Sulfated zirconia: an efficient solid acid catalyst for esterification of myristic acid with short chain alcohols†
K.
Saravanan
,
Beena
Tyagi
* and
H. C.
Bajaj
Discipline of Inorganic Materials and Catalysis, Council of Scientific and Industrial Research (CSIR), Central Salt and Marine Chemicals Research Institute (CSMCRI), G. B. Marg, Bhavnagar, Gujarat 364 002, India. E-mail: btyagi@csmcri.org; Fax: +91-278-2566970
First published on 31st July 2012
Abstract
Sulfated zirconia (SZ) catalysts prepared by a two-step sol–gel method and calcined at 600–700 °C were evaluated for esterification of myristic acid with methanol using varied acid to alcohol ratio, reaction temperature and catalyst concentration. An exceptionally small concentration of SZ catalysts (0.125–0.5 wt% to acid) exhibited 98–100% conversion of myristic acid with methanol at 60 °C after 5 h. The conversion was decreased with an increase in the alkyl chain of alcohol from methanol to butanol, however, similar conversion was achieved by increasing the reaction temperature to 90 °C. The ester formation was selective, irrespective of alcohol and other reaction variables. The calcination temperature has strong influence on the structural, textural and acidic features and thus on the activity and re-usability of SZ catalysts. The reaction is sensitive to moisture present in methanol or reaction mixture. The studied reaction is Brönsted acid catalyzed and the SZ catalyst having higher number of Brönsted acid sites was re-used successfully without significant loss in activity; whereas the SZ catalyst having lower number of Brönsted acid sites showed a decrease in activity (∼28%) after five reaction cycles. The results clearly indicated the necessity of higher number of Brönsted acid sites for better performance and recycling of the SZ catalysts for esterification of myristic acid with methanol under the conditions studied.
1. Introduction
Myristic acid is a saturated fatty acid (n-tetradecanoic acid, C14H28O2) naturally occurring in nutmeg Myristica fragrans. It is also found in palm kernel oil, coconut oil, butter fat and animal fats, which are used to produce fatty acid alkyl esters (FAAEs), used as biodiesel.1 It increases low density lipoprotein cholesterol making it one of the most hypercholesterolemic of the saturated fatty acids.2 Alkyl myristate of short chain alcohols namely methanol, ethanol, propanol and butanol are used in perfumes, flavors, cosmetics, personal care products, food additives, detergents, soaps, lubricants for textiles, plasticizers etc.3 Besides, methyl myristate has been approved as a new active constituent in veterinary chemical products by Australian pesticides and veterinary medicines authority4 and ethyl myristate is used as a marker of excessive ethanol consumption that can be isolated from the hair of an alcoholic individual.5Conventionally, FAAEs have been prepared by homogeneous acid catalyzed esterification of the corresponding acid with an alcohol or homogeneous base catalyzed transesterification of oils and fats. Industrial esterification processes are carried out in the presence of homogeneous Brönsted acid catalysts such as sulfuric, p-toluene sulfonic or phosphoric acid.6 To overcome the disadvantages associated with homogeneous catalysts, a number of solid acid catalysts such as inorganic metal oxides, heteropoly acids and sulfonic acid based resins etc. are being studied to produce FAAEs via esterification of fatty acids (FAs) or transesterification of oil with alcohols.6,7 Among metal oxide based solid acid catalysts, sulfated zirconia (SZ) has exhibited significant activity in both the reactions.6–13 Kiss et al.9 have screened various solid acids including zeolites, ion exchange resins and metal oxides for the esterification of lauric acid with different alcohols and found SZ as the most active solid acid catalyst; zeolites were not suitable due to their microporous nature having diffusion limitations of large fatty acid molecules, whereas, ion exchange resins have low thermal stability. SZ is a potential solid acid catalyst for alkane isomerization at mild temperature14 and many other commercially important organic transformations due to its strong acid properties.15–17
Few studies have been reported for the esterification of myristic acid using SZ18,19 and niobia20 metal oxide catalysts. Zeolites21 and acidic ion exchange resins namely Amberlyst-15 and silica based resin having sulfonic acid groups22 were not found effective as compared with metal salts or homogenous p-TSA. Among various metal salts, zirconium sulfate showed significant activity.21 MCM-48 supported tungstophosphoric acid23 and ZrOCl2·8H2O24 have been studied with long chain alcohols such as cetyl alcohol in sc-CO2 and other organic solvents. However, most of these studies have been done for various long chain saturated and unsaturated FAs or along with triglycerides at high temperature (120–180 °C)18,19,23,24 in the presence of higher concentration of solid catalysts (5–15 wt%).19–21 To the best of our knowledge, no detailed study has been reported for the esterification of myristic acid with methanol and other short chain alcohols over SZ catalysts.
