Vitalizing fuel cells with vitamins: pyrolyzed vitamin B12 as a non-precious catalyst for enhanced oxygen reduction reaction of polymer electrolyte fuel cells†
Sun-Tang
Chang
ab,
Chen-Hao
Wang
*b,
He-Yun
Du
ad,
Hsin-Cheng
Hsu
b,
Chih-Ming
Kang
e,
Chia-Chun
Chen
e,
Jeffrey C. S.
Wu
d,
Shi-Chern
Yen
d,
Wen-Fei
Huang
f,
Li-Chyong
Chen
*c,
M. C.
Lin
f and
Kuei-Hsien
Chen
*ac
aInstitute of Atomic and Molecular Sciences, Academia Sinica, Taipei, 10607, Taiwan. E-mail: chenkh@pub.iams.sinica.edu.tw
bDepartment of Materials Science and Engineering, National Taiwan University of Science and Technology, Taipei, 10617, Taiwan. E-mail: chwang@mail.ntust.edu.tw
cCenter for Condensed Matter Sciences, National Taiwan University, Taipei, 10607, Taiwan. E-mail: chenlc@ntu.edu.tw
dDepartment of Chemical Engineering, National Taiwan University, Taipei, 10607, Taiwan
eDepartment of Chemistry, National Taiwan Normal University, Taipei, 11677, Taiwan.
fDepartment of Applied Chemistry, National Chao Tung University, Hsinchu, 30010, Taiwan.
First published on 17th November 2011
Abstract
The limited natural abundance and high cost of Pt has been a major barrier in its applications for hydrogen or methanol fuel cells. In this work, based on the pyrolyzed corrin structure of vitamin B12 (py-B12/C), it is reported to produce superior catalytic activity in the oxygen reduction reaction (ORR) with an electron transfer number of 3.90, which is very close to the ideal case of 4. The H2–O2fuel cell using py-B12/C provides a maximum power density of 370 mW cm−2 and a current density of 0.720 A cm−2 at 0.5 V at 70 °C. Calculations based on density functional theory suggests that the corrin complex with a low-symmetric structure offers a much preferable path for the ORR, which is not applicable to the porphyrin with a high-symmetric structure. The long-term stability and high ORR activity of py-B12/C make it a viable candidate as a Pt-substitute in the ORR.
Broader contextA polymer electrolyte fuel cell (PEFC) is an electrochemical device that converts chemical energy to electrical energy by the redox reaction of hydrogen and air (oxygen), which are fed into the anode and the cathode, respectively. The oxygen reduction reaction (ORR) in the cathode is much slower than the hydrogen oxidation reaction in the anode, and therefore, to accelerate the ORR, a high loading of Pt/C is used in the cathode, making the PEFC expensive. Non-precious metal catalysts of the oxygen reduction reaction are extremely important for a PEFC owing to their relatively low cost. This study investigates non-precious metal catalysts with low cost and high ORR activity using pyrolyzed vitamin B12 supported by carbon black (py-B12/C). Py-B12/C has a promising performance and high stability for use in the cathode of PEFC. From density functional theory simulation, Py-B12/C with a low symmetry of the Co–corrin centre has a preferable ORR pathway, providing four-electron transfer (direct pathway). |
Introduction
The oxygen reduction reaction (ORR) is one of the essential reactions pertinent to the majority of living beings, which is slow in nature and requires enzymes and/or catalysts assistance. For example, the rate limiting reaction in a polymer electrolyte fuel cell (PEFC) is the ORR at the cathode.1 A large amount of Pt catalysts of nanometres in diameter have been utilized to overcome the slow reaction of the ORR, adding to the cost and making worse the agglomeration problem of the catalyst during operation. Searching for efficient, durable and most importantly, inexpensive non-precious metal catalysts has become the Holy Grail in catalysis owing to its importance in hydrogen energy applications.Since Jasinski investigated cobalt phthalocyanine as an ORR catalyst for fuel cells in 1964,2 various studies using transition metal macrocyclic compounds, such as porphyrin,3–7phthalocyanine,8,9 and tetraazannulene,10 have been reported.
Among them, iron- and cobalt-based macrocyclic compounds have been demonstrated to exhibit the highest catalytic activity for the ORR.11–13 Okada et al. concluded that the cobalt-based macrocyclic compounds as cathode catalysts were affected by the number of ligands surrounding the central cobalt, the closed- or opened-ring structure, the bonding strength between the central cobalt and ligand, the bonding strength between the carbon black and the macrocyclic compound, and the facial orientation of the macrocyclic compounds.14 Bashyam et al. used a cobalt–polypyrrole composite for the cathode in an H2–O2 PEFC and demonstrated a maximum power density of 150 mW cm−2 at 80 °C, with no significant degradation for over 100 h.15 Lefevre et al. reported microporous carbon supported iron-based catalysts with active sites coordinated by pyridinic nitrogen functionalities that exhibited performances comparable to that of precious metals.16 Wang et al. presented alternative non-metal ORR catalysts using ordered mesoporous carbons by NH3 heat treatment, showing high activity and good stability.17 Chung et al. used cyanamide as the nitrogen source for a metal–nitrogen–carbon type catalyst at a pyrolysis temperature of 1050 °C, showing good performance.18 Recently, Wu et al. utilized polyaniline as a precursor to a carbon–nitrogen template in the high-temperature synthesis of catalysts that incorporated iron and cobalt that exhibited high activity and remarkable stability.19 Wu et al. used multi-step pyrolysis of iron phthalocyanine and phenolic resin to prepare highly active and highly stable ORR catalysts.20 Other materials, such as TiOx, TiNx and RuSex, were employed in the ORR to improve the catalyst stability.21–23 However, all these alternatives are still inadequate for the ORR in terms of their activity and stability.11,24 Recently, some papers reviewed the classification, mechanism, activity and performance of Pt-free catalysts.25–28 Jaouen et al. reviewed the metal/nitrogen/carbon (Me/N/C) catalysts obtained through the pyrolysis of iron and/or cobalt precursors, nitrogen- and carbon-precursors for the ORR.28 Additionally, they reported a highly active Me/N/C catalyst using the new synthesis methods, which used a metal salt, phenanthroline or NH3 gas or cyanamide, and a microporous carbon black as the metal-, nitrogen-, and carbon-precursors in the pyrolysis. Chen et al. reviewed several kinds of non-precious metal catalysts over the past 40 years, and they considered that the most promising catalysts were pyrolyzed transition metal nitrogen-containing complexes supported on carbon materials (pyrolyzed M–Nx/C).26 From their review, the activity and stability of the pyrolyzed M–Nx/C are affected by several factors, including transition metal type and loading, carbon support surface properties and nitrogen content, and heat treatment conditions and duration.
