Bimetallic IrNi core platinum monolayer shell electrocatalysts for the oxygen reduction reaction†
Kurian A.
Kuttiyiel
a,
Kotaro
Sasaki
a,
YongMan
Choi
a,
Dong
Su
b,
Ping
Liu
a and
Radoslav R.
Adzic
*a
aChemistry Department, Brookhaven National Laboratory, Upton, NY 11973. E-mail: adzic@bnl.gov
bCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973
First published on 28th October 2011
Abstract
We synthesized a low-Pt content electrocatalyst consisting of a Pt monolayer placed on carbon-supported thermally treated IrNi core–shell structured nanoparticles using galvanic displacement of a Cu monolayer deposited at underpotentials. The Pt mass activity of the PtML/IrNi/C electrocatalyst obtained in a scale-up synthesis is approximately 3 times higher than that of the commercial Pt/C electrocatalyst. The electronic and geometrical effects of the IrNi substrate on the Pt monolayer result in its higher catalytic activity than that of Pt nanoparticles. The structure and composition of the core–shell nanoparticles were verified using transmission electron microscopy and in situX-ray absorption spectroscopy, while a potential cycling test was employed to confirm the stability of the electrocatalyst. Our experimental results, supported by the density functional calculations using a sphere-like model, demonstrate an effective way of using Pt that can resolve key problems of cathodic oxygen reduction hampering fuel cell commercialization.
Broader contextThe proton exchange membrane fuel cell (PEMFC) is widely considered as a potentially highly efficient source of clean energy systems. The last decade has seen considerable advances made in fuel cell electrocatalysis, yielding improved electrocatalysts, and increasing our understanding of the kinetics of the oxygen-reduction reaction (ORR). However, some technological difficulties hampering the automotive applications of PEMFCs, such as high Pt loading, low ORR activity, and poor stability, have not been removed. Here, we report on a newly developed high performance ORR electrocatalyst synthesized by depositing a Pt monolayer on IrNi core nanoparticles by the galvanic displacement of a Cu monolayer. A Cu monolayer is obtained by underpotential deposition on IrNi nanoparticles having two monolayers of Ir covering a Ni core. The ORR activity of the PtML/IrNi/C electrocatalyst is much higher than that of the commercial Pt/C electrocatalyst. Long-term fuel cell tests demonstrated its good stability. The electronic, geometrical, and segregation effects of the IrNi substrate on the Pt monolayer result in its higher catalytic activity than that of Pt nanoparticles. Our experimental results, supported by DFT calculations, demonstrated a promising way of using the Pt monolayer that can resolve key problems of cathodic oxygen reduction hampering PEMFC. |
1. Introduction
Although the potential benefits of fuel cells as power sources can be significant, many technical and economic challenges still exist that prevent their widespread application. Inadequate efficiency of energy conversion and the cost associated with Platinum (Pt) catalysts remain the main causes.1–3 Despite extensive research work in finding low-cost electrocatalysts, platinum is still the only single-metal electrode material that can fulfil demands in fuel cells working in acid media. Numerous studies have reported that Pt alloys (mostly with transition metals such as Co, Ni, and Fe) are more active for the ORR than pure Pt.4–6 However, the Pt loading in these alloys is very high and moreover the non-noble metals in these alloys are usually etched easily in the acid environment of the PEMFC. Therefore, it is desirable to design catalysts that have higher ORR activity, lower Pt loading, and enhanced stability.We recently inaugurated platinum monolayer electrocatalysts, comprised of a Pt monolayer on nanoparticles of a suitable metal, or alloy substrate. This approach was verified as an attractive way of designing improved ORR electrocatalysts, and also reducing platinum loadings in fuel-cell electrodes.7–10 Using non-noble metal–noble metal core–shell nanoparticles as supports facilitates a further reduction of the content of the noble metal in the ORR electrocatalysts. In addition, by properly selecting the noble-metal shell, the activity of the Pt monolayer can be heightened through electronic and/or geometric effects.11,12 This new approach represents a viable way of designing electrocatalysts with enhanced activity and stability and with very low platinum loading.
Using the Pt monolayer approach we have devised a new electrocatalyst consisting of one monolayer of Pt on a carbon supported Iridium-Nickel core–shell structured nanoparticle substrate. Iridium is one of the most stable metals and its activity towards the ORR is slightly enhanced by alloying it with other transition metals.13,14 Density functional theory (DFT) slab calculations for single crystals of Ir show that the Pt monolayer on Ir (111) surfaces causes too strong a contraction in Pt–Pt bonding and the binding energy of oxygen to Pt becomes too small, in agreement with the experiment.15 However, the ligand effect which arises due to the presence of other metals around a metal atom changes its electronic environment, giving rise to modifications of its electronic structure and consequently, its chemical properties.16,17 Herein, we report on our newly developed high performance ORR electrocatalyst synthesized by depositing a Pt monolayer on IrNi core nanoparticles by the galvanic displacement of a Cu monolayer obtained by underpotential deposition. Density functional theory (DFT) and experimental calculations show that mixing Ni with Ir induces geometric, electronic, and segregation effects which weaken the binding energy of oxygen with Pt resulting in its enhanced electrocatalytic activity.
