suggesting that these nanorods are composed of nanocrystal subunits.21Fig. 1b and c show SEM images of rutile TiO2 mesocrystals synthesized at 50 °C for 7 days, which suggested that the nanorod-like product was of uniform morphology with a cuspate end and had a proclivity to bundling. The TEM image in Fig. 1d shows the unique TiO2 mesocrystals, which exhibited a well-defined morphology. The lengths and diameters were mostly found to be ca. 300 nm and 60–80 nm, respectively. Fig. 1e shows a typical TEM image of a single nanorod, clearly showing that the nanorod was composed of ultrathin nanowire subunits (left inset). As depicted in Fig. 1e (right inset), the related selected area electron diffraction (SAED) pattern with “single-crystal-like” diffraction from the whole nanorod indicated that a mesocrystal structure was formed. This suggested that these regular nanorods were composed of crystallographically ordered rutileTiO2 nanowires arranged along the [001] direction. Fig. 1f shows a HRTEM image of the end of a single nanorod, confirming that these TiO2 nanowires were oriented along the [001] direction. It was also found that these nanowires were highly crystalline and had a diameter of ca. 3–5 nm. The lattice fringes were approximately 0.30 and 0.32 nm, respectively, for the d001 and d110 spacing. It can be seen from Fig. 1f that the boundaries among the nanowires were clear and the nanowires aggregated together by perfect or imperfect attachments, as highlighted in the inset. More detailed TEM and HRTEM images of the rutile TiO2 mesocrystals are shown in Fig. S2†. It is well documented in the literature27,34–36 that aggregation of nanowires with the same crystallographic orientations as well as the presence of defects are strong evidence of oriented attachment (OA). Therefore, the mesocrystals were constructed from ultrathin rutileTiO2 nanowires and have been successfully fabricated without additives using a low-temperature synthetic route.
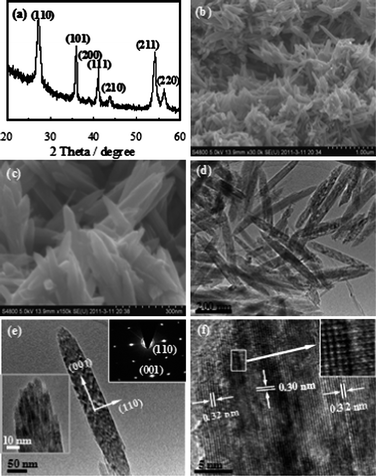 | ||
| Fig. 1 Rutile TiO2 mesocrystals obtained at 50 °C for 7 days: (a) XRD pattern, (b and c) low and high magnification SEM images, (d and e) TEM and (f) HRTEM images. The lower left inset in (e) shows an enlarged TEM image, and the upper right inset is the SAED pattern of the whole nanorod-like mesocrystal. The inset in (f) is an enlarged HRTEM image. | ||
To shed light on the formation mechanism of the nanorod-like rutile TiO2 mesocrystals, a series of samples were synthesized for different periods of time; the results are shown in Fig. 2. As shown in Fig. 2a, numerous nanowires and some short nanorod-like products were observed when the reaction was executed for 2 days. The TEM image in Fig. 2b further confirmed that the short nanorod-like products were formed by the aggregation of ultrathin nanowires. An HRTEM image taken from two adjacent nanowires is presented in the inset in Fig. 2b. The lattice fringes were found to be ca. 0.73 and 0.32 nm, respectively, corresponding to the d200 spacing for the titanate precursor and the d110 spacing for the rutile TiO2. These results indicated that the transfer reaction from the titanate precursor to rutile TiO2 was incomplete after a short time, and that the aggregation of the titanate nanowires happened prior to the transformation of the crystal rutile phase. Fig. 2c depicts the HRTEM image of the rutileTiO2 nanowire, revealing that these ultrathin nanowires were oriented along the [001] direction, as highlighted by the fast Fourier transform of the image in Fig. 2c (inset). When the reaction time was further increased to 5 days, a large number of nanorods were formed beside the residual nanowires, as shown in Fig. 2d. As shown in Fig. 2e, the TEM image revealed that the nanorods obtained were larger and more regular. The inset in Fig. 2e is an HRTEM image of a residual nanowire, which indicated that the precursor titanate nanowires were not transformed completely into rutile TiO2 even though the reaction time had been increased to 5 days. Fig. 2f shows a TEM image of a single nanorod aggregated by ultrathin nanowires. The related SAED pattern in the inset (upper) of Fig. 2f suggested that the rutile nanorods adopted a single-crystal-like mesoscopic structure arranged along the [001] direction. The HRTEM image in the inset of Fig. 2f (lower left) displays the rutile mesocrystal, and the boundary could be observed clearly among the nanowires. These imperfect attachments further confirm that the formation of the rutile mesocrystals was dominated by the OA mechanism. Although the OA mechanism has been proposed for the growth of rutileTiO2 nanocrystals and nanorods composed of smaller nanocrystal subunits,37,38 evidence of mesocrystalline nature has not been given.