Zagal et al. investigated the electrochemical behaviours of cyanocobalamin (vitamin B12) adsorbed on a graphite electrode, in which the cyanocobalamin catalyzed the ORR via both the two-electron and the four-electron transfer routes, with the four-electron transfer occurring at a low pH value and at a more negative potential.29–31 Therefore, this finding manifests that cyanocobalamin may be suitable for cathode catalysts in PEFC. We extended this idea and investigate the use of pyrolyzed cyanocobalamin (vitamin B12) supported by carbon black (py-B12/C) for the ORR in PEFC.
Experimental section
Preparation of pyrolyzed B12/C (py-B12/C)
A 0.1g mass of cyanocobalamin (99%, Aldrich), one kind of vitamin B12 with a molecular structure as depicted in Fig. 1a, was dissolved in 10 mL of de-ionized water with constant stirring for 30 min at room temperature. Then, 0.4 g of carbon black (Vulcan XC-72R) was added to the cyanocobalamin solution, again with constant stirring for 30 min at room temperature. The solution was heated using steam to 80 °C for 60 min to eliminate water. After this step, the suspension was totally dry. Subsequently, it was dried at room temperature under vacuum for 12 h to obtain the slurry. For the pyrolysis, the slurry was loaded into a fused aluminium oxide boat, which was introduced into a quartz tube furnace. The temperature of the furnace was raised to 700 °C at a rate of 20 °C per minute, and then the slurry was pyrolyzed in a nitrogen atmosphere at 700 °C for 2 h. Following the pyrolysis, the furnace was cooled to room temperature by natural convection. The mass of the as-prepared py-B12/C was 0.455 g, representing a total weight loss of around 9.0% throughout the process.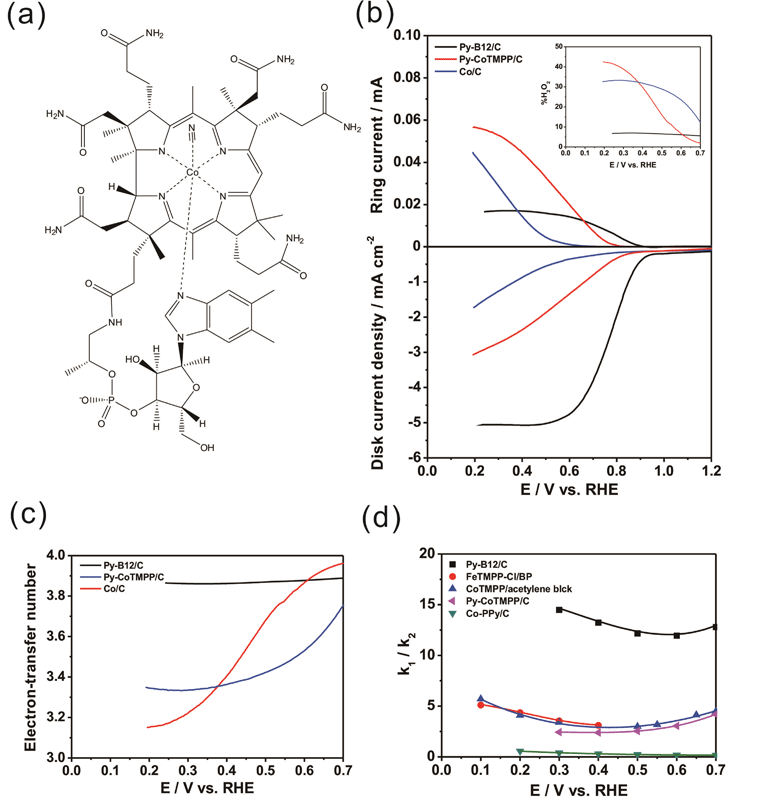 | ||
| Fig. 1 (a) The molecular structure of B12; (b) ORR curves for py-B12/C, py-CoTMPP/C and Co/C obtained by the RRDE method in oxygen-saturated 0.1 M HClO4 solution at rotation rates of 1600 rpm and scan rates of 10 mV s−1; (c) the electron-transfer number of the catalysts dependence on disk potentials (d) the k1/k2 ratios of py-B12/C, FeTMPP-Cl/BP,33CoTMPP/acetylene black,34 and Co-PPy/C35 as a function of applied potential. | ||
To obtain X-ray diffraction, Raman spectroscopy, Fourier transform infrared (FTIR) spectroscopy and X-ray absorption spectroscopy (XAS) of py-B12 without the background associated with carbon black, the cyanocobalamin solution without additional carbon black was loaded into a silica boat for the pyrolysis, yielding py-B12.
Preparation of pyrolyzed CoTMPP/C and Co/C
Pyrolyzed CoTMPP/C (py-CoTMPP/C) and Co/C were prepared to compare their electrochemical activity and PEFC performance with those of py-B12/C. Py-CoTMPP/C was prepared as follows: 0.1 g of cobalt(II) tetramethoxyphenylporphyrin (CoTMPP, 99%, Aldrich) and 0.4 g of carbon black (Vulcan XC-72R) were dispersed in N,N-dimethylmethanamide (DMF, 99.7%, Alfa Aesar) solution. The mixed solution was ultrasonically stirred for 30 min to yield a homogeneous solution, which was filtered to remove the solvent, leaving a mixed catalyst precipitate on the filter paper. The catalyst mixture was then dried using a vacuum heater at 60 °C for 24 h. After the residual solvent had been removed, a fused aluminium oxide boat that contained the catalyst mixture was introduced into a quartz tube, which was placed in a tubular furnace. The catalyst mixture was pyrolyzed in a nitrogen atmosphere at 600 °C for 2 h. Following the pyrolysis, the furnace was cooled to room temperature by natural convection. The mass of the as-prepared pyrolyzed py-CoTMPP/C was 0.425 g, representing a total weight loss of around 15.0% throughout the process.The Co/C was prepared as follows: 0.22 g of dried CoCl2 and 0.4 g of carbon black (Vulcan XC-72R) were dispersed in de-ionized water. The mixed solution was ultrasonically stirred for 30 min to yield a homogeneous solution. The solution was steam heated to 80 °C for 60 min to eliminate water followed by drying in a vacuum heater at 60 °C for 24 h. After the residual solvent had been removed, a fused aluminium oxide boat that contained the catalyst mixture was introduced into a quartz tube, which was placed in a tubular furnace. The catalyst mixture was reduced in a 5% hydrogen atmosphere at 200 °C for 2 h. Following the reduced reaction, the furnace was cooled to room temperature by natural convection.