2. Experimental section
The carbon supported Iridium (Ir) and Nickel (Ni) electrocatalyst was prepared by mixing an equal molar ratio of (NH4)2IrCl6, Ni(HCO2)2H2O salts with high area Vulcan XC72R carbon black to obtain 20% total metal. The synthesis and the microstructure of IrNi core–shell nanoparticles has been reported recently.18 Briefly, carbon supported IrNi core–shell nanoparticles when synthesized by chemical reduction and subsequent thermal annealing form Ir-rich shells on IrNi solid solution alloy cores. The average size of the IrNi nanoparticles is 4.7 nm and the Ni atoms inside the cores were completely protected by the Ir shell from oxidation or dissolution under elevated potentials. The mole ratio of Ir to Ni of the resultant IrNi sample was determined to be 1 to 0.56 by energy dispersive X-ray spectroscopy (EDX).2.1 Synthesis on glassy carbon electrode
A micro-gram quantity of PtML/IrNi/C electrocatalyst was prepared by depositing a Pt monolayer on an IrNi substrate electrochemically on a glassy carbon electrode. Catalyst inks of IrNi/C nanoparticles were prepared by mixing 5 mg of the catalysts with 5 mL of 18 MΩ water. The solution was ultrasonicated until a dark, uniform ink was achieved. Thin-film electrodes with IrNi/C nanoparticles were prepared by placing 10 μl of nanoparticle suspension onto a flat glassy carbon electrode (5-mm diameter, Pine Instrument). After drying in a vacuum, the electrode was covered with 10 μl of a dilute Nafion solution (2 μg/5 μl) and dried again. The Pt monolayer was placed on the IrNi/C thin-film electrode by galvanic displacement of a Cu monolayer formed by under potential deposition (upd) with Pt. The Cu monolayer was deposited in a 50 mM H2SO4 + 50 mM CuSO4 solution. The electrode covered with a Cu monolayer was rinsed and immersed in a 1.0 mM K2PtCl4 + 50 mM H2SO4 solution to displace the Cu with Pt. For comparison of the ORR activity, carbon supported Ir (E-tek 10%) nanoparticles were deposited with a Pt monolayer using the same method and henceforth are denoted as PtML/Ir/C. A leak-free reference electrode (Ag/AgCl/KCl (3M)) was used and a platinum wire served as the counter electrode. All the potentials are given with respect to a reversible hydrogen electrode (RHE). The electrolytes were prepared from Optima sulfuric acid and perchloric acid (Fisher), and MilliQ UV-plus water. The Pt content in the RDE samples was derived from the Cu upd charge and/or the H adsorption/desorption charge.2.2 Scale-up synthesis
The gram-scale sample (1 gm batch) was synthesized in a specially designed electrochemical cell consisting of a Ti cylinder that acts as a cell wall and working electrode, platinized Pt foil as the counter electrode and an Ag/AgCl/KCl (3M) leak-free electrode as the reference electrode. A detailed explanation of the method and synthesis procedure can be found elsewhere.19 After reducing IrNi nanoparticle surface oxides by applying potentials in 50 mM H2SO4 solution, CuSO4 solution (50mM) is added in the cell. Purging continuously with Ar gas, a constant potential is then applied to form upd Cu adatoms on the IrNi nanoparticle surfaces. At a constant potential, the solution is intermittently stirred to disperse the IrNi/C particles in the electrolyte, and therefore to ensure the formation of upd Cu monolayers on the whole surface. The Cu upd process is carried out until the current becomes a steady value near zero. After the Cu upd formation, a Pt2+ solution (K2PtCl4 in a 50mM H2SO4 solution) in a separate reservoir is injected slowly into the cell. To allow Pt atoms to replace Cu monolayers on IrNi surfaces galvanically the solution is vigorously stirred. The resultant PtML/IrNi/C electrocatalyst is washed repeatedly by filtering. The Pt content in the scale-up synthesis sample (4.4 wt. %) was determined by assay elemental analysis using inductively coupled plasma (ICP) emission spectroscopy.2.3 Characterization