 | ||
| Fig. 2 SEM images (a and d) and TEM images (b, c, e and f) of the samples obtained at 50 °C for different reaction times: (a–c) 2 and (d and f) 5 days. The insets: (a and d) enlarge SEM images, (b and e) HRTEM images, (c) related fast Fourier transform image, lower left in (f) the HRTEM image and upper right in (f) the SAED pattern. | ||
In order to follow the phase transformation process, Raman spectroscopy was used to measure the samples obtained over different reaction times, and the results are shown in Fig. 3. The spectra of the precursor titanate nanowires (the 0 d black line in Fig. 3) with clearly identifiable peaks at 190, 276, 443 and 653 cm−1 were similar to those reported in the literature.32 The peaks of the titanate nanowires were significantly weakened in 2 days and disappeared altogether in 5 days. At the same time, the peaks at 229, 438 and 600 cm−1 corresponding to rutile TiO2 (ref. 39 and 40) merged in 2 days and their intensity increased gradually with the increasing reaction time, indicating the phase transition from titanate to rutile TiO2.
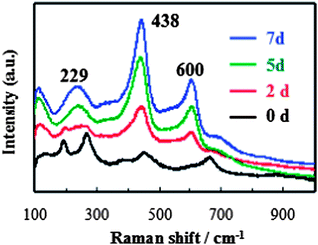 | ||
| Fig. 3 Raman spectra of the samples collected at different reaction times. | ||
In all synthesis of mesocrystals in the literature, polymer additives are usually used,5–7 except the most recent work by Yeet al.21 on the synthesis of anatase TiO2 mesocrystals. The role of polymer additives in the formation of mesocrystals could be considered as a structure-directing agent, similar to that in the formation of synthetic zeolites,41 which assists the crystal growth in zeolites and the assembly of nanocrystals in mesocrystals. The presence of polymer additives would also hinder the diffusion of growth species or nanocrystals so as to allow the attachment or assembly of newly arrived growth species or nanocrystals to the lowest energy state: perfect crystal growth or homoepitaxial aggregates. In the absence of polymer additives, a mechanism for the formation of rutile TiO2 mesocrystals through homoepitaxial aggregation of hydrogen titanate single crystal nanowires is proposed based on the experimental results and is depicted in Scheme 1, similar to the “nanoslab” hypothesis proposed for the zeolite synthesis.42 The principal attractive force between two objects is the van der Waals attraction; the Brownian motion would bring two titanate nanowires in contact. The Hamaker constant of hydrogen titanate in aqueous solution is small as the dielectric constant of water is more than an order of magnitude larger than that of hydrogen titanate and, thus, the attraction force between two hydrogen titanate nanowires would be rather weak, allowing better alignment of nanowires during the homoepitaxial aggregation process, so that the formation of mesocrystals becomes more favorable. In addition, the acidic condition would ensure a relatively slow condensation reaction between two surfaces of hydrogen titanate or titania nanowires,43 and further permit better crystal orientation alignment when aggregation occurs.