We have studied SZ catalysts for various reactions mainly acetylation,25a,b isomerization,25c,d Pechmann,25e,f condensation26a and recently esterification of caprylic acid.26b Herein, we report a systematic detailed study of the esterification of myristic acid with methanol at lower temperature in the presence of small concentration of SZ catalysts. The effect of various reaction parameters such as acid to alcohol ratio, catalyst concentration and reaction temperature along with other short chain alcohols namely ethanol, n-propanol and n-butanol has been investigated. The effect of calcination temperature on the acidity and activity, re-usability and deactivation of SZ catalysts has also been addressed. In our earlier report25c we have observed that selectivity of the isomerized products can be varied depending upon the acidity of SZ catalysts in terms of Brönsted (B) to Lewis (L) acid site concentration ratio. Herein, we have found that the acidic properties in terms of Brönsted acid site concentration, B/L ratio and total surface acidity play a major role in the activity and deactivation of SZ catalysts for esterification of myristic acid with methanol under the conditions studied. The present study gives novel insight into the influence of the acidic properties on the catalytic performance of fresh and re-used SZ catalysts and the use of a very small amount of the catalyst resulting in maximum conversion of acid at lower temperature.
2. Results and discussion
2.1 Catalyst characterization
The partial characterization of SZ catalysts has been reported in our previous paper.26a In the context of the present work few salient features are mentioned briefly to understand the structure–activity relationship.A powder X-ray diffraction (PXRD) pattern of SZ catalysts showed the presence of tetragonal crystalline phase after calcination at 600–700 °C. SZ-600 was less crystalline and the crystallinity of the samples increased with increasing calcination temperature (Fig. S1, ESI†). The catalysts were found to have nano-crystallite size in the range of 11–16 nm (Table 1). The bulk sulfur content before calcination was 3.7 wt%, which successively decreased after calcination at 600–700 °C (Table 1). The TG/DTA profile (Fig. S2, ESI†) of the catalyst also confirmed the crystallization of the SZ sample at >600 °C.
| Catalyst | Crystallite sizea (nm) | Sulfura (wt%) | S BET (m2 g−1) | Average pore volume (cm3 g−1) | BJH pore diameter (nm) | Cyclohexanol conversion (%) | NH3-TPD | DRIFTb | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Temp. (°C) | Acid sites (mmol g−1) | Total acid sites (mmol g−1)a | B acid site (%T cm−1) | B/L ratio | |||||||
| a Data from ref. 26a. b B and L = Brönsted and Lewis acid sites, at 150 °C. | |||||||||||
| SZ-600 | 11 | 2.6 | 75 | 0.12 | 6.5 | 91 | 719 | 2.50 | 2.5 | 3690 | 2.07 |
| SZ-650 | 15 | 1.04 | 74 | 0.09 | 5.7 | 87 | 116 | 0.80 | 0.99 | 2939 | 1.66 |
| 564 | 0.19 | ||||||||||
| SZ-700 | 16 | 0.87 | 54 | 0.08 | 7.1 | 85 | 112 | 0.38 | 0.67 | 2738 | 1.21 |
| 400 | 0.07 | ||||||||||
| 627 | 0.14 | ||||||||||
| 793 | 0.08 | ||||||||||
A TEM micrograph of SZ-600 (Fig. 1) showed the aggregates of SZ crystallites varying from 6.9 to 10.2 nm with an average crystallite size of 7.3 nm, which was in agreement with crystallite size calculated by PXRD. The high-resolution TEM image (inset, Fig. 1) exhibited the lattice fringes, the width between two fringes (∼2.86 Å) also agreed with d-spacing (∼2.93 Å) of characteristic peaks of tetragonal phase of zirconia (2θ = 30.22) shown by PXRD.