The as synthesized electrocatalyst was dispersed in water and one drop of the slurry was deposited on a carbon covered copper grid (EMS, Hatfield, PA). The electrocatalyst was characterized using a Hitachi aberration-corrected scanning transmission electron microscope (HD-2700C) at the Center for Functional Nanomaterials (CFN), Brookhaven National Laboratory (BNL). A 1.4 Å electron probe with probe current ∼50 pA and an electron source with an energy resolution of 0.35 eV were used in this study. The microscope was equipped with a cold field emission electron source and a high resolution Gatan Enfina energy-loss spectrometer. Electrochemical studies were conducted in a three-electrode glass cell in 0.1 M HClO4 solution at room temperature using a CHI 700B electrochemical analyzer. For the X-ray absorption spectroscopy (XAS) measurements, the electrocatalyst was pressed and sealed in an electrochemical cell as described in refs 19 and 20. All the XAS measurements were carried out at the National Synchrotron Light Source (NSLS), Brookhaven National Laboratory (BNL) using Beam Line X19A. The measurements were carried out at the Pt L3 edge (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564 eV), Ir L3 edge (11215 eV) and Ni K edge (8333 eV) in 1 M HClO4 at different potentials at room temperature. The data were processed and analyzed by Athena and Artemis software.21
564 eV), Ir L3 edge (11215 eV) and Ni K edge (8333 eV) in 1 M HClO4 at different potentials at room temperature. The data were processed and analyzed by Athena and Artemis software.21
2.4 Computational methods
Spin-polarized DFT calculations were carried out with the Vienna ab initio simulation package (VASP) code,22,23 using only the Γ point for k sampling, and a cut-off energy of 400 eV. We applied the projector augmented wave method (PAW)24 with the generalized gradient approximation (GGA) using the revised Perdew–Burke–Ernzerhof (RPBE) functional.25 We employed sphere-like nanoparticle models to simulate the core–shell nanoparticles studied in our experiments, wherein all atoms were allowed to relax fully. The binding energy of atomic oxygen on the (111) plane of a nanoparticle (BE–O) was calculated, and considered as a descriptor for scaling the ORR activity. Our previous studies showed that DFT calculated BE–O based on the hemisphere-like model was able to well describe the experimentally measured ORR activity of core–shell nanoparticles.26,27 Here, BE–O is defined as BE–O = E[O–NP] − E[NP] − E[O], where E[O–NP], E[NP], and E[O], respectively, are the calculated electronic energies of an adsorbed oxygen-atom on a nanoparticle, a clean nanoparticle, and a triplet oxygen atom. In addition, to examine geometric effects, we predicted surface strains according to (a − a0)/a0 where a and a0, respectively, are the averaged Pt–Pt distance in the Pt monolayer, and the calculated Pt–Pt distance of bulk Pt (2.824 Å).3. Results and discussion
3.1 STEM/EELS profile analysis
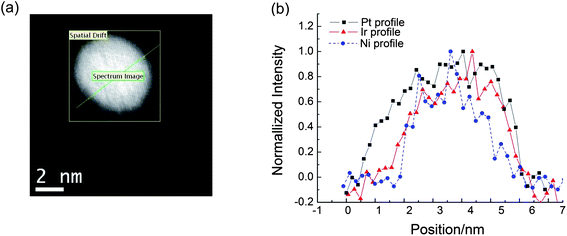
From the scaled-up synthesized sample, numerous individual particles of different sizes were imaged, and their Pt shell thicknesses were analyzed simultaneously by element-sensitive EELS spectroscopy. All signals from the Pt M (2122 eV), Ir M (2040 eV) and Ni L (855 eV) edges were detected by EELS mapping. Fig. 1 illustrates the line profile analysis by STEM/EELS showing the distribution of Pt, Ir and Ni components in a single nanoparticle. Fig. 1(a) shows a high angle annular dark field (HAADF) image of a representative single nanoparticle. A STEM-EELS line scan of the Pt and Ir M-edge and Ni L-edge scan can be done by moving the electron probe along the line indicated in Fig. 1(a) and their intensities are as shown in Fig. 1(b). It