Synthesis and structural characterization of a ternary palladium–ruthenium–tin nanoalloy supported on graphene nanosheets for methanol electrooxidation in alkaline medium
Rahul
Awasthi
and
Ravindra Nath
Singh
*
Department of Chemistry, Centre of Advanced Study, Faculty of Science, Banaras Hindu University, Varanasi-221005, India. E-mail: rnsbhu@rediffmail.com; Fax: +91-542-2368127; Tel: +91-542-6701596
First published on 13th August 2012
Abstract
A graphene-supported ternary Pd–Ru–Sn nanoalloy with a face centered cubic structure has been synthesized by a microwave-assisted polyol reduction method and investigated for electrooxidation of methanol in 1 M KOH at 25 °C. The electrocatalytic activity of the novel ternary alloy electrode is more than 2 times higher than that of the Pd/GNS (base) electrode. It is observed that the percentage increase in catalytic activity of the ternary alloy electrode in comparison with the base electrode increases with the electrolysis time during the chronoamperometry study.
Introduction
Increased interest has been shown during recent years towards the development of alkaline direct methanol fuel cells (DMFCs). This is because of the fact that as compared to acid DMFCs, the alkaline DMFCs show improved kinetics of the methanol oxidation reaction (MOR) and metals other than Pt can be employed as catalysts.1 Among several non platinum metals, Pd seems to be a promising alternative.2 Attempts are continued to improve its catalytic activity and stability by obtaining it in dispersed form on high surface area supports, namely carbon nanopowders,3 multiwalled carbon nanotubes (MWCNTs),4,5 nanowire arrays (NWAs),6 carbonized TiO2 nanotubes,7 and graphene nanosheets (GNS),8–11 and through alloying with other metals like Ni, Ag, Zn–Ni, etc.12–14 Recently, Wang et al.13 prepared bimetallic Pd–Ag/C catalysts with different Ag loadings by borohydride reduction method and investigated for MOR. The Pd–Ag (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/C catalyst exhibited the greatest catalytic activity among the series. Yi et al.15 prepared titanium-supported binary Pd–Ru particles in different atomic ratios by a hydrothermal method and investigated their electrocatalytic activities for the ethanol oxidation reaction (EOR) in alkaline medium. Among the electrocatalysts investigated, the Pd87Ru13 catalyst displayed the greatest electrocatalytic activity towards EOR in alkaline medium. He et al.16 prepared carbon supported Pd4Au and Pd2.5Sn electrocatalysts by chemical reduction method and observed that the Pd4Au alloy nanoparticles displayed a better catalytic activity towards EOR in alkaline media. Singh et al.17,18 prepared Pd–0.5wt%C–xwt%Ru (x = 1, 2, 5, 10, 20, 30 and 50) composites by the borohydride reduction method and investigated their electrocatalytic activities toward MOR and EOR in 1 M KOH. Among the series, the Pd–0.5wt%C–20wt%Ru composite exhibited the greatest catalytic activity.
:1)/C catalyst exhibited the greatest catalytic activity among the series. Yi et al.15 prepared titanium-supported binary Pd–Ru particles in different atomic ratios by a hydrothermal method and investigated their electrocatalytic activities for the ethanol oxidation reaction (EOR) in alkaline medium. Among the electrocatalysts investigated, the Pd87Ru13 catalyst displayed the greatest electrocatalytic activity towards EOR in alkaline medium. He et al.16 prepared carbon supported Pd4Au and Pd2.5Sn electrocatalysts by chemical reduction method and observed that the Pd4Au alloy nanoparticles displayed a better catalytic activity towards EOR in alkaline media. Singh et al.17,18 prepared Pd–0.5wt%C–xwt%Ru (x = 1, 2, 5, 10, 20, 30 and 50) composites by the borohydride reduction method and investigated their electrocatalytic activities toward MOR and EOR in 1 M KOH. Among the series, the Pd–0.5wt%C–20wt%Ru composite exhibited the greatest catalytic activity.