BET surface area and pore volume were found to decrease with an increase in calcination temperature; however SZ-700 showed higher pore diameter due to collapsing of pore walls at higher temperature (Table 1). The N2 adsorption isotherms (Fig. S3, ESI†) showed the increase in adsorption at higher relative pressure indicating the presence of larger size of mesopores in the samples.27 The inflection point at P/Po = ∼0.4–0.5 was not sharp, which reflected that the pores have varied pore size distribution (Fig. 2).
 | ||
| Fig. 2 Pore size distribution profiles of SZ catalysts calcined at different temperatures. | ||
IR-spectra of the catalysts showed the presence of sulfate groups in the range of 1245–900 cm−1 (Fig. S4, ESI†). SZ-600 showed the presence of a broad intense peak showing shoulders at 1230, 1138, 1040 and 990 cm−1, however, highly crystalline SZ-650 and SZ-700 clearly exhibited the peaks at 1230–1245, 1136–1142, 1047–1056 and 988 cm−1, which are assigned to asymmetric and symmetric stretching frequencies of S![[partial double bond, top dashed]](https://www.rsc.org/images/entities/char_e12f.gif) O and S–O bonds and are characteristic of inorganic chelating bidentate sulfate. The partially ionic nature of the SO bond is responsible for the Brönsted acid sites in sulfated zirconia.28 The intensity of the sulfate peaks was reduced in SZ-700 due to loss of sulfate species at higher calcination temperature.
O and S–O bonds and are characteristic of inorganic chelating bidentate sulfate. The partially ionic nature of the SO bond is responsible for the Brönsted acid sites in sulfated zirconia.28 The intensity of the sulfate peaks was reduced in SZ-700 due to loss of sulfate species at higher calcination temperature.
The dehydration of cyclohexanol over SZ-600 showed 91% conversion of cyclohexanol with selective formation of cyclohexene indicating the presence of higher Brönsted acidity in SZ-600, which slightly decreased in SZ-650 and SZ-700 (Table 1). Similarly, the total surface acidity analyzed by NH3-TPD showed the highest number of acid sites (2.5 mmol g−1) in SZ-600, which decreased ominously in SZ-650 and SZ-700 (Table 1). The acidity in sulfated zirconia is attributed to the presence of sulfate groups; as SZ-600 has the highest sulfur content, it showed the highest number of acid sites, the decrease in sulfur content with increasing calcination temperature resulted in a decrease in acidity. Furthermore, NH3 desorption occurred at relatively higher temperature in the SZ-600, indicating the presence of only strong acid sites (719 °C), whereas SZ-650 and SZ-700 showed the presence of weak (112–116 °C), moderate (400–500 °C) to strong (>600 °C) acid sites (Table 1).