Characterization
High-energy X-ray diffraction (XRD) spectra were obtained using the beam line 01C1 (at the National Synchrotron Radiation Research Center, Hsinchu, Taiwan) with Mo Kα1 radiation (λ = 0.70930 Å) and an energy of 25 KeV in a limited range of angles at room temperature. The 2θ scans were performed at a scan rate of 10° per minute, with steps of 0.05° from 5° to 45°. All XRD spectra throughout this work were referenced to Cu Kα1 radiation (λ = 1.54056 Å). Raman spectroscopy (with excitation using a He–Ne laser of 632.8 nm wavelength, Jobin-Yvon LabRAM HR800-Confocal micro-Raman spectroscope) and FTIR spectroscopy (Thermo Nicolet Nexus 6700) were carried out to elucidate the changes in catalysts caused by the pyrolysis.X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) at the Co K-edge were recorded using the beam line 17C1 (at the National Synchrotron Radiation Research Center, Hsinchu, Taiwan), which is based on a multi-pole wiggler source with a critical energy of 2.7 KeV. The electron storage ring is operated at an energy of 1.5 GeV with a beam current of 120–200 mA. The beamline employs a double Si(1 1 1)-crystal monochromator for energy selection with a resolution (ΔE/E) better than 2 × 10−4 in the energy range of 5–15 keV. All spectra were recorded at room temperature in a transmission mode in which the intensities of incident and transmitted X-ray beams were measured by gas-filled ionization chambers. The catalyst powder was pressed into a slot of the stainless-steel holder and then placed in an in situcell for treatment under desired conditions. A standard compound, Co foil, was measured simultaneously by using the third ionization chamber so that energy calibration could be performed scan by scan in real time. The XANES and EXAFS data, including background subtraction, normalization with respect to the edge jump, Fourier transformation, and curve fitting, were processed by the computer programs which were implemented in the software package of IFEFFIT. In addition, the phase shifts and backscattering amplitudes for specific atom pairs were theoretically calculated by using the FEFF7 code.32 It is difficult to carry out precise simulations for fitting the EXAFS data for the macrocyclic compound due to its mixed phase structure. Therefore, for pristine B12 and py-B12, only the cobalt-containing corrin ring is fitted in this study.
Electrochemical measurement
Electrochemical measurements were made in a three-electrode cell, which were conducted by a potentiostat/galvanostat instrument (Biologic Bi-stat). The working electrode was a rotating-ring disk electrode (RRDE, PINE AFE7R9GCPT) with a glassy carbon (GC) disk (diameter = 5.61 mm) and a ring made of platinum (outer diameter = 7.92 mm and inner diameter = 6.25 mm). The counter electrode and reference electrode were a Pt foil and a saturated calomel electrode (0.242 V vs.NHE), respectively. All potentials in this work are referenced to the reversible hydrogen electrode (RHE). The electrolyte in the ORR test was oxygen-saturated 0.1 M HClO4 solution.Catalyst ink was made by mixing 160 mg of catalyst with 20 mL deionized water. 40 μL of the ink and 5 μL of 0.1 wt% Nafion® solution were dropped onto the GC disk, which was then left to dry in air at room temperature. Before the catalyst ink was deposited onto the GC disk, the GC disk had been polished to a mirror-like finish using 1.0 and then 0.05 μm alumina slurry. Linear scan voltammetry (LSV) was applied to the catalysts at the specified scan rates. The ORR curves at the GC were obtained at a low scan rate of 10 mV s−1 to reduce the substantial non-Faradic current that would otherwise have been produced by the catalysts. To determine the yield of hydrogen peroxide in the ORR that was catalyzed by the catalysts on the GC disk, 1.35 V was applied to the ring to produce a current that oxidized the hydrogen peroxide.
Accelerated durability test
The accelerated durability test (ADT) of py-B12/C follows the method demonstrated by Colón-Mercado et al.25 The ADT cell consists of a three-electrode system, including a reference electrode (Hg/HgSO4, 0.65 V vs.NHE), a counter electrode of Pt foil and a working electrode of py-B12/C coated on carbon cloth (catalyst loading: 6 mg cm−2). During the ADT, the electrode was immersed in a 0.3 M H2SO4 solution at a fixed potential of 0.68 V (vs. RHE), and the solution was extracted periodically for examination. The concentration of dissolved Co was measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES, PerkinElmer ICP-OES Optima 3000).Fuel cell test
A membrane-electrode-assembly (MEA) with an area of 5 cm2 was prepared by hot-pressing two electrodes on both sides of a Nafion® 212 (H+, DuPont) membrane at 135 °C and 130 kg cm−2 for 2 min. For the preparation of specific cathodes, as-prepared catalysts (i.e.py-B12/C and py-CoTMPP/C) were dispersed in a 5 wt% Nafion® solution as a cathode catalyst ink, with a catalysts![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nafion® solution weight ratio of 1:10. Thus, the weight ratio of catalysts to dry Nafion® is 2:1. The cathode catalyst ink was hand-painted on carbon cloth, with a catalyst loading of 6 mg cm−2, and then the cathode was dried at room temperature in a vacuum for 6 h. The commercial electrode (E-TEK), Pt/C with a metal loading of 0.25 mg cm−2, was employed as the anode of the MEA. A polarization experiment on PEFC was performed at 70 °C, using hydrogen and oxygen through the anode and the cathode, respectively, at flow rates of 0.1 and 0.15 slpm. Hydrogen and oxygen were passed through humidifiers at 70 °C before they entered an MEA. The back pressure gauges of the anode and the cathode sides were set to 1 atm. The PEFC performance was measured using a fuel cell test station (Asia Pacific Fuel Cell Technologies, Ltd.) by recording the cell voltage and current after they had reached steady values. The cell resistance is the sum of the proton-conduction resistances and the various electronic resistances. The measurements of electrochemical impedance spectroscopy (EIS) at 20 kHz determined the cell resistances as a function of current densities, which yielded the typical cell resistance of about 0.25 Ω cm2. The 100-hour durability tests of PEFC using py-B12/C as the cathode catalyst was performed at H2-air feeding to the anode and the cathode, respectively, at flow rates of 0.1 and 0.75 slpm. The cell voltage was held at 0.4 V for 100 h and current recorded by the fuel cell test station. The back pressure gauges of the anode and the cathode sides were set to 1 atm.
:Nafion® solution weight ratio of 1:10. Thus, the weight ratio of catalysts to dry Nafion® is 2:1. The cathode catalyst ink was hand-painted on carbon cloth, with a catalyst loading of 6 mg cm−2, and then the cathode was dried at room temperature in a vacuum for 6 h. The commercial electrode (E-TEK), Pt/C with a metal loading of 0.25 mg cm−2, was employed as the anode of the MEA. A polarization experiment on PEFC was performed at 70 °C, using hydrogen and oxygen through the anode and the cathode, respectively, at flow rates of 0.1 and 0.15 slpm. Hydrogen and oxygen were passed through humidifiers at 70 °C before they entered an MEA. The back pressure gauges of the anode and the cathode sides were set to 1 atm. The PEFC performance was measured using a fuel cell test station (Asia Pacific Fuel Cell Technologies, Ltd.) by recording the cell voltage and current after they had reached steady values. The cell resistance is the sum of the proton-conduction resistances and the various electronic resistances. The measurements of electrochemical impedance spectroscopy (EIS) at 20 kHz determined the cell resistances as a function of current densities, which yielded the typical cell resistance of about 0.25 Ω cm2. The 100-hour durability tests of PEFC using py-B12/C as the cathode catalyst was performed at H2-air feeding to the anode and the cathode, respectively, at flow rates of 0.1 and 0.75 slpm. The cell voltage was held at 0.4 V for 100 h and current recorded by the fuel cell test station. The back pressure gauges of the anode and the cathode sides were set to 1 atm.