Recently, we have prepared 20wt%Pd nanoparticles (NPs) dispersed on GNS/MWCNT by a microwave-assisted polyol reduction method8,9 and investigated them as electrocatalysts for the MOR in alkaline medium;8 the results have shown that the catalytic activity of Pd was improved by ∼30% when GNS was used as the support material in place of MWCNT. The catalytic activity of the 20wt%Pd NPs/GNS electrode was improved further by ∼45% with 2wt%Sn introduction.9 This result prompted us to prepare a series of ternary nanocomposites of Pd, Ru and Sn, aiming to produce a highly active electrocatalyst for the MOR. Among the series, the 40wt%Pd–5wt%Ru–2wt%Sn is found to exhibit the best performance. The present paper describes the details of the results of structural and electrocatalytic properties of the novel active 40wt%Pd–5wt%Ru–2wt%Sn/GNS and 40wt%Pd/GNS electrodes towards methanol electrooxidation in 1 M KOH at 25 °C.
Experimental
Catalyst preparation
The catalyst support, GNS, was prepared through the reduction of GO (graphite oxide) by NaBH4 reduction method.8,19 The oxide (GO) was prepared by a modified Hummers and Offenmans method.8,20 The base Pd catalyst, 40wt%Pd/GNS, and the composite, 40wt%Pd–5wt%Ru–2wt%Sn/GNS, were prepared by a microwave-assisted polyol reduction method.8,9 In a typical procedure, 10.6 mg of GNS was dispersed in 40 mL of ethylene glycol (Merck) solvent by ultrasonification for 1 h and then to this, a mixed solution comprising 102.5 μL of 0.096 M RuCl3 (Sigma-Aldrich), 6.6 mL of 0.01 M PdCl2 (Merck) and 380 μL of 8.86 mM SnCl2.2H2O (Merck) was added with ultrasonification. Solutions of RuCl3 and PdCl2 were prepared in 0.05 M HCl (Merck). The pH of this mixture was adjusted to 10 by adding 0.8 M KOH (Merck). The resulting mixture was kept in a microwave oven for 5 min at 800 watt power and was then centrifuged, washed with acetone and dried in a vacuum oven at 100 °C overnight. Similarly, the base catalyst (40wt%Pd/GNS) was also prepared. For simplicity in representation, 40wt%Pd/GNS and 40wt%Pd–5wt%Ru–2wt%Sn/GNS catalysts have been represented as Pd/GNS and Pd–Ru–Sn/GNS in the text.Catalyst electrode preparation
First of all, the catalyst ink was prepared by dispersing 3 mg of the catalyst powders in 600 μL of ethanol–water (2:1) mixture mechanically and then ultrasonicating for 30 min. 30 μL of the ink, so obtained, was placed on the pretreated glassy carbon (0.5 cm2) plate (GC) and dried in air. 10 μL of 1% Nafion solution (Alfa Aesar) was then dropped over the catalyst film on GC. The catalyst loading (metal catalyst and GNS) on GC was 0.3 mg cm−2. The pretreatment of GC, electrical contact with the catalyst over-layer and electrode mounting were carried out as described previously.21
Catalyst characterization
Structural characterization of the catalyst was completed by recording the X-ray diffraction (XRD) patterns (X-ray diffractometer, Thermo Electron) and transmission electron microphotographs (TEM: TECNAI G2 FEI) of the catalysts. CuKα was used as the radiation source (λ = 1.541841 Å). To obtain TEM pictures, the catalyst was dispersed in methanol and a drop of this suspension was placed onto a carbon coated copper grid, and dried. The metal loading of the ternary nanoalloy catalyst was analyzed by inductively coupled plasma spectroscopy (ICP).Electrochemical studies, namely cyclic voltammetery (CV) and chronoamperometry (CA), have been carried out in a three-electrode single-compartment Pyrex glass cell using a potentiostat/galvanostat Model 273A (PARC, USA). A pure Pt-foil (∼8 cm2) and Hg/HgO/1 M KOH were used as auxiliary and reference electrodes, respectively. The potentials of the working electrode were measured using the reference, Hg/HgO/1 M KOH (E° = 0.098 V vs. SHE), but values given in the text are against the reversible hydrogen electrode, RHE (E° ∼ −0.83 V vs. SHE).17 The CV of each electrocatalyst has been recorded at a scan rate of 50 mV s−1 between 0.13 and 1.13 V vs. RHE in 1 M KOH with and without methanol at 25 °C. Before recording the final voltammogram, each electrode was cycled for several runs at the potential scan rate of 50 mV s−1 in 1 M KOH. All electrochemical experiments were performed in an Ar deoxygenated solution. The electrochemical activity data given in the text and table are average ones and have been obtained with triplicate electrodes of each catalyst under identical experimental conditions.