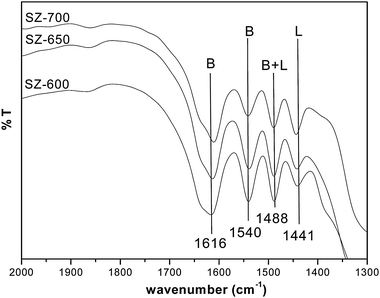
The DRIFT spectra of pyridine adsorbed SZ catalysts exhibited the characteristic peaks of pyridinium ions (Brönsted acid sites) at 1540 cm–1 and of covalently bonded pyridine (Lewis acid sites) at 1441 cm–1 (Fig. 3) along with peaks at 1616 cm–1 and 1488 cm–1 representing the Brönsted and total acid sites respectively.25e,29 Both acid sites were observed to be strong enough as they were present even after heating at 450 °C; though the intensity of the peaks was decreased after successive heating (Fig. S5, ESI†). SZ-600 and SZ-650 showed intense peaks for Brönsted acid sites, whereas, in SZ-700 the peak for the Lewis acid site was intense. The Brönsted acid site concentration and B/L ratio, calculated from the characteristic peak area at 150 °C, were in the descending order from SZ-600 to SZ-700 (Table 1).
2.2 Catalytic activity
Initially, esterification of myristic acid was studied with methanol over the SZ-600 catalyst to optimize acid to alcohol ratio, catalyst concentration and temperature to achieve the maximum conversion of myristic acid and selectivity for methyl myristate.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 1:20 over catalyst concentration in the range of 0.125–0.5 wt% to acid. The varying concentration of the catalyst (0.125–0.5 wt%) resulted in 98–100% conversion of myristic acid having 100% selectivity for methyl myristate using an acid to methanol molar ratio of 1:20 at 60 °C after 7 h (Table 2). By decreasing the acid to methanol molar ratio to 1:10, the catalyst showed 47% conversion with 0.125 wt% of catalyst, which enhanced upon increasing the catalyst concentration. By further decreasing the acid to methanol ratio to 1:5, 82% conversion was observed with 0.5 wt% catalyst concentration. The results indicated the requirement of higher acid to alcohol ratio to obtain complete conversion of FAs as also reported in the literature.6
:5 to 1:20 over catalyst concentration in the range of 0.125–0.5 wt% to acid. The varying concentration of the catalyst (0.125–0.5 wt%) resulted in 98–100% conversion of myristic acid having 100% selectivity for methyl myristate using an acid to methanol molar ratio of 1:20 at 60 °C after 7 h (Table 2). By decreasing the acid to methanol molar ratio to 1:10, the catalyst showed 47% conversion with 0.125 wt% of catalyst, which enhanced upon increasing the catalyst concentration. By further decreasing the acid to methanol ratio to 1:5, 82% conversion was observed with 0.5 wt% catalyst concentration. The results indicated the requirement of higher acid to alcohol ratio to obtain complete conversion of FAs as also reported in the literature.6
Theoretically, esterification reaction requires one mole of alcohol for one mole of acid; however, in practice, a molar ratio higher than stoichiometric ratio is needed to complete the reaction having acceptable reaction rates. The higher amount of methanol shifted the equilibrium to the right side, thus achieving the maximum conversion even with lower catalyst amount (0.125 wt%). On the other hand, higher concentration (0.5 wt%) of the catalyst increased the availability of acid sites, which favour the accessibility of more number of reactant molecules to these acid sites and thus enhanced the conversion in the presence of lower acid to alcohol ratio. Though a very high acid to alcohol ratio has been used to achieve maximum acid conversion of FAs, e.g., 1:50,30 1:4031 to 1:10011 and 1:20032 in the presence of HPA, SZ and WZ catalysts, respectively; lower amount of alcohol is more desirable in the industrial synthesis of FAAEs, and thus we preferred an acid to methanol ratio of 1:10 along with 0.5 wt% of catalyst (protocol A) for rest of the reactions. However, for comparison, we have also studied few reactions with an acid to methanol ratio of 1:20 in the presence of 0.125 wt% of catalyst (protocol B). It is worth to be noted here that the amount of SZ catalyst used was exceptionally small (0.125–0.5 wt% to myristic acid, i.e., acid:SZ (wt/wt) = 800–200:1) and to the best of our knowledge, not reported earlier. Similar good activity was also found for caprylic acid with 0.5 wt% of catalyst.26b
To optimize the reaction temperature, we have studied the reaction kinetics both at 50 and 60 °C. Though the final conversion of myristic acid was similar (98–100%) after 7 h and remained steady afterwards both at 50 and 60 °C (Fig. 4), the rate of increase in conversion was higher at 60 °C. Therefore, 60 °C temperature was chosen as optimum reaction temperature for rest of the studies.