is clearly observed that both the intensities of Ir and Ni are approximately constant around the center of the nanoparticle and Ni is depleted at both edges of the nanoparticles. In our previous study, the EXAFS analyses demonstrated that the IrNi nanoparticles comprise two-layer Ir shells and IrNi random alloy cores18 and this EELS observation is in line with the analyses. The differences in the Pt and the Ir profile, measured at the half value of the intensities are from 0.27 to 0.33 nm, corresponding to approximately one monolayer Pt shell on the IrNi alloy core. Thus, the formation of one monolayer of a Pt shell over an IrNi solid solution core18 is confirmed by EELS.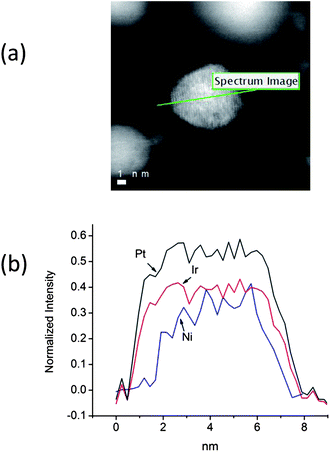 | ||
| Fig. 1 (a) A HAADF-STEM image of a PtIrNi nanoparticle, and (b) a comparison of the EELS intensities for the Pt and Ir M-edge and Ni L-edge along the scanned line (as indicated in Fig. 1(a)). | ||
3.2 In situ XAS analysis
In situ XANES of Pt L3, Ir L3 and Ni K edges were measured from the nanoparticles in 1 M HClO4 at various potentials. As shown in Fig. 2(c), Ni in the nanoparticles is considered to be metallic and alloying with Ir may have changed the electronic states of the Ni atoms.18 Also, the in situXANES of the Ir L3 edge (Fig. 2(b)) from the nanoparticles does not show any shift in the Edge energy at various potentials indicating that the Ir atoms in the IrNi core material retain the same metallic form and that no appreciable amount of oxygen-containing species adsorb on the Ir sites in the potential range of 0.41–1.11 V. These results indicate that the Pt layers deposited on IrNi nanoparticles completely block Ir oxide formation in acidic environments to very high potentials (>0.9 V), further confirming the formation of a complete Pt shell on an IrNi alloy core. On the other hand, the XANES spectra of the Pt L3 edge from PtML/IrNi/C (Fig. 2(a)) indicate a very small potential dependence. This effect is more evident in the relative change of the X-ray absorption peak intensity of the Pt L3 edge spectra for PtML/IrNi/C and Pt/C as a function of potential (Fig. 2(d)). The increase in the intensity of the absorption edge peak for the PtML/IrNi/C electrocatalyst commences at considerably higher potentials than does that for the Pt/C catalyst. The difference in absorption properties between PtML/IrNi/C and pure Pt is due to modification of the electronic properties of the surface atoms by the underlying metal via geometric strain and ligand interactions.15,28 The high Pt oxidation of the PtML/IrNi/C electrocatalyst (significant delay in PtOH formation) clearly indicates the better stability of the electrocatalyst, which is also discussed further in this paper. The Pt and Ir mole ratio was 3:7 which was determined by the ex situXAS analysis (ESI, Fig. S1†). So, from combined XAS and EDX/TEM elemental analysis we get the final product molar ratio as Pt Ir2.29 Ni1.29.
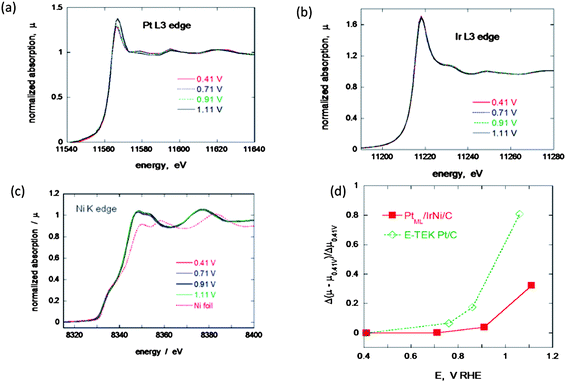 | ||
| Fig. 2 In situ XANES for the Pt monolayer IrNi electrocatalyst at various potentials, (a) Pt L3 edge, (b) Ir L3 edge and (c) Ni K edge. (d) A comparison of the change of the adsorption peak as a function of potential for PtML/IrNi/C and Pt/C. | ||
3.3 ORR activity of Pt monolayer on IrNi nanoparticle electrocatalyst