Results and discussion
XRD
The X-ray diffraction patterns of Pd/GNS and Pd–Ru–Sn/GNS are shown in Fig. 1. The catalysts exhibit typical diffraction peaks corresponding to the (111), (220) and (200) planes of the face centered cubic phase of Pd NPs.13 The diffraction peak at 2θ ≈ 25° corresponds to the (002) plane of the carbon support.13 The observation of diffractograms for the ternary and the base catalysts indicates that the diffraction peaks for Pd in the case of the ternary catalyst slightly shift towards a higher 2θ angle (≈0.4°), compared to the base catalyst and that the diffraction peaks for Ru/Sn or their oxides are absent. Thus, the results show that there is an alloy formation between Pd and Ru–Sn NPs. However, the possibility of the presence of Ru, Sn or their oxides as an amorphous phase cannot be ignored. The XRD peaks corresponding to Pd in the ternary alloy NPs are broader compared to those observed in Pd/GNS, indicating thereby that the crystallite size of Pd NPs is smaller in the ternary nanoalloy catalyst compared to that in the Pd/GNS catalyst. The crystallite size of Pd was determined by using the Scherrer formula and applying the Gaussian line fit to the most intense diffraction (111) peak.13 Estimates of the Pd NP crystallite size in Pd–Ru–Sn/GNS and Pd/GNS were 3.8 and 7.5 nm, respectively. Thus, the crystallite size of Pd is greatly reduced with the introduction of Sn and Ru. | ||
| Fig. 1 XRD patterns of Pd/GNS and Pd–Ru–Sn /GNS catalysts. | ||
TEM
Fig. 2a–d show TEM images of the Pd/GNS and Pd–Ru–Sn/GNS catalysts. The Pd NPs are dispersed on the GNS surface with some agglomeration (Fig. 2a and b). However, dispersion of the NPs seems to improve in the ternary catalyst with reduced particle size (Fig. 2c and d). Estimates of the mean particle sizes of the NPs are ∼9.0 and ∼5.3 nm in Pd/GNS and Pd–Ru–Sn /GNS, respectively. | ||
| Fig. 2 TEM images of (a),(b) the Pd/GNS and (c),(d) the Pd–Ru–Sn catalysts. | ||
Compositional analyses
The composition of the Pd–Ru–Sn nanoalloy for Pd, Ru and Sn content was determined by ICP analysis and found to be 22.11 (≈38.8 wt%), 3.53 (≈5.8 wt%) and 1.19 (≈2.3 wt%) at.%, respectively. Thus, the final metal contents in the material are close to the initial amounts used in the synthesis procedure.Cyclic voltammetery
In order to know the maximum anodic potential up to which the home-made GNS and commercial nanocarbon powder (NC) are stable, CVs have been recorded at a slow scan rate of 1 mV s−1 in 1 M KOH and are shown in Fig. 3. Only the anodic part of the CV curves are reproduced in Fig. 3. From Fig. 3 it is clear that both forms of the carbon are stable up to an anodic potential of ∼1.3 V vs. RHE. | ||
| Fig. 3 Cyclic voltammograms (only anodic part) of (a) GNS and (b) NC in 1 M KOH at 1 mV s−1. Carbon loading = 0.3 mg cm−2, geometrical area = 0.5 cm2, T = 25 °C. | ||
CVs of the Pd/GNS and Pd–Ru–Sn/GNS electrodes were recorded in the potential region from 0.13 to 1. 13 V vs. RHE at the scan rate of 50 mV s−1 in Ar-saturated 1 M KOH and 1 M KOH + 1 M CH3OH at 25 °C. The curves so obtained are shown in Fig. 4 and 5. Features of the voltammograms of the electrodes shown in Fig. 4 were quite similar regardless of the nature of the electrode material. The observed cathodic peaks in the potential region from ∼0.43 to ∼0.13 V vs. RHE and anodic peaks in the potential region from ∼0.13 to ∼0.54 V vs. RHE are due to the adsorption–desorption of hydrogen. A gradual increase in the anodic current from a potential > ∼ 0.60 V vs. RHE on the positive-going scan has been noticed, which can be attributed to a slow transformation of the palladium metal present in the surface film into palladium(II) oxide (PdO). It is generally accepted that the adsorbed hydroxyl species