 | ||
| Fig. 4 Conversion of myristic acid with time at (a) 50 °C and (b) 60 °C, (c) effect of fresh methanol (at 60 °C). Reaction conditions: acid:methanol = 1:10; SZ = 0.5 wt%. | ||
Under (B) reaction variables, decrease in conversion was observed over both SZ-650 and SZ-700 catalysts (Table 4). Though, the SZ-650 catalyst showed similar conversion to SZ-600 under (A) reaction conditions, it reduced to 80% under (B) reaction conditions. Because, its lower acidity and Brönsted acid site concentration (compared to SZ-600) were sufficient to achieve >98% conversion when its concentration was higher in reaction (A), however, with lower concentration along with higher amount of methanol in reaction (B), the number of acid sites available was insufficient to interact with the reactant molecules and thus to obtain complete conversion of acid. The decreasing catalytic activities of SZ catalysts were in good agreement with their decreasing Brönsted acid site concentration as well as B/L ratio and total surface acidity with an increase in calcination temperature (Table 1). These results clearly revealed that the acidity and activity of a SZ catalyst are strongly influenced by the temperature at which it has been calcined before undergoing the reaction along with the reaction parameters. The pure ZrO2 sample, calcined at 400–600 °C, showed only 23–28% conversion of myristic acid (Table 4) thus confirming the enhanced catalytic activity of SZ catalysts.
 | ||
| Fig. 5 Esterification of myristic acid with different alcohols over the SZ-600 catalyst. Reaction conditions: acid:methanol = 1:10; SZ = 0.5 wt%. | ||
A similar trend was found with caprylic acid26b and also has been reported for SZ catalyzed transesterification of triglycerides10 and esterification of carboxylic acids of different chain lengths with methanol using sulfuric acid and nafion–silica composite33 suggesting that the lower reaction rates are due to steric hindrance effects of the larger alkyl chains either of alcohols or acids. However, in the present study the reduced conversion of myristic acid with propanol and butanol was enhanced by increasing the reaction temperature to 90 °C. The results clearly envisage that methanol having a small carbon chain is more economical for the esterification of fatty acid owing to its lower boiling point that consumes lower thermal energy and also in terms of comparatively inexpensive. The selectivity for alkyl myristate was 100% irrespective of alcohol and temperature. As excess of alcohol may lead to dehydration or etherification in the presence of acid catalyst, the selectivity of ester was further confirmed by checking the formation of side products during the reaction of methanol with a SZ-600 catalyst at 60 °C for 5–10 h. No dehydrated or ether product was detected by GC analysis under the conditions studied.
2.3 Re-usability of SZ catalysts
The used SZ-600 and SZ-650 catalysts were recovered from the reaction mixture, washed with methanol, dried and activated at 450 °C for 2 h before their re-use for further reaction cycle under the similar reaction conditions (A).The SZ-600 catalyst exhibited similar activity for myristic acid esterification till five reaction cycles without showing any significant decrease in the conversion (only a slight decrease to 94%). However, the activity of SZ-650 was decreased to 86% after one re-cycle, which further decreased (71%) after five reaction cycles (Fig. 6). The sulfur content of the re-activated SZ-600 and SZ-650 catalysts after five reaction cycles was decreased; 1.84 wt% and 0.604 wt%, respectively, compared to fresh catalysts. However, IR spectra of re-activated catalysts were found to be similar to the fresh ones (Fig. 7). Furthermore, the absence of any adsorbed organic species in the IR spectra of re-activated SZ-600 and SZ-650 catalysts indicated that the deactivation of the catalyst did not occur due to the deposition of the product molecules on the active acid sites of the catalyst.
 | ||
| Fig. 6 Re-usability of SZ-600 and SZ-650 catalysts after different runs. | ||
 | ||
| Fig. 7 IR spectra of (i) fresh and (ii) re-activated (after 5 cycles) (a) SZ-600 and (b) SZ-650 catalysts. | ||
We tried to find out the possible reasons for decreased activity of the SZ-650 catalyst as deactivation of a SZ catalyst is supposed to be its major disadvantage though having strong super acidity.