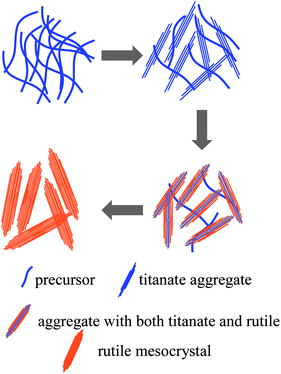 | ||
| Scheme 1 Schematic of a tentative mechanism for the formation of rutile TiO2 mesocrystals. | ||
In the current reaction system, it is proposed that the precursor hydrogen titanate nanowires gradually transformed to rutile TiO2 under acidic conditions in conjunction with the homoepitaxial aggregation, in a good agreement with the literature that the layered titanate would be transferred to rutile TiO2 under a relatively concentrated acidic condition.44 At the same time, the oriented attachment of the rutile nanowires would readily occur when the nanowires all arranged along the [001] direction, leading to the formation of ordered rutile TiO2 aggregates. It would also be worthy of notice that the oriented attachment along the [001] direction permits the maximal attachment and, thus, the most reduction in surface energy. Subsequently, the phase transformation of the precursor titanate nanowires would proceed continuously and would then be coherent to such rutile TiO2 aggregates by the same faces, resulting in the formation of nanorod-like rutile TiO2 mesocrystals with a single-crystal-like structure. It is suggested that the slow phase transformation at such a low temperature accounted for the stability of the ultrathin nanowires against immediate single crystal. The oriented attachment by two identical crystal faces has previously been observed in the formation of mesocrystals.7,27,30
Recently, there has been a great deal of interest in using nanostructured rutile TiO2 as anode materials in rechargeable lithium-ion batteries.45–49 The rutile TiO2 mesocrystals synthesized in this study offer much large specific surface area and shorter transport distance, and thus would promise better lithium-ion insertion properties. Fig. 4a shows representative CV curves of the rutile TiO2 mesocrystals at a scanning rate of 1 mV s−1 between 1.0 and 3.0 V. The results are in good agreement with the literature data.50,51 The cathodic peak at appropriately 1.35 V is attributed to the lithium-ion insertion into the rutile structure.45–49 Its associated anodic reaction can be identified as a broad peak at about 1.85 V. Interestingly, the peak intensity increased gradually with increased cycling numbers, which may be indicative of a possible activation process in the electrode material or an improved surface wetting of electrolyte. As shown in Fig. 4b, it exhibited the charge–discharge profiles of the rutile TiO2 mesocrystals at a current density of 1 C (1 C = 170 mA g−1) for the initial two cycles in the potential of 1.0–3.0 V. A large discharge capacity of 298.7 mA h g−1 and a reversible charge capacity of 207.9 mA h g−1 were obtained at the first cycle, which indicated that appropriately 0.89 mol Li-ion could be inserted into TiO2 (Li0.89TiO2) and 0.62 mol Li-ion could be reversibly extracted (Li0.62TiO2) in the initial cycle. The initial discharge capacity was much larger than that of the previously reported nanosized rutile TiO2.45 It can be noted that there was a voltage plateau near 1.05 V in the first discharge curve. A similar result was also reported in the literature, and this can be attributed to the irreversible change in the structure of the rutile TiO2 upon deeper Li-ion insertion (between 1.4 and 1.0 V).45 In addition, the sloped discharge curve in the second cycle might be ascribed to the irreversible formation of ‘nanocomposite’ crystalline grains and amorphous regions.52–54Fig. 4c presents the rate capability of the rutile TiO2 mesocrystals from 0.5 to 5 C for 10 cycles at each current rate. It is noteworthy that the rutile TiO2 mesocrystals had a reversible capacity of ∼199.8 mA h g−1 after 10 cycles at 0.5 C and retained a capacity of ∼100 mA h g−1 even at a current rate as high as 5 C, indicating that the rutile TiO2 mesocrystals deliver a high-rate performance. A remarkably large capacity of ∼201.3 mA h g−1 was achieved when the current rate was returned to 0.5 C even after 40 cycles at different current rates. Fig. 4d shows the cycling behavior of a cell made of the rutile TiO2 mesocrystals at a current rate of 1 C. It clearly demonstrated that the rutile TiO2 mesocrystals had an excellent performance up to 100 cycles with a reversible capacity of 171.3 mA h g−1 retained, better than that of cells composed of nanosized rutile,45 or porous rutileTiO2 nanorod microspheres.48 A specific surface area (BET) of 38.5 m2 g−1 and a low volume of mesopores were observed from the nitrogen adsorption/desorption isotherm (Fig. S3†). Such enhanced lithium-ion intercalation property can be attributed to the mesoscopic structure of the rutile TiO2 mesocrystals constructed from ultrathin nanowires, which offer large specific surface area and short transport distance, and thus facilitate lithium-ion intercalation reaction at the interface, and short distance for both mass and charge transport, as well as freedom for volume change accompanied by lithium-ion intercalation.