is formed on the Pd surface in the initial stage of the oxide formation, which overlaps with the hydrogen desorption peak, and higher valance PdO are then formed at higher anodic potentials.15 Further, in the negative-going scan, a strong reduction peak observed at around ∼0.70 and 0.63 V vs. RHE is ascribed to the reduction of Pd(II) oxide into elemental Pd. Similar voltammograms in KOH solutions were also reported for Pd or Pd-based alloys/composites.2–6,8,9,11 Due to the penetration of hydrogen into Pd and Pd-based catalysts,22 the electrochemically active surface areas (EASAs) of the electrodes are estimated by determining the charge used in the reduction of palladium(II) oxide into palladium metal. The EASA of the electrode was estimated using the relation EASA = Q/S, where Q is the coulombic charge (in mC) and S is the proportionality constant.9 A charge value of 0.405 mC cm−2 is considered for the reduction of a PdO monolayer.9,22,23 The estimates of the EASA were ∼85 and ∼105 cm2 for Pd/GNS and Pd–Ru–Sn/GNS, respectively.
 | ||
| Fig. 4 Cyclic voltammograms of Pd/GNS and Pd–Ru–Sn/GNS catalysts at a scan rate of 50 mV s−1 in 1 M KOH at 25 °C. | ||
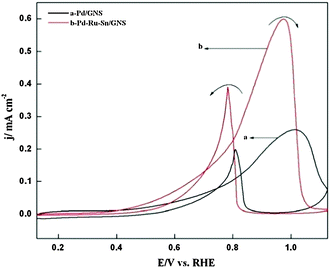 | ||
| Fig. 5 Cyclic voltammograms of Pd/GNS and Pd–Ru–Sn/GNS catalysts at a scan rate of 50 mV s−1 in 1 M KOH + 1 M CH3OH at 25 °C. | ||
Fig. 5 shows the CVs of electrocatalysts recorded at 50 mV s−1 in 1 M KOH + 1 M CH3OH at 25 °C. Each CV curve exhibits two characteristic oxidation peaks observed, respectively, in the positive (Ip,f) and the negative going scans (Ip,b).2–5,8,9,11,12 It is observed that on the positive going scan the MOR commences on Pd/GNS at a potential ∼0.39 V vs. RHE and then the oxidation current increases progressively with potential, attains an optimum value and declines rapidly thereafter. The decrease in current has been attributed to the poisoning of the electrode surface by adsorbed methanol oxidation intermediates such as CO molecules and to the formation of a palladium(II) oxide surface film.11,17 The palladium(II) oxide surface film gets reduced to active Pd metal under cathodic condition and the methanol oxidation starts again. As the potential region for MOR overlaps with the potential region for electroreduction of PdO (Fig. 4 and 5), the cathodic peak corresponding to the electroreduction of PdO does not appear in the presence of methanol in the electrolyte. However, some authors have considered that the oxidation peak (Ip,b) is caused by the oxidation of carbonaceous species that are not completely oxidized under the anodic conditions.3,11,12 The onset potential (Eop), the peak current density (jp = Ip,f, mA/EASA, cm2) and the corresponding peak potential (EP) for the methanol oxidation reaction have been determined from Fig 5 based on the positive-going scan and are given in Table 1. The onset potentials given in Table 1 have been determined by extending the base current of the methanol oxidation current peak of the forward scan (Fig. 5) towards the higher potential and noting the potential from which the methanol oxidation current begins to increase.17 The value of the onset potential for MOR on Pd–Ru–Sn/GNS is ∼50 mV lower than the one observed on Pd/GNS. This indicates that the ternary electrode, Pd–Ru–Sn/GNS, is more active than the Pd/GNS electrode, which is also evident from the values of the jp based on the positive going scan. As the values of Ep shown in Table 1 are not the same, for activity comparison, values of the current density (j = I, mA/EASA, cm2) for both the electrodes are determined at 0.60 V vs. RHE from Fig. 5 and are listed in Table 1. The activity data summarized in Table 1 show that the Pd–Ru–Sn/GNS electrode is ∼2 times more active than the Pd/GNS electrode.