The cyclic voltammetry curves for the thin-film electrodes of the PtML/IrNi/C and the IrNi/C nanoparticles in 0.1 M HClO4 solution show no anodic currents that can be ascribed to the oxidation/dissolution of Ni, demonstrating that Ni is protected by the Ir shell (ESI, Fig. S2†). Furthermore, the apparent peaks of upd of Cu imply that the surface mainly consists of Ir, since upd does not take place on a Ni surface. The Pt loading is 8.65 nmol (8.61 μg cm−2), calculated from the Cu upd. Fig. 3(a) compares the currents obtained during the ORR in the anodic sweep cycle on the PtML/IrNi/C, PtML/Ir/C and Pt/C (E-tek 20%) electrocatalyst at 1600 rpm in 0.1 M HClO4 solution. Though the kinetic currents for Pt/C (10 μg cm−2 (10 nmol) Pt loading) are slightly higher than PtML/Ir/C (3.6 μg cm−2Pt), there is not much change in the ORR activity. Moreover, alloying Ni with Ir (PtML/IrNi/C) one can clearly observe the increases in diffusion current and activity for the ORR. This enhancement in activity may be due to electronic modifications induced by strain, ligand and segregation effects from bimetallic IrNi cores. It has always been difficult to separate the role of strain and ligand effects in ORR activity enhancements as both play a cumulative effect on the modification of chemical properties of these surfaces.30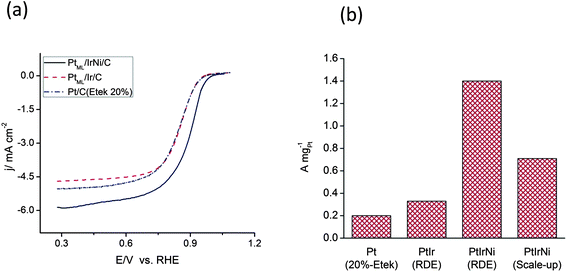 | ||
| Fig. 3 (a) Polarization curves for the ORR of the PtML/IrNi/C and PtML/Ir/C and Pt/C (E-tek 20%) nanoparticle electrocatalysts at 1600 rpm in oxygen-saturated 0.1 M HClO4. (b) A comparison of Pt mass activities for Pt/C (E-tek 20%), PtML/Ir/C and PtML/IrNi/C synthesized using RDE and scale-up synthesis method. | ||
Using the polarization curves of the PtML/IrNi/C electrocatalyst in 0.1 M HClO4 solution purged by O2 at different rotation speeds at a scan rate of 10 mV s−1, the inverse current density (1/j) was plotted as a function of the inverse of the square root of the rotation rate (ω1/2), also known as a Koutecky–Levich plot (ESI, Figs S3 and S4, respectively†). The linearity and parallelism of the Koutecky- Levich plots suggest first-order kinetics with respect to molecular oxygen.29Fig. 3(b) compares the Pt mass activities at 0.9 V for Pt/C (20%E-tek), PtML/Ir/C and PtML/IrNi/C nanoparticles. The Pt mass activities for our scaled-up PtML/IrNi/C electrocatalyst and RDE synthesized PtML/IrNi/C electrocatalyst are, respectively, three to six times that of the Pt/C electrocatalyst. The Pt specific activities of the PtML/IrNi/C, PtML/Ir/C, and Pt/C electrocatalysts at 0.9 V/RHE are 0.60, 0.13, and 0.24 mA cm−2, respectively.
We undertook DFT calculations to elucidate the observed enhancement of activity of PtML/IrNi/C compared to that of Pt/C and PtML/Ir/C electrocatalysts. To simulate the nanoparticles properly, and to save computational time, we constructed sphere-like nanoparticle models of ∼1.7 nm based on a truncated octahedron (Fig. 4). Since our model nanoparticle is smaller than the average size of the experimental ones (∼5.0 nm),18 we could not use the experimental mole ratio of Ni/Ir = 0.56 with the two Ir monolayers for the PtMLIrNi model.18 Thus, we used only the mole ratio of the IrNi subcore (Ni/Ir = 0.37). As Fig. 4(d) shows, while the IrNi core has one Ir submonolayer (60 Ir atoms), it retains the Ir/Ni ratio of ∼0.36 by replacing 14 of the 33 Ir atoms in the subcore with Ni atoms.
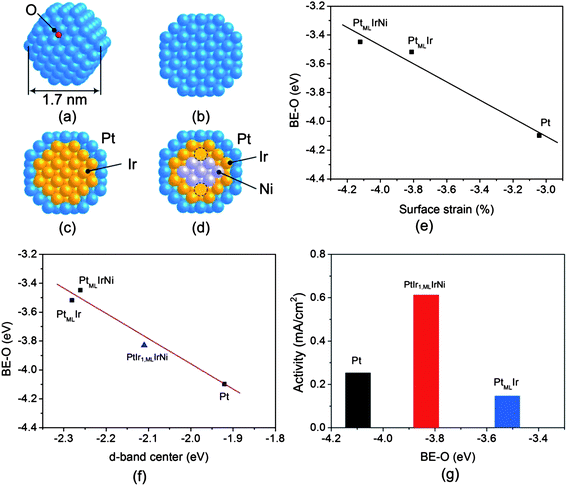 | ||
| Fig. 4 (a) A schematic of a sphere-like nanoparticle considered in DFT calculations representing adsorption of atomic oxygen at the fcc site. Cross sectional views of nanoparticle models of (b) Pt, (c) PtMLIr, and (d) PtMLIrNi with 1.7 nm. The dotted circles represent Ir in sub-core. (e) The DFT calculated binding energy of oxygen (BE–O) as a function of strain on PtMLIrNi, PtMLIr and Pt using the nanoparticle models. (f) The DFT calculated binding energy of oxygen (BE–O) as a function of the average d-band center of metals interacting with O on PtIr1,MLIrNi, PtMLIrNi, PtMLIr, and Pt. (g) The Pt specific activity against BE–O on PtIr1,MLIrNi, PtMLIr and Pt. “ML” is the monolayer. | ||