Results and discussion
Instead of directly absorbing the material on a graphite support as reported by Zagal et al.,30 the vitamin B12 in this study was loaded on a carbon support, here after denoted as py-B12/C for brevity, and pyrolyzed at 500–900 °C in nitrogen ambient prior to electrochemical measurements. Fig. S1† demonstrates the ORR curves of py-B12/C with pyrolysis temperatures of 300, 500, 700 and 900 °C, which shows the optimal ORR activity at a pyrolysis temperature of 700 °C. Thus, py-B12/C pyrolyzed at 700 °C is employed in this work. Fig. S2† presents the cyclic voltammetry (CV) curves of py-B12/C, py-B12 and pyrolyzed carbon blacks in N2-purged 0.1 M HClO4 solution with scan rates of 50 mV s−1. The slight redox peak at 0.5 V could be attributed to the poly-aromatic carbon-based materials. Zagal et al. used ordinary pyrolytic graphite electrodes with pre-adsorbed B12, which yielded the redox peak of Co(II)/Co(III) at 0.55 V with respect to normal hydrogen electrode in a medium purged with N2 at pH = 1.30 Since only ∼1% Co exists in the py-B12/C, the redox peak of Co(II)/Co(III) may be overwhelmed by the carbon-like capacitance.The ORR pathway is mainly composed of the following reactions.
| Reaction 1: O2 + 4H+ + 4e− → 2H2O E0 = 1.229 V. |
| Reaction 2: O2 + 2H+ + 2e− → H2O2E0 = 0.695 V. |
| Reaction 3: H2O2 + 2H+ + 2e− → 2H2O E0 = 1.763 V. |
Reaction 1 is a direct ORR pathway that involves transfer of four electrons, while Reaction 2 follows an H2O2 pathway with two-electron transfer. Since a direct ORR pathway yields a higher thermodynamically reversible potential than an H2O2 pathway, Reaction 1 is preferable over Reaction 2 for the ORR in PEFC. Fig. 1b shows the ORR behaviors of py-B12/C, py-CoTMPP/C and Co/C, determined using the rotating-ring disc electrode (RRDE) method. The lower and upper parts of Fig. 1b display the disk current (Id) and the ring current (Ir), respectively, as a function of applied potential. The typical ORR curve obtained in acid media reveals three dominant potential regions – viz. the kinetic range (>0.8 V), the mixed range (0.8–0.6 V), and the mass-transfer range (<0.6 V). The Id curve for py-B12/C is clearly above those of py-CoTMPP/C and Co/C in all potential regions, indicating that py-B12/C has a much higher ORR activity. The nominal Co wt% in py-B12/C, py-CoTMPP/C and Co/C are 0.95, 1.74 and 20%, respectively. The fact that py-B12/C has the lowest Co loading, and yet exhibits the highest ORR activity, attests to its superb catalytic activity. Fig. 1c and the inset in Fig. 1b plot the electron-transfer number (n value) and hydrogen peroxide yield (%H2O2) during the ORR, respectively, as a function of the potential of the glassy carbon (GC) disk. (See ESI† for the calculations of the n value and %H2O2.) At a large overpotential of 0.3 V, the n values for py-CoTMPP/C and Co/C are reduced to 3.22 and 3.15, respectively. For py-B12/C, on the other hand, the n values and %H2O2 remain at around 3.90 and 5.0%, respectively, over a wide range of overpotentials up to 0.7 V, which may suggest that the ORR of py-B12/C proceeds preferentially along the four-electron direct ORR pathway, as implied by the plot of k1/k2 ratio in Fig. 1d, where k1 and k2 are the forward rate constants of Reactions 1 and 2, respectively. (See ESI† for the calculation of k1/k2 ratio.) The k1/k2 ratio for py-B12/C ranges between 12 to 14.5, which is the highest among the reported values, such as those of Pearls carbon-supported FeTMPP-Cl (FeTMPP-Cl/BP),33acetylene black-supported CoTMPP (CoTMPP/acetylene black),34 and carbon-supported cobalt polypyrrole (Co-PPy/C).35 Alternatively, a sequential two-electron reduction or a two-electron reduction with subsequent H2O2 decomposition cannot be excluded by the RRDE technique, especially at high catalyst loading. New technique development is underway to further differentiate these processes.
In order to investigate the structure of the product after the pyrolysis, X-ray diffraction (XRD) patterns of py-B12, pristine B12 and the reference patterns of vitamin B12 (JCPDS, PDF# 050029), β-Co (JCPDS, PDF# 150806) and α-Co (JCPDS, PDF# 050727) were depicted in Fig. S3.† Pristine B12 exhibits the same characteristic peaks as those in the vitamin B12 reference pattern. However, py-B12 does not display any diffraction peak, suggesting that py-B12 is likely transformed into an amorphous structure and very little metallic cobalt is formed during the pyrolysis under the sensitivity of XRD. This finding drastically differs from that of transition metal macrocyclic compounds (M–N4, M: Co and Fe) for the ORR, in which the M–N4 moiety was partially or completely decomposed during the pyrolysis, yielding metallic particles (Co and Fe).13,36–38 These Co- and Fe-metallic particles can be easily corroded in an acidic medium under a high potential. In contrast, py-B12/C with less metallic Co formation is significantly more durable as a catalyst for the ORR.
Fig. 2 shows the ADT of py-B12/C for 100 h. The current initially increases. Colón-Mercado et al. suggested that the initial increase was caused by the initial wetting and saturation of the thin Nafion layer covering the catalyst particles.25 After 15 h, the current reaches steady-state. After 100 h, the concentration of dissolved cobalt is 0.03 ppm, which means only 1.30% of Co loss.
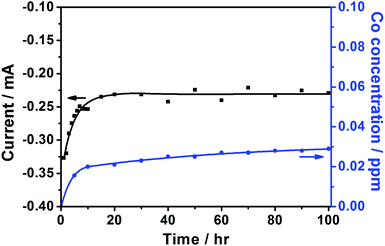 | ||
| Fig. 2 The ADT test of py-B12/C at 0.68 V (vs. RHE) for 100 h. | ||
To verify the performance of py-B12/C as the cathode catalyst in a fuel cell, a PEFC was constructed, comprising of the abovementioned cathode and a commercial Pt/C anode (E-TEK, 30 wt% 0.25 mg cm−2). This cell was operated at 70 °C, at H2 and O2 back pressure of 1 atm, and Nafion 212 as the electrolyte. The polarization curve of py-B12/C, presented in Fig. 3, shows an open-cell-potential of 0.843 V and the highest power density of 370 mW cm−2, and yields 0.720 A cm−2 at 0.5 V. For comparison, another PEFC comprised of the py-CoTMPP/C cathode catalyst was operated under the same conditions, and the results are also plotted in Fig. 3.