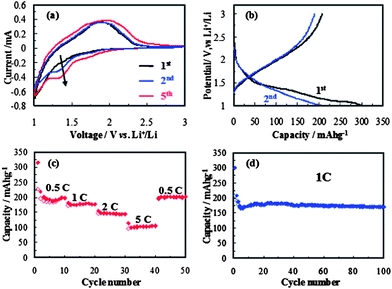 | ||
| Fig. 4 The electrochemical properties of rutile TiO2 mesocrystals: (a) cyclic voltammograms between 1.0 and 3.0 V with a scan rate of 1 mV s−1, (b) charge–discharge profiles at a current density of 1 C, (c) rate capability from 0.5 to 5 C, (d) cycling performance at a constant current density of 1 C. | ||
4. Conclusions
Unique nanorod-like mesocrystals constructed from ultrathin rutileTiO2 nanowires were successfully fabricated by a low-temperature synthetic route without using an additive. It was shown that rutile TiO2 mesocrystals with a single-crystal-like structure were formed by the face-to-face oriented attachment of the ultrathin nanowiresvia homoepitaxial aggregation, accompanied and promoted by a simultaneous phase transformation from precursor hydrogen titanate to rutile TiO2. It is hypothesized that the relatively lower Hamaker constant and slower surface condensation may be required for the homoepitaxial aggregation in the absence of polymer additives. The rutile TiO2 mesocrystals were used for the first time as the electrode material in rechargeable lithium-ion batteries which demonstrated a large reversible charge–discharge capacity and excellent cycling performance, which is attributed to the intrinsic characteristics of the mesoscopic structure of rutile TiO2 mesocrystals constructed from loosely packed ultrathin nanowires.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (NSFC 21073039 and 21173049), Fujian Province Fund (JA10016), Ministry of Education (200803860004) and Key Laboratory of Novel Thin Film Solar Cells, CAS. GZC would like to acknowledge the financial support by National Science Foundation (NSF, CMMI-1030048).References
- A. Dong, J. Chen, P. M. Vora, J. M. Kikkawa and C. B. Murray, Nature, 2010, 466, 474 CrossRef CAS.
- S. Srivastava, A. Santos, K. Critchley, K. S. Kim, P. Podsiadlo, K. Sun, J. Lee, C. Xu, G. D. Lilly, S. C. Glotzer and N. A. Kotov, Science, 2010, 327, 1355 CrossRef CAS.
- M. J. Bierman, Y. K. A. Lau, A. V. Kvit, A. L. Schmitt and S. Jin, Science, 2008, 320, 1060 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591 CrossRef CAS.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576 CrossRef.
- L. Zhou and P. O'Brien, Small, 2008, 4, 1566 CrossRef CAS.
- R. Q. Song and H. Cölfen, Adv. Mater., 2010, 22, 1301 CrossRef CAS.
- H. Van Bekkum, E. M. Flanigen, P. A. Jacobs and J. C. Jansen, Introduction to Zeolite Science and Practice, Elsevier, Amsterdam, 2nd edn, 2001 Search PubMed.
- J. V. Smith, Chem. Rev., 1988, 88, 149 CrossRef CAS.
- T. Wang, H. Cölfen and M. Antonietti, J. Am. Chem. Soc., 2005, 127, 3246 CrossRef CAS.
- A. N. Kulak, P. Iddon, Y. Li, S. P. Armes, H. Cölfen, O. Paris, R. M. Wilson and F. C. Meldrum, J. Am. Chem. Soc., 2007, 129, 3729 CrossRef CAS.
- X. Geng, L. Liu, J. Jiang and S.-H. Yu, Cryst. Growth Des., 2010, 10, 3448 CAS.
- S. Kumar, T. Ito, Y. Yanagihara, Y. Oaki, T. Nishimura and T. Kato, CrystEngComm, 2010, 12, 2021 RSC.
- M. Huang, U. Schilde, M. Kumke, M. Antonietti and H. Cölfen, J. Am. Chem. Soc., 2010, 132, 3700 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- Z. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588 CrossRef CAS.
- Y. Ren, L. J. Hardwick and P. G. Bruce, Angew. Chem., Int. Ed., 2010, 49, 2570 CrossRef CAS.
- J. S. Chen, Y. L. Tan, C. M. Li, Y. L. Cheah, D. Luan, S. Madhavi, F. Y. C. Boey, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 6124 CrossRef CAS.
- J. Ye, W. Liu, J. Cai, S. Chen, X. Zhao, H. Zhou and L. Qi, J. Am. Chem. Soc., 2011, 133, 933 CrossRef CAS.
- D. Liu, Y. Zhang, P. Xiao, B. B. Garcia, Q. Zhang, X. Zhou, Y. H. Jeong and G. Cao, Electrochim. Acta, 2009, 54, 6816 CrossRef CAS.
- D. Liu and G. Cao, Energy Environ. Sci., 2010, 3, 1218 CAS.
- B. Liu and H. C. Zeng, Chem. Mater., 2008, 20, 2711 CrossRef CAS.