As Fig. 4 illustrates, we calculated the BE–O that serves as a descriptor for scaling the ORR activity.26Fig. 4(e) shows that partially replacing Ni in the core weakens the BE–O, and introduces more contraction therein (−3.45 eV and −4.12%, respectively) compared to that of PtMLIr (−3.52 eV and −3.81%, correspondingly). Fig. 4(f) plots the BE–O variations against the d-band center, in which the trend from Pt to PtMLIr nanoparticles agrees with that reported for the extended surfaces (Pt(111) and PtMLIr(111)).31 In addition, this figure also shows that using Ir or IrNi as a core shifts the d-band center of Pt in the shell away from the Fermi level, thereby lowering the BE–O.10 Interestingly, the partial replacement of Ir with Ni entails a slightly weaker BE–O than that of PtMLIr (∼0.1 eV) (Fig. 4(f)). Therefore, it is likely that extending the partial replacement of Ir by Ni in PtMLIrNi should augment surface strain, and accordingly, lower the BE–O; expectedly, there should be a maximum for this due to the low stability of the Ni core under the ORR conditions.32,33 To verify the trend of weakening of the BE–O upon mixing Ni with Ir, we calculated the extreme case with a pure Ni core (PtMLNi); this led to the most strained surface and the weakest BE–O (−5.60% and −3.12 eV, respectively). Careful structural examination showed that PtMLNi is slightly collapsed inward, causing more surface contraction than the other nanoparticles do. Based on the calculated strain, d-band center, and BE–O, a descriptor for the ORR activity,26 it is anticipated that PtMLIr and PtMLIrNi may have similar ORR activities, which cannot fully support experimental findings. Thus to gain a better understanding of the experimentally measured ORR activity, we also considered the possibility of anti-segregation under the ORR condition. Possibly, the Ir in the core might be exchanged with the Pt in the shell, driven by a stronger Ir–O interaction than that of Pt–O. To examine anti-segregation effects, we interchanged one Ir atom in the core with its interconnected Pt atom in the shell (in our notation, PtIr1,MLIrNi for PtMLIrNi, and PtIr1,MLIr for PtMLIr. In agreement with a previous study by others on extended surfaces,10 our calculations clearly show that for bare clusters, Ir preferentially remains in the core due to the energy cost of such transitions, i.e., 0.64 eV for PtMLIr → PtIr1,MLIr, and 0.53 eV for PtMLIrNi → PtIr1,MLIrNi. In addition, we noted that it is energetically more costly to pull out Ir from the PtMLIr core and move it to the shell than it is when Ir is in a PtMLIrNi core. Therefore, we considered only the anti-segregation of Ir of the latter, rather than the former. On exposure to oxygen, as shown in Fig. 4(f), O predominantly moves to the Pt–Pt–Ir hybrid hollow site, and the corresponding BE–O is negatively shifted, −3.83 eV. Consequently, the cost of pulling one Ir atom out to the shell declines from 0.53 eV to 0.14 eV. That is, on interacting with oxygen, the anti-segregation of Ir is energetically more likely than that on the bare cluster, so strengthening the O–PtMLIrNi interaction. Fig. 4(g) illustrates the calculated BE–Os versus the measured specific activities for Pt/C, PtMLIr/C, and PtMLIrNi/C. We note that the BE–O for PtMLIrNi corresponds to the strength of O binding after the exchange of one Ir atom in the core with one Pt atom in the shell (PtIr1,MLIrNi). Pt nanoparticles bind O too strongly to be removed from the catalyst, while PtMLIr does so too weakly to dissociate oxygen; hence, it lowers the ORR activity more than Pt does. In contrast, the partial replacement of Ir in the core with Ni favors the anti-segregation of Ir to the shell, and PtMLIrNi supports a moderate binding of oxygen between Pt and PtMLIr, so facilitating the efficient breakage of the O–O bond, and allowing the facile removal of O under fuel cell conditions. Overall, it is evident that the geometric, electronic, and segregation effects under fuel cell conditions have a key role in rationalizing the increase in the ORR activity of PtMLIrNi/C compared to that of PtMLIr/C and Pt/C.