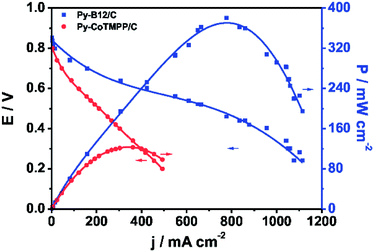 | ||
| Fig. 3 Polarization curves of the H2–O2 PEFCs using py-B12/C and py-CoTMPP/C as cathodes. Operation temperature: 70 °C; back pressure of H2 and O2: 1 atm; anode catalysts: 30 wt% Pt/C with the metal loading of 0.25 mg cm−2 (E-TEK); electrolyte: Nafion® 212 (H+, DuPont). | ||
It is evident from Fig. 3 that the performance of the py-B12/C cell is far better than that of the py-CoTMPP/C cell. The Py-B12 cell is significantly higher than the reported values using Co-based catalysts in the literature. For example, Olson et al. used 4 mg cm−2CoTMPP/C with a 1:1 mass ratio of Nafion to catalyst loaded in the cathode, which generated about 0.17 A cm−2 at 0.5 V.39 Pylypenko et al. studied CoTPP supported by amorphous fumed silica with the catalyst loading of 4.0 mg cm−2 as the cathode catalyst for the H2–O2 PEFCs at 80 °C, which generated about 0.28 A cm−2 at 0.5 V.37 Ma et al. investigated the influences of the pre-treatments of BP2000 carbon supports by 6 M HNO3 and 30 wt% H2O2 on the catalytic activity.40 They used CoTMPP/BP (without pre-treatment), CoTMPP/BP(6 M HNO3) and CoTMPP/BP(30 wt% H2O2) with the catalyst loading of 10.0–12.0 mg cm−2 as the cathode catalysts for H2–O2 PEFC at 50 °C, which generated about 0.21, 0.19 and 0.15 A cm−2, respectively. Bashyam et al. reported a Co–polypyrrole composite for H2–O2 PEFC at 80 °C, which generated about 0.28 A cm−2 at 0.5 V.15
Compared to Fe-based and Fe–Co-based catalysts, the py-B12 cell is higher in output current density than most of the reported values in the literature. Wu et al. used polyaniline–Fe–C with the catalyst loading of 2.0 mg cm−2 as the cathode catalyst for H2–O2 PEFC at 80 °C, which generated about 0.40 A cm−2 at 0.5 V.19 Chung et al. used a cyanamide-derived Fe-based catalyst with the catalyst loading of 4.0 mg cm−2 as the cathode catalyst for H2–O2 PEFC at 80 °C, which generated about 0.60 A cm−2 at 0.5 V.18 Choi et al. investigated a Fe/Co–Nx composite on nano-porous carbon black with ethylenediamine as a nitrogen precursor with the catalyst loading of 4.0 mg cm−2 as the cathode catalyst for H2-O2 PEFC at 80 °C, which generated about 0.68 A cm−2 at 0.5 V.41 Wu et al. used a multi-step pyrolysis of iron phthalocyanine and phenolic resin to prepare Fe-based catalysts as the cathode catalysts for H2–O2 PEFC at 80 °C, which gave the best catalytic performance at about 0.95 A cm−2 at 0.5 V.20 Liu et al. reported Fe supported by N-doped ordered porous carbon as the cathode catalyst for H2–O2 PEFC at 75 °C, which generated about 0.60 A cm−2 at 0.5 V.42 To compare with the performance of Fe-based catalysts reported by Lefevre et al.,16 the performance of the py-B12/C cell is corrected by the iR compensation, which yields 1.10 A cm−2 at 0.5 ViR-free. (See ESI† for the calculation of iR compensation.) It is significantly higher than the 0.75 A cm−2 at 0.5 ViR-free obtained by Lefevre et al. Nallathambi et al. studied Co–Fe–N chelate complexes of FeNx, CoNx, Co1Fe3Nx, Co3Fe1Nx and Co1Fe1Nx with the catalyst loading of 6.0 mg cm−2 as the cathode catalyst for H2–O2 PEFC at 80 °C, which generated about 0.65, 0.50, 0.30, 0.26 and 0.25 A cm−2 at 0.5 V, respectively.43
Among the non-precious metal catalysts, py-B12/C outperforms even the Pd-based catalysts such as Pd–Co–Au alloy (0.2 A cm−2 at 0.5 V, 60 °C, O2 back pressure of 1.36 atm),44 Pd–Co–Mo (0.2 A cm−2 at 0.5 V, 60 °C, O2 back pressure 1.36 atm) and Pd–Ti (0.1 A cm−2 at 0.5 V, 60 °C, O2 back pressure of 1.36 atm).45
In order to study the evolution of electronic and bonding structures of py-B12/C catalyst in pristine, pyrolyzed, and related samples, Raman and FTIR measurements were applied. The Raman spectra of pristine B12 and py-B12, presented in Fig. 4, show an absence of the Co–CN stretching (481 cm−1) mode in py-B12, indicating that cleavage of the –CN species changes the Co oxidation state. In Fig. 4, the two strong peaks at 1330 and 1580 cm−1 are attributed to D- and G-peaks, respectively, of the carbon-like materials, suggesting that py-B12 forms a network structure of poly-aromatic hydrocarbons.46 Similar comparison of the FTIR spectra in Fig. 5 shows four characteristic peaks (A to D) for pristine B12, which are attributed to the cyanide stretching frequency at 2130 cm−1 (band A), acetamide and propionamide side chains between 1620 and 1690 cm−1 (band B), and breathing modes of the corrin ring at 1575 and 1545 cm−1 (bands C and D).47 After the pyrolysis, peaks A and B disappear while peaks C and D remain. Meanwhile, new peaks can be seen to appear between 1620 and 1650 cm−1, corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C conjugated with CC, conjugated CH2CH, and vinyl group –CHCH2.48 The disappearance of peak A is associated with the cleavage of –CN during pyrolysis, which is also confirmed in the Raman study.
C conjugated with CC, conjugated CH2CH, and vinyl group –CHCH2.48 The disappearance of peak A is associated with the cleavage of –CN during pyrolysis, which is also confirmed in the Raman study.