| Catalyst | E op/V | In the positive going scan | ||
|---|---|---|---|---|
| E p/V | j p/mA cm−2 | j (at E = 0.60 V)/mA cm−2 | ||
| Pd/GNS | 0.39 | 1.02 | 0.259 | 0.034 |
| Pd–Ru–Sn/GNS | 0.34 | 0.99 | 0.598 | 0.070 |
The catalytic activity of the ternary Pd–Ru–Sn/GNS electrode prepared in this study (I ≈ 123 mA mg−1Pd) for MOR is also found to be better than those of recently reported bi metallic Pd–Ag (1:1)/CNTs13 (I ≈ 46 mA mg−1Pd) and PdAu41/C24 (I ≈ 27 mA mg−1Pd) electrodes under similar experimental conditions (electrolyte: 1 M KOH + 1 M CH3OH, scan rate = 50 mV s−1 and E = 0.60 V vs. RHE).
Chronoamperometry
Chronoamperograms of the electrocatalysts in 1 M KOH + 1 M CH3OH have been recorded for 2 h at E = 0.63 V vs. RHE and at 25 °C (Fig. 6). This figure shows that the activity (j) of the Pd–Ru–Sn/GNS electrode towards MOR is greater than the base electrode during the entire period of the CA study. It is observed that the percentage increase in activity of the ternary electrode gradually increases with the passage of the electrolysis time. Estimates of the percentage increase in the activity in relation to the base electrode are found to be 29.7, 44.9, 55.2, 57.1, 62.6 and 64.9% at 5, 10, 20, 40, 60, and 120 min, respectively. Thus, the results show that the performance of the ternary electrocatalyst is much superior to the base electrocatalyst, Pd/GNS. | ||
| Fig. 6 Chronoamperograms of Pd/GNS and Pd–Ru–Sn/GNS catalysts at E = 0.63 V in 1 M KOH + 1 M CH3OH at 25 °C. | ||
It is known that Ru does not influence the electrocatalysis of methanol electrooxidation directly.17,18 It, however, interacts with water molecules, producing OHads at relatively lower potential.17,18 The latter intermediate species can promote the desorption of adsorbed methanol residues12,17 and hence the catalytic activity. The role of Sn can be understood by considering the electronegativities of Pd and Sn. As Pd is more electronegative than Sn, the electrons will be shifted towards Pd, which thereby weakens the Pd–CO bond.9,12,24 Thus, both Ru and Sn additions seem to improve the activity of the catalyst for the methanol electrooxidation.
Conclusions
The study demonstrates that the Pd–Ru–Sn/GNS catalyst, obtained by microwave-assisted polyol method, is greatly active for MOR. At E = 0.60 V vs. RHE, the Pd–Ru–Sn/GNS catalyst exhibited ∼2 times higher catalytic activity than the base electrocatalyst under similar experimental conditions. Further, the increase in the activity of the ternary electrode (with reference to base electrode) gradually increases with time during the chronoamperometry experiment, the observed increase in activity being 64.9% during 2h of the experiment.Acknowledgements
Financial support received from the Council of Scientific and Industrial Research (CSIR), Government of India, New Delhi through a research project (ref. no. 01(2320)/09-EMR-II) and a Senior Research Fellowship (grant no. 09/013(0126)/2007-EMR-I) to carry out the investigation is thankfully acknowledged. Authors also thank Dr S. K. Tiwari, National Metallurgical laboratory, Jamshedpur, India for the ICP analyses.Notes and references
- E. Antolini and E. R. Gonzalez, J. Power Sources, 2010, 195, 3431 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183 CrossRef CAS.