2.4 Deactivation of SZ catalysts
The successive decrease in the activity of SZ catalysts may be due to (i) deactivation of active acid sites by water molecules formed during the esterification reaction or/and (ii) leaching of SO42− species from the catalyst in polar alcohol medium during the course of reaction. Both possibilities have been debated in the following sections.The effect of water was also studied by adding a small amount of water (2000 ppm) to the reaction mixture. The reaction showed lower conversion (90–91%) of myristic acid after 5–7 h (Fig. 8). Water has higher affinity to interact with the active acid sites and thus lowers the interaction of methanol with active sites resulting in a decrease in the conversion of acid. A similar retarding effect of water has been observed in homogeneous acid catalyzed esterification of FAs and also base catalyzed transesterification of triglycerides, however, acid catalyzed esterification is more tolerant to water compared to the latter.34 Kiss et al.35 have successfully used a reactive distillation process to remove the water, however, the reaction was done at higher temperature (130–160 °C). The results indicated the sensitivity of the esterification reaction of FAs towards the water molecules present in the reaction system.
 | ||
| Fig. 8 Effect of addition of water on the conversion of myristic acid with methanol. Reaction conditions: acid:methanol = 1:10; SZ = 0.5 wt% | ||
We have studied the leaching of SO42− ions of the catalyst by employing two methods. (i) In one experiment, it was tested during the esterification reaction. After 10 min of the reaction, the catalyst was removed from the reaction mixture; and the reaction was continued with the remaining solution without having any catalyst. The plot of concentration of myristic acid with time (Fig. 9) clearly showed an insignificant decrease in concentration of myristic acid after removal of the catalyst as compared to a linear decrease in myristic acid concentration in the presence of catalyst. The autocatalysis (a blank test in the absence of catalyst), under the similar reaction conditions, showed nil conversion till 45 min and increased from 2.99% (at 60 min) to 4.87% after 120 min (19% after 7 h, not shown in figure). The initial rate of autocatalysis was significantly lower (0.5 μmol min−1) as compared to the catalytic reaction (12.9 μmol min−1). The rate after removal of the catalyst (2.4 μmol min−1) was observed to be five times lower than the reaction done in the presence of catalyst. These results showed that no significant leaching of SO42− ions of SZ catalysts occur under the experimental conditions studied.
 | ||
| Fig. 9 Concentration of myristic acid (★) without catalyst, (○) catalyst removed after 10 min and (▲) with catalyst. Reaction conditions: acid:methanol = 1:10; SZ = 0.5 wt% | ||
(ii) In a second experiment, a fresh SZ-600 catalyst was stirred in methanol and distilled water separately (25 ml of each) and pH was measured (Toshniwal, CL54) during 24 h. The pH of the reaction mixture was found to decrease significantly after 1 h of stirring both in methanol and water, however, it was not much affected further till 24 h (Fig. S6, ESI†). Initially, the pH of the reaction mixture was higher in water (4.3) compared to methanol (2.7), however, the decrease in pH was observed in a similar range (38.4% in water and 38.6% in methanol) after 1 h of stirring.
It appeared from the decrease in pH by treatment with water or methanol that the sulfate groups may be hydrolyzed in the presence of –OH groups on the catalyst surface9 or the sulfate groups may be leached out during the initial first hour,10 which may not be strongly bonded with the zirconia surface; however strongly bonded sulfate groups did not leach out till further 24 h of stirring. To further confirm this, the SZ-700 catalyst having less sulfur content and highly crystalline nature was checked for the same study, which also showed a decrease in pH from 4.74 to 3.63 during the first hour of stirring in water. The pure ZrO2 sample having no sulfate species also showed a decrease in pH from 5.76 to 4.88–5.04 during 1–2 hours of stirring in water, though it was lower compared to SZ. Therefore, the decrease in pH (as also reported by others) does not seem to be purely due to the leaching of sulfate species; it may be due to the hydrolysis of sulfate species and also due to the acidic metal oxide matrix.