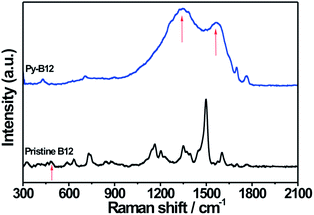 | ||
| Fig. 4 Raman spectra of py-B12 and pristine B12. | ||
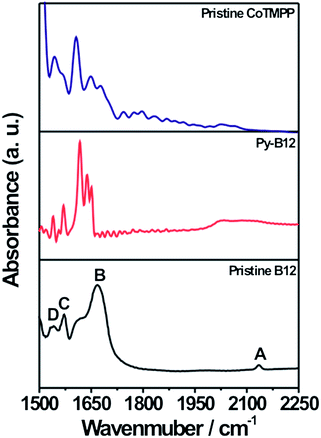 | ||
| Fig. 5 FTIR spectra of pristine CoTMPP, py-B12 and pristine B12. | ||
For a transition metal macrocyclic compound, the ORR activity is dominated by the oxidation state of the central transition metal, the redox potential of M(II)/M(III) and the effect of the ligand.49,50 The oxidation state of the central cobalt in pristine vitamin B12 has been reported to be Co(III).51,52 X-ray absorption near edge spectroscopy (XANES) is employed to compare Co oxidation states in CoTMPP, pristine B12, py-B12/C and CoO powder, the results of which are shown in Fig. 6a. After detailed analysis and fitting of the spectra, the energy value, E0, was derived as shown in Table S1† and it was concluded that the oxidation states for pristine CoTMPP, pristine B12, py-B12/C and CoO powder are 2+, 3+, 2+ and 2+, respectively. This clearly shows that the pyrolysis changes the oxidation state of vitamin B12 from Co(III) to Co(II).
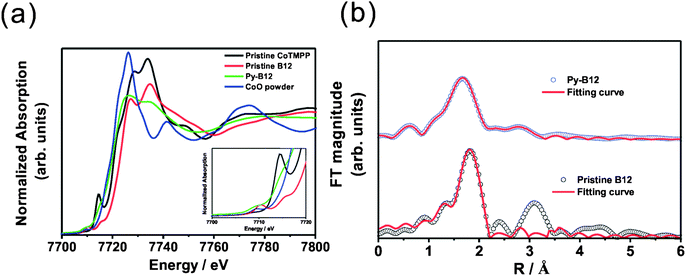 | ||
| Fig. 6 (a) XANES spectra of pristine CoTMPP, pristine B12, py-B12 and CoO powder; (b) Fourier transforms of k3-weighted EXAFS data at the Co K-edge for pristine B12 and py-B12. | ||
One important issue is the coordination number of the central cobalt in py-B12. Pristine B12 is a six-fold coordinated octahedral species, while py-B12 with Co(II) may be a five-coordinated species, with one vacant axial position (β site), or a four-coordinated species, with both axial positions vacant (α and β sites). (See ESI† for the definitions of α and β.) Since a five-coordinated species is highly sensitive to oxygen,53 the high stability of py-B12 as reported here indicates that it is unlikely to be a five-coordinated octahedral species. Instead, it is conjectured that the central Co in py-B12 is a four-coordinated species, which will further be supported by the following observations. In the pre-edge region in XANES spectra between 7700 to 7720 eV, shown in the inset of Fig. 6a, the characteristic peak at around 7710 eV is strong for CoO powder as well as for pristine B12, both of which possess six-coordinated octahedral species. However, this signal at around 7710 eV is very weak for py-B12, and is completely absent for pristine CoTMPP. In contrast, another characteristic peak at around 7714 eV is strong for pristine CoTMPP, somewhat lower but still appreciable for py-B12 and pristine B12, and absent for CoO powder. Note that pristine CoTMPP consists of a four-coordinated square-planar structure. It has been reported by Scheuring et al. that the six- and five-coordinated B12 species yielded strong XANES peaks at 7710 eV, but the four-coordinated B12 species yielded a strong peak at 7714 eV.54 Thus, the central Co in py-B12 is most likely to be a four-coordinated structure since the XANES signal at 7714 eV is more pronounced than its counterpart at 7710 eV.
To resolve the Co coordination number issue, further analysis using extended X-ray absorption fine structure (EXAFS) was performed on the Co K-edge spectra, which yields precise metal–ligand distances and the coordination number of the central Co in py-B12. As shown in Fig. 6b, Fourier transformation of k3-weighted Co K-edge EXAFS data for pristine B12 and py-B12 show clear differences. The structural parameters obtained from curve fitting55,56 to these spectra are listed in Table S2,† which clearly suggests that Co in pristine B12 has a coordination number of 6 while that in py-B12 has a coordination number of 4. Therefore, based on the XANES and EXAFS results, the core in the py-B12 catalyst is a four-coordinated species.
The present findings corresponding to the evolution of vitamin B12 during pyrolysis are also supported by other reports in the literature. Yang et al. performed voltammetric studies of B12 and concluded that Co(III) was directly reduced to Co(II) by the transfer of a single electron associated with –CN cleavage.57,58 Okada et al. studied four-, five- and six-coordinated Co(II)- or Co(III)-based compounds, and identified the four-coordinated Co(II)-based compounds with a coplanar chelate structure to be the best catalyst for the ORR.14 Moreover, the four-coordinated Co(II) species has been identified in several enzyme active sites in the literature.59–61 All the aforementioned reports support the existence of a stable four-coordinated Co(II) compound as proposed in this work.
The 100-hour durability test of py-B12/C as the cathode catalyst in the fuel cell by flowing H2 and air fed into the anode and the cathode, respectively, is demonstrated in Fig. 7. The fuel cells using Fe–N4/C and Co–PPy/C, demonstrated by Lefevre et al. and Bashyam and Zelenay,15,16 are also plotted in the figure under the same conditions of H2–air. Lefevre et al. used Fe–N4/C as the cathode catalyst with the catalyst loading of 5.6 mg cm−2 and Nafion 117 as the membrane in the PEFC, which was operated at a cell temperature 80 °C and a back pressure 30 psi.16 Bashyam and Zelenay used Co–PPy–C as the cathode catalyst with the Co loading of 0.06 mg cm−2 and Nafion 117 as the membrane in the PEFC, which was operated at a cell temperature 80 °C and a back pressure 2 atm.15 The initial current density of the fuel cell using py-B12/C is much higher than the others and maintains after the 100-hour period, with the final current density of 0.573 A cm−2, compared to the 0.360 and 0.140 A cm−2 of Fe–N4/C and Co–PPy/C, respectively. Relative to the 43% decay of Fe–N4/C and stable performance of Co–PPy/C, py-B12/C has about a 15% decay, which is mainly caused by water flooding. The continued research focuses on improving the mass-transport property of the catalyst layer using py-B12/C, such as additional PTFE. It is emphasized that the metal-loadings of each non-precious metal catalyst are 79 μg cm−2-Co (py-B12/C), 95 μg cm−2-Fe (Fe–N4/C) and 60 μg cm−2-Co (Co–PPy/C), indicating that py-B12/C has the higher mass activity than the others.