- L. Li and C. Liu, CrystEngComm, 2010, 12, 2073 RSC.
- J. Feng, M. Yin, Z. Wang, S. Yan, L. Wan, Z. Li and Z. Zou, CrystEngComm, 2010, 12, 3425 RSC.
- R. O. Da Silva, R. H. Goncalves, D. G. Stroppa, A. J. Ramirez and E. R. Leite, Nanoscale, 2011, 3, 1910 RSC.
- L. Zhou, D. Smyth Boyle and P. O'Brien, Chem. Commun., 2007, 144 RSC.
- L. Zhou, D. Smyth-Boyle and P. O'Brien, J. Am. Chem. Soc., 2008, 130, 1309 CrossRef CAS.
- S.-J. Liu, J.-Y. Gong, B. Hu and S.-H. Yu, Cryst. Growth Des., 2009, 9, 203 CAS.
- D. Zhang, G. Li, F. Wang and J. C. Yu, CrystEngComm, 2010, 12, 1759 RSC.
- X. Meng, D. Wang, J. Liu, B. Lin and Z. Fu, Solid State Commun., 2006, 137, 146 CAS.
- Z. Hong, X. Zheng, X. Ding, L. Jiang, M. Wei and K. Wei, Energy Environ. Sci., 2011, 4, 1886 CAS.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969 CrossRef CAS.
- R. L. Penn and J. F. Banfield, Geochim. Cosmochim. Acta, 1999, 63, 1549 CrossRef CAS.
- D. G. Stroppa, L. A. Montoro, A. Beltran, T. G. Conti, R. O. da Silva, J. Andres, E. R. Leite and A. J. Ramirez, Chem. Commun., 2011, 47, 3117 RSC.
- C. Ribeiro, C. Vila, D. B. Stroppa, V. R. Mastelaro, J. Bettini, E. Longo and E. R. Leite, J. Phys. Chem. C, 2007, 111, 5871 CAS.
- J.-M. Wu and B. Qi, J. Am. Ceram. Soc., 2008, 91, 3961 CrossRef CAS.
- J. L. Gole, S. M. Prokes and O. J. Glembocki, J. Phys. Chem. C, 2008, 112, 1782 CAS.
- C. P. Kumar, N. O. Gopal, T. C. Wang, M.-S. Wong and S. C. Ke, J. Phys. Chem. B, 2006, 110, 5223 CrossRef CAS.
- S. L. Burkett and M. E. Davis, J. Phys. Chem., 1994, 98, 4647 CrossRef CAS.
- C. E. A. Kirschhock, R. Ravishankar, L. V. Looveren, P. A. Jacobs and J. A. Martens, J. Phys. Chem. B, 1999, 103, 4972 CrossRef CAS.
- C. J. Brinker and G. W. Scherer, Sol–Gel Science: the Physics and Chemistry of Sol–Gel Processing, Academic Pr, 1990 Search PubMed.
- D. V. Bavykin, J. M. Friedrich, A. A. Lapkin and F. C. Walsh, Chem. Mater., 2006, 18, 1124 CrossRef CAS.
- Y. S. Hu, L. Kienle, Y. G. Guo and J. Maier, Adv. Mater., 2006, 18, 1421 CrossRef CAS.
- J. S. Chen and X. W. Lou, J. Power Sources, 2010, 195, 2905 CrossRef CAS.
- J. Liu, G. Cao, Z. Yang, D. Wang, D. Dubois, X. Zhou, G. L. Graff, L. R. Pederson and J.-G. Zhang, ChemSusChem, 2008, 1, 676 CrossRef CAS.
- H. Qiao, Y. Wang, L. Xiao and L. Zhang, Electrochem. Commun., 2008, 10, 1280 CrossRef CAS.
- D. Wang, D. Choi, Z. Yang, V. V. Viswanathan, Z. Nie, C. Wang, Y. Song, J.-G. Zhang and J. Liu, Chem. Mater., 2008, 20, 3435 CrossRef CAS.
- N. A. Milne, M. Skyllas-Kazacos and V. Luca, J. Phys. Chem. C, 2009, 113, 12983 CAS.
- L. Kavan, D. Fattakhova and P. Krtil, J. Electrochem. Soc., 1999, 146, 1375 CrossRef CAS.
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 5215 RSC.
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621 CrossRef CAS.
- H. Furukawa, M. Hibino and I. Honma, J. Electrochem. Soc., 2004, 151, A527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ee02551a |
| This journal is © The Royal Society of Chemistry 2012 |