3.4 Stability of Pt monolayer on IrNi nanoparticle electrocatalyst
The long-term stability of ORR electrocatalysts is another critical requirement for applying them in fuel cells. We evaluated the stability of the Pt monolayer electrocatalyst by an accelerated test involving potential cycling between 0.6 V and 1.0 V/RHE at a scan rate of 50 mV s−1 in an air-saturated HClO4 solution. Table 1 summarizes the loss in Pt and noble metal mass activity for the PtML/IrNi/C electrocatalyst prepared on the glassy carbon electrode and in the scale-up synthesis method. Voltammetry was used to determine the Pt surface area of the electrodes by measuring H adsorption before and after potential cycling. Integrating the charge between 0 and 0.33 V associated with H adsorption for PtML/IrNi/C shows a small loss of Pt surface area (ESI, Fig. S5 inset†). The table also shows the percentage loss in the initial surface area and the half wave potential after the accelerated cycling test. A decrease in H adsorption and desorption charges was observed after the potential cycling test compared with those before cycling. The catalyst made on a glassy carbon electrode showed a 45% loss in the surface area and 18 mV loss in half-wave potential after 30000 cycles. The catalyst activity was much more stable for the sample obtained using the scale-up synthesis method. The Pt mass activity of the PtML/IrNi/C electrocatalyst after 50000 cycles is 0.57 A mg−1Pt at 0.9 V/RHE. The polarization curves at 1600 rpm on the PtML/IrNi/C sample before and after 50000 potential cycles in 0.1 M HClO4 show no change in activity (ESI, Fig. S5†). The catalyst made by the scale-up synthesis method showed only a 23% loss in the Pt surface area and no loss in half-wave potential was observed even after 50000 cycles. This demonstrates the advantages of applying the galvanic displacement of a Cu upd layer to the large-scale synthesis of catalysts with well-defined core–shell structures.
| Synthesis on glassy carbon electrode | Scale-up synthesis method | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mass activity (jk/A mg−1) | Half-wave potential (mV) | % loss in surface area | Mass activity (jk/A mg−1) | Half-wave potential (mV) | % loss in surface area | |||||
| Initial (0.9 V) | After 30000 cycles (0.9 V) | Initial | After 30000 cycles |
After 30000 cycles |
Initial (0.9 V) | After 50000 cycles (0.9 V) |
Initial | After 50000 cycles |
After 50000 cycles |
|
| Pt | 1.4 | 0.48 | 890 | 872 | 45 | 0.71 | 0.57 | 851 | 851 | 23 |
| PGM | 0.78 | 0.27 | 0.22 | 0.17 | ||||||
There have always been questions about Ni leaching out upon potential cycling in an acidic environment, so to check the hypothesis we carried out STEM/EELS analysis on numerous individual particles obtained after potential cycling. Fig. 5(b) shows the distribution of Pt and IrNi components in a single representative nanoparticle after 50000 cycles accelerated potential cycling. Fig. 5(a) shows a HAADF image of a single nanoparticle after 50000 potential cycles. The intensity of the spectra for the Pt M-edge, Ir M-edge and Ni L-edge are integrated along the line for a given energy region and the integrated values along the scanned line (as indicated in Fig. 5(a) and shown in Fig. 5(b)). It is clearly shown that the core–shell structure of the IrNi nanoparticle is maintained even after 50000 cycles of potential cycling. The Pt and Ir shell protects Ni from leaching out. The Pt shell on IrNi nanoparticles is also intact which clearly justifies that the IrNi core is a very stable substrate for the Pt monolayer.
 | ||
| Fig. 5 (a) A representative HAADF-STEM image of a PtML/IrNi/C nanoparticle after potential cycling test. (b) A comparison of the corresponding HAADF Pt (black), Ir (red) and Ni (blue) EELS intensity profiles in a line scan indicated. | ||
4. Conclusions
We demonstrated the formation of a new class of core–shell electrocatalysts for the ORR, consisting of a Pt monolayer shell and bimetallic IrNi core. Thus, we coupled highly stable, inexpensive core–shell nanoparticles with a Pt monolayer to produce electrocatalysts with high activity and very high stability. The results show that the activity of Pt monolayer electrocatalysts for the ORR is strongly substrate metal-dependent and our IrNi core–shell structured substrate is a new very suitable core for Pt monolayer electrocatalysts. The electronic structure of substrates is an important factor, because it can alter the catalytic activity of deposited Pt. Our DFT calculations using a sphere-like model clearly demonstrate that mixing Ni with Ir (PtML/IrNi/C) induces geometric, electronic, and segregation effects and thus weakens the binding energy of oxygen, resulting in higher activity than pure Pt/C and PtML/Ir/C electrocatalysts. The galvanic displacement of a Cu upd layer demonstrated through gram-scale synthesis of Pt-shell on IrNi core catalysts seems very promising in scale-up synthesis of highly active, cost-effective nanoparticles with a core–shell structure suitable for specific catalytic reaction requirements. We consider that these data confirm that the Pt monolayer approach holds excellent potential for creating efficient fuel cell electrocatalysts ready for commercialization.Acknowledgements
This work is supported by U.S. Department of Energy, Divisions of Chemical and Material Sciences under the Contract No. DE-AC02-98CH10886. AIF acknowledges support by DOE BES Grant DE-FG02-03ER15476. Beamlines X19A at the NSLS are supported in part by the Synchrotron Catalysis Consortium, U. S. Department of Energy Grant No DE-FG02-05ER15688.We thank the National Energy Research Scientific Computing (NERSC) Center, BNL's Center for Functional Nanomaterials (CFN), and Prof. M. C. Lin for CPU time.References
- W. Vielstich, A. Lamm, H. A. Gasteiger, Handbook of Fuel Cells – Fundamentals, Technology and Applications, John Wiley & Sons, Chichester, 2003 Search PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, B 56, 9 CrossRef.