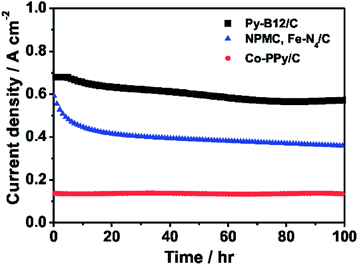 | ||
| Fig. 7 The 100-hour durability tests of H2–air PEFC using py-B12/C, Fe–N4/C,16 and Co–PPy/C15 as the cathode at 0.40 V. The operation conditions of py-B12/C PEFC: operation temperature: 70 °C; back pressure of H2 and air: 1 atm; anode catalysts: 30 wt% Pt/C with the metal loading of 0.25 mg cm−2 (E-TEK); electrolyte: Nafion® 212 (H+, DuPont). | ||
To understand the mechanism of the ORR under the catalytic activity of the central Co–corrin structure of py-B12, the geometries and the energies of the reactants, products, intermediates, and transition states on the potential energy surface of the corrin + O2 system are calculated by DFT at the B3LYP level62–64 with the LANL2DZ basis set.65–67 All the stationary points are identified for local minima and transition state by vibrational analysis. Final energies with zero-point-energy (ZPE) corrections were calculated at the same level. All electronic structure calculations are performed with the Gaussian 09 program.68
The potential energy surface of the ORR is shown in Fig.8. First, O2(g) can undergo adsorption on corrin forming OO–Co(a) with an exothermicity of 21.5 kcal mol−1. The decomposition of OO–Co(a) giving O–Co,O–C(a) has to overcome a high energy barrier at TS1, 73.1 kcal mol−1, with an endothermicity of 43.1 kcal mol−1. Significantly, compared to the neutral reaction (black line), the decomposition of a negatively charged O2-intermediate, OO–Co−(a), as depicted by the red line, can occur spontaneously. In principle, the singly negatively charged intermediate can be formed by the adsorption of O2− on corrin, capturing an electron by the neutral intermediate OO–Co(a) or the adsorption of a neutral O2 on corrin−, which is expected to exist in abundance in the cathode under the ORR conditions. The second possibility is equally likely; however, the formation of O2− in the gas phase prior to adsorption is expected to be less likely. The O2 adsorption energy on corrin− is 28.8 kcal mol−1, which is 7.3 kcal mol−1 larger than the neutral case. The OO–Co−(a) decomposition barrier at TS2 is 53.9 kcal mol−1 lower than that in the neutral case, TS1. Hence OO–Co−(a) is not only more stable, it also decomposes more readily to give O–Co,O–C−(a). We have also considered the two-electron charged case, but the formation of doubly charged species is energetically unfavorable. (See the ESI,† Fig. S5). In summary, the formation and decomposition of the singly negatively charged O2-corrin intermediate producing O–Co,O–C−(a) can take place spontaneously. The reaction of such a dissociated product with H atoms on the cathode can readily give two adsorbed hydroxyl radicals, HO–Co,HO–C−(a), which can further decompose to give H2O and/or H2O2, whose relative yield depends on their transition state structures and energies. Similarly, reactions of both neutral and charged O2-adsorbates with H atoms could lead to the formation of H2O and H2O2.
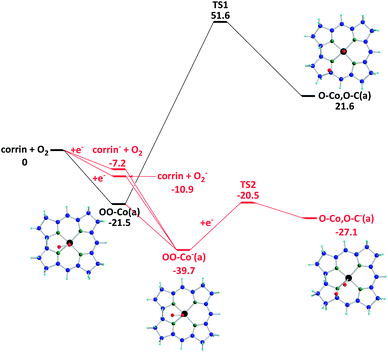 | ||
| Fig. 8 The potential energy profiles of oxygen decomposition reactions are shown with ZPE correction. The red, black, green, blue, and light blue atoms represent the oxygen, cobalt, nitrogen, carbon, and hydrogen atoms, respectively. (Unit: kcal mol−1). | ||
From a Mulliken charge analysis, Co losses 0.262e in corrin. We also calculate another popular ORR material, porphyrin, with B3LYP‖LANL2DZ. In porphyrin, the Co atom has a loss of 0.631e. Thus in the corrin system, the electron density of Co is greater and should interact more strongly with O2. The O2 adsorption energy on corrin is larger than that on porphyrin (21.5 and 39.7 kcal mol−1vs. 3.7 and 12.7 kcal mol−1 for the neutral and the singly negatively charged case, respectively).
Conclusions
Py-B12/C is shown to exhibit a high activity for the ORR, high durability after 100 h, and favourable performance for fuel cell application. The PEFC composed of a py-B12/C cathode demonstrates a preferential four-electron direct ORR pathway, and yields a high current density and high power density. Calculations based on the DFT at the B3LYP level conclude that the O2–Co–corrin complex with one additional electron goes through a much preferable path for the ORR, which isn't applicable to the case of porphyrin with higher symmetry. Py-B12 with a low symmetry of Co–corrin structure has a preferable pathway of the ORR, which is viable for Pt-substitute in the cathode of PEFC.Acknowledgements
The authors would like to thank the Ministry of Education, Academia Sinica, the National Synchrotron Radiation Research Center (Beam-line 17C1), and the National Science Council of Taiwan for financially supporting this research under Contract No. NSC 100-2218-E-011-013. Technical support from NanoCore, the Core facilities for nanoscience and nanotechnology at Academia Sinica in Taiwan, is acknowledged. This work was dedicated to Prof. Jerry C. Huang, who initiated this work during the last years of his life.References
- A. J. Appleby, J. Electroanal. Chem., 1993, 357, 117–179 CrossRef CAS.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CAS.
- N. A. Savastenko, V. Bruser, M. Bruser, K. Anklam, S. Kutschera, H. Steffen and A. Schmuhl, J. Power Sources, 2007, 165, 24–33 CrossRef CAS.
- P. Bogdanoff, I. Herrmann, M. Hilgendorff, I. Dorbandt, S. Fiechter and H. Tributsch, J. New Mater. Electrochem. Syst., 2004, 7, 85–92.
- C. Mocchil and S. Trasatti, J. Mol. Catal. A: Chem., 2003, 204–205, 713–720 Search PubMed.
- X.-Y. Xie, Z.-F. Ma, X. Wu, Q.-Z. Ren, X. Yuan, Q.-Z. Jiang and L. Hu, Electrochim. Acta, 2007, 52, 2091–2096 Search PubMed.
- H. Liu, C. Song, Y. Tang, J. Zhang and J. Zhang, Electrochim. Acta, 2007, 52, 4532–4538 CrossRef CAS.
- W. Jingjie, T. Haolin, P. Mu, W. Zhaohui and M. Wentao, Electrochim. Acta, 2009, 54, 1473–1477 Search PubMed.
- Y. Lu and R. G. Reddy, Electrochim. Acta, 2007, 52, 2562–2569 CrossRef CAS.
- P. Convert, C. Coutanceau, P. Crouigneau, F. Gloaguen and C. Lamy, J. Appl. Electrochem., 2001, 31, 945–952 CrossRef CAS.
- C. W. B. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- A. A. Serov, M. Min, G. Chai, S. Han, S. J. Seo, Y. Park, H. Kim and C. Kwak, J. Appl. Electrochem., 2009, 39, 1509–1516 Search PubMed.