2.5 Reaction rate
The kinetic profile of esterification of myristic acid with fresh methanol (Fig. 10a) showed the continuous decrease in concentration of myristic acid with time. The myristic acid concentration [CA] was calculated by subtracting the consumed concentration from the initial concentration of myristic acid. A fast linear decrease in myristic acid concentration was observed till 60 min. The reaction rate (ν) was determined by applying linear analysis to the plot of decreasing [CA] with time by the equation, ν = −d[CA]/dt. The initial rate was 12.9 μmol min−1. Turnover frequency (TOF, moles of fatty acid converted per mole of acid site concentration per hour) at 100% conversion was 4.44 per hour. | ||
| Fig. 10 Esterification of myristic acid with fresh methanol (a) conversion/concentration of myristic acid with time (b) apparent induction period. Reaction conditions: acid:methanol = 1:10; SZ = 0.5 wt% | ||
The kinetic experiments with <10 min reaction time were performed to have a view on apparent induction period of the reaction. The kinetic profile in Fig. 10b shows the conversion of myristic acid within 1 min of the reaction, a fast decrease in [CA] between 1 and 2 min, which extended to 4 min and after that it became slow. It is to be noted that while performing the reaction, the catalyst was added after mixing the acid and methanol by stirring at ambient temperature. This time was taken as zero time. After that the reaction mixture was heated at the desired temperature (60 °C), which was already maintained at 60 °C (to avoid the shooting of temperature (∼4–5 °C) beyond 60 °C that normally occurs during the heating of the system). This was kept in mind because of low boiling point of methanol (64–65 °C). Thus, the apparent induction period of the studied reaction is just the heating of the system; it will be slightly extended during the heating of the system to the desired temperature. The initial reaction rate of this period was determined to be significantly higher (38.8 μmol min−1).
2.6 Comparison of catalytic performance with other acids
Based on literature reports9,11–13,36 we have focused on SZ in the present study. However, we have also carried out the reaction with concentrated H2SO4 and heterogeneous ion exchange resin Amberlyst-15 to have a comparable view on the catalytic activity. The results (Fig. S7, ESI†) showed the activity in the order of H2SO4 > SZ > Amberlyst-15. Concentrated H2SO4, used as a benchmark catalyst, is not eco-friendly and cannot be readily re-cycled. To overcome the disadvantages associated with liquid acid catalysts, heterogeneous catalysts are the sustainable alternatives, though having lower catalytic performance. The initial rate of esterification reaction was slow with SZ catalysts (12.9 μmol min−1) as compared to conc. H2SO4 (14–15 μmol min−1), the final conversion after 2 h was in a comparable range. These results clearly showed the potential of SZ solid acid catalyst near to the benchmark catalyst. Amberlyst-15 showed significantly lower activity (5–6 μmol min−1) under the reaction conditions studied and was comparable with esterification of oleic acid with methanol at 80 °C.38A comparison of catalytic activity of SZ used in the present study with reported studies of SZ or other solid acid catalysts gave fairly good performance (716 μmol min−1 g−1) as compared to reported SZ (125 μmol min−1 g−1) and SZ incorporated on SBA-15 (175 μmol min−1 g−1) for methyl palmate and laurate formation at 68 °C.39 TOF of the present reaction was also found to be higher (4.44 per hour) as compared to SZ (1.9–2.5 per hour) and SZ incorporated on SBA-15 (2.6–3.4 per hour) for methyl palmate and laurate (calculated from data given in ref. 39). Similarly, the activity was observed to be higher (716 μmol min−1