- R. R. Adzic, in Frontiers in Electrochemistry, Vol. 5, Electrocatalysis, ed. J. Lipkowski and P. N. Ross, VCH Publishers, New York, 1998; p 197 Search PubMed.
- S. Mukerjee, S. Srinivasan, M. P. Soriaga and J. Mcbreen, J. Phys. Chem., 1995, B 99, 4577 CrossRef.
- S. Chen, P. J. Ferrira, W. Sheng, N. Yabuuchi, L. Allard and Y. Shao-Horn, J. Am. Chem. Soc., 2008, 130, 13818 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493 CrossRef CAS.
- R. R. Adzic, J. Zhang, K. Sasaki, M. B. Vukmirovic, M. Shao, J. X. Wang, A. U. Nilekar, M. Mavrikakis and F. Uribe, Top. Catal., 2007, 46, 249 CrossRef CAS.
- M. H. Shao, K. Sasaki, N. S. Marinkovic, L. Zhang and R. R. Adzic, Electrochem. Commun., 2007, 9, 2848 CrossRef CAS.
- J. Zhang, F. H. B. Lima, M. H. Shao, K. Sasaki, J. X. Wang, J. Hanson and R. R. Adzic, J. Phys. Chem. B, 2005, 109(48), 22701–22704 CrossRef CAS.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71 CrossRef CAS.
- J. A. Rodriguez, Surf. Sci. Rep., 1996, 24, 225 CrossRef.
- W. Zhou, K. Sasaki, D. Su, Y. Zhu, J. Wang and R. R. Adzic, J. Phys. Chem. C, 2010, 114, 8950–8957 CAS.
- K. Lee, L. Zhang and J. Zhang, J. Power Sources, 2007, 170, 291 CrossRef CAS.
- J. Qiao, R. Lin, B. Li, J. Ma and J. Liu, Electrochim. Acta, 2010, 55, 8490–8497 CrossRef CAS.
- F. H. B. Lima, J. L. Zhang, M. H. Shao, K. Sasaki, M. B. Vukmirovic, E. A. Ticianelli and R. R. Adzic, J. Phys. Chem. C, 2007, 111, 404–410 CAS.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS.
- M. Mavrikakis, B. Hammer and J. K. Norskov, Phys. Rev. Lett., 1998, 81, 2819 CrossRef.
- K. Sasaki, K. Kuttiyiel, L. Barrio, D. Su, A. I. Frenkel, N. Marinkovic, D. Mahajan and R. Adzic, J. Phys. Chem. C, 2011, 115, 9894–9902 CAS.
- K. Sasaki, J. X. Wang, H. Naohara, N. Marinkovic, K. More, H. Inada and R. R. Adzic, Electrochim. Acta, 2010, 55, 2645–2652 CrossRef CAS.
- J. McBreen, W. E. O'Grady, K. I. Pandya, R. W. Hoffman and D. E. Sayers, EXAFS Study of the Nickel-Oxide Electrode, Langmuir, 1987, 3, 428–433 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953–17979 CrossRef.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter, 1999, 59, 7413–7421 CrossRef.
- J. X. Wang, H. Inada, L. Wu, Y. Zhu, Y. Choi, P. Liu, W.-P. Zhou and R. R. Adzic, J. Am. Chem. Soc., 2009, 131, 17298–17302 CrossRef CAS.
- K. Sasaki, H. Naohara, Y. Cai, Y. Choi, P. Liu, M. B. Vukmirovic, J. Wang and R. R. Adzic, Angew. Chem., Int. Ed., 2010, 49, 8602–8607 CrossRef CAS.
- Ma Yuguang and Perla B. Balbuena, J. Electrochem. Soc., 2010, 157, B959–B963 CrossRef.
- N. A. Anastasijevic, V. Vesovic and R. R. Adzic, J. Electroanal. Chem., 1987, 229, 317 CrossRef CAS.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 15 CrossRef.
- J. L. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2132 CrossRef CAS.
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, J. Power Sources, 2006, 160, 957 CrossRef CAS.
- M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura and P. Stonehart, J. Electrochem. Soc., 1994, 141, 2659 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ee02067f |
| This journal is © The Royal Society of Chemistry 2012 |