- C.-H. Wang, H.-C. Hsu, S.-T. Chang, H.-Y. Du, C.-P. Chen, J. C.-S. Wu, H.-C. Shih, L.-C. Chen and K.-H. Chen, J. Mater. Chem., 2010, 20, 7551–7557 RSC.
- T. Okada, S. Gotou, M. Yoshida, M. Yuasa, T. Hirose and I. Sekine, J. Inorg. Organomet. Polym., 1999, 9, 199–219 Search PubMed.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS.
- M. Lefevre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef CAS.
- X. Wang, J. S. Lee, Q. Zhu, J. Liu, Y. Wang and S. Dai, Chem. Mater., 2010, 22, 2178–2180 CrossRef CAS.
- H. T. Chung, C. M. Johnston, K. Artyushkova, M. Ferrandon, D. J. Myers and P. Zelenay, Electrochem. Commun., 2010, 12, 1792–1795 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS.
- L. Wu, Y. Nabae, S. Moriya, K. Matsubayashi, N. M. Islam, S. Kuroki, M.-a. Kakimoto, J.-i. Ozaki and S. Miyata, Chem. Commun., 2010, 46, 6377–6379 RSC.
- J.-H. Kim, A. Ishihara, S. Mitsushima, N. Kamiya and K.-I. Ota, Electrochim. Acta, 2007, 52, 2492–2497 CrossRef CAS.
- B. Avasarala, T. Murray, W. Li and P. Haldar, J. Mater. Chem., 2009, 19, 1803–1805 RSC.
- P. J. Kulesza, K. Miecznikowski, B. Baranowska, M. Skunik, S. Fiechter, P. Bogdanoff and I. Dorbandt, Electrochem. Commun., 2006, 8, 904–908 CrossRef CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 RSC.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 RSC.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 RSC.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- J. H. Zagal and M. A. Paez, Electrochim. Acta, 1997, 42, 3477–3481 Search PubMed.
- J. H. Zagal, M. J. Aguirre and M. A. Paez, J. Electroanal. Chem., 1997, 437, 45–52 CrossRef CAS.
- J. H. Zagal, M. Paez and C. Paez, J. Electroanal. Chem., 1987, 237, 145–148 Search PubMed.
- J. J. Rehr, R. C. Albers and S. I. Zabinsky, Phys. Rev. Lett., 1992, 69, 3397–3400 CrossRef CAS.
- S. L. Gojkovic, S. Gupta and R. F. Savinell, Electrochim. Acta, 1999, 45, 889–897 CrossRef CAS.
- M. R. Tarasevich, L. A. Beketaeva, B. N. Efremov, N. M. Zagudaeva, L. N. Kuznetsova, K. V. Rybalka and V. E. Sosenkin, Russ. J. Electrochem., 2004, 40, 542–551 Search PubMed.
- K. Lee, L. Zhang, H. Lui, R. Hui, Z. Shi and J. Zhang, Electrochim. Acta, 2009, 54, 4704–4711 CrossRef CAS.
- M. Manzoli and F. Boccuzzi, J. Power Sources, 2005, 145, 161–168 CrossRef CAS.
- S. Pylypenko, S. Mukherjee, T. S. Olson and P. Atanassov, Electrochim. Acta, 2008, 53, 7875–7883 CrossRef CAS.
- C.-H. Wang, S.-T. Chang, H.-C. Hsu, H.-Y. Du, J. C.-S. Wu, L.-C. Chen and K.-H. Chen, Diamond Relat. Mater., 2011, 20, 322–329 Search PubMed.
- T. S. Olson, K. Chapman and P. Atanassov, J. Power Sources, 2008, 183, 557–563 Search PubMed.
- Z.-F. Ma, X.-Y. Xie, X.-X. Ma, D.-Y. Zhang, Q. Ren, N. Hess-Mohr and V. M. Schmidt, Electrochem. Commun., 2006, 8, 389–394 CrossRef CAS.
- J.-Y. Choi, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2010, 114, 8048–8053 CrossRef CAS.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Appl. Catal., B, 2009, 93, 156–165 CrossRef CAS.
- V. Nallathambi, J.-W. Lee, S. P. Kumaraguru, G. Wu and B. N. Popov, J. Power Sources, 2008, 183, 34–42 CrossRef CAS.
- V. Raghuveer, P. J. Ferreira and A. Manthiram, Electrochem. Commun., 2006, 8, 807–814 CrossRef CAS.
- V. Raghuveer, A. Manthiram and A. J. Bard, J. Phys. Chem. B, 2005, 109, 22909–22912 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- K. S. Taraszka, E. Chen, T. Metzger and M. R. Chance, Biochemistry, 1991, 30, 1222–1227 CrossRef CAS.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, Wiley, Chichester, 2001 Search PubMed.
- F. Beck, J. Appl. Electrochem., 1977, 7, 239–245 CAS.
- J. H. Zagal, Coord. Chem. Rev., 1992, 119, 89–136 CrossRef CAS.
- H. Haraguchi, K. Fujiwara and K. Fuwa, Chem. Lett., 1975, 4, 409–414 Search PubMed.
- S. Mebs, J. Henn, B. Dittrich, C. Paulmann and P. Luger, J. Phys. Chem. A, 2009, 113, 8366–8378 CrossRef CAS.
- M. Wolak, G. Stochel and R. van Eldik, J. Am. Chem. Soc., 2003, 125, 1334–1351 CrossRef CAS.
- E. M. Scheuring, W. Clavin, M. D. Wirt, L. M. Miller, R. F. Fischetti, Y. Lu, N. Mahoney, A. Xie, J.-j. Wu and M. R. Chance, J. Phys. Chem., 1996, 100, 3344–3348 CrossRef CAS.
- I. Sagi, M. D. Wirt, E. Chen, S. Frisbie and M. R. Chance, J. Am. Chem. Soc., 1990, 112, 8639–8644 Search PubMed.
- I. Sagi and M. R. Chance, J. Am. Chem. Soc., 1992, 114, 8061–8066 CrossRef CAS.
- N. Yang, Q. Wan and X. Wang, Electrochim. Acta, 2005, 50, 2175–2180 CrossRef CAS.
- D. Lexa and J. M. Saveant, J. Am. Chem. Soc., 1976, 98, 2652–2658 Search PubMed.
- M. D. Liptak, S. Datta, R. G. Matthews and T. C. Brunold, J. Am. Chem. Soc., 2008, 130, 16374–16381 CrossRef CAS.
- T. A. Stich, M. Yamanishi, R. Banerjee and T. C. Brunold, J. Am. Chem. Soc., 2005, 127, 7660–7661 Search PubMed.
- T. A. Stich, N. R. Buan, J. C. Escalante-Semerena and T. C. Brunold, J. Am. Chem. Soc., 2005, 127, 8710–8719 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. Heyd, J. E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, P. V. Parandekar, N. J. Mayhall, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and J. Cioslowski, Gaussian, Inc., Wallingford, CT, 2009.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ee01962g |
| This journal is © The Royal Society of Chemistry 2012 |