- R. Awasthi, Anindita and R. N. Singh, Open Catal. J., 2010, 3, 62 CrossRef.
- V. Bambagioni, C. Bianchini, A. Marchionni, J. Filippi, F. Vizza, J. Teddy, P. Serp and M. Zhiani, J. Power Sources, 2009, 190, 241 CrossRef CAS.
- R. N. Singh, A. Singh and Anindita, Int. J. Hydrogen Energy, 2009, 34, 2052 CrossRef CAS.
- F. Cheng, H. Wang, Z. Sun, M. Ning, Z. Cai and M. Zhang, Electrochem. Commun., 2008, 10, 798 CrossRef CAS.
- F. Hu, F. Ding, S. Song and P. K. Shen, J. Power Sources, 2006, 163, 415 CrossRef CAS.
- R. N. Singh and R. Awasthi, Catal. Sci. Technol., 2011, 1, 778 Search PubMed.
- R. Awasthi and R. N. Singh, Int. J. Hydrogen Energy, 2012, 37, 2103 CrossRef CAS.
- Z. Wen, S. Yang, Y. Liang, W. He, H. Tong, L. Hao, X. Zhang and Q. Song, Electrochim. Acta, 2010, 56, 139 CrossRef CAS.
- H. Li, G. Chang, Y. Zhang, J. Tian, S. Liu, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Catal. Sci. Technol., 2012, 2, 1153 CAS.
- Z. Qi, H. Geng, X. Wang, C. Zhao, H. Ji, C. Zhang, J. Xu and Z. Zhang, J. Power Sources, 2011, 196, 5823 CrossRef CAS.
- Y. Wang, Z. M. Sheng, H. Yang, S. P. Jiang and C. M. Li, Int. J. Hydrogen Energy, 2010, 35, 10087 CrossRef CAS.
- V. Bambagioni, C. Bianchini, J. Filippi, W. Oberhauser, A. Marchionni, F. Vizza, R. Psaro, L. Sordelli, M. L. Foresti and M. Innocenti, ChemSusChem, 2009, 2, 99 CrossRef CAS.
- Q. Yi, F. Niu, L. Song, X. Liu and H. Nie, Electroanalysis, 2011, 23, 2232 CrossRef CAS.
- Q. He, W. Chen, M. S. Chen and F. Laufek, J. Power Sources, 2009, 187, 298 CrossRef CAS.
- Anindita, R. Awasthi, Madhu and R. N. Singh, Open Catal. J., 2011, 4, 83 Search PubMed.
- Anindita, R. Awasthi, Madhu and R. N. Singh, Open Catal. J., 2011, 4, 43 CrossRef.
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514 CrossRef CAS.
- L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2009, 131, 1043 CrossRef CAS.
- R. N. Singh, T. Sharma, A. Singh, Anindita, D. Mishra and S. K. Tiwari, Electrochim. Acta, 2008, 53, 2322 CrossRef CAS.
- G. F. Alvarez, M. Mamlouk, S. M. S. Kumar and K. Scott, J. Appl. Electrochem., 2011, 41, 925 CrossRef CAS.
- Y. Zhao, L. Zhan, J. Tian, S. Nie and Z. Ning, Electrochim. Acta, 2011, 56, 1967 CrossRef CAS.
- Y. Z. Su, M. Z. Zhang, X. B. Liu, Z. Y. Li, X. C. Zhu, C. W. Xu and S. P. Jiang, Int. J. Electrochem. Sci., 2012, 7, 4158 CAS.
| This journal is © The Royal Society of Chemistry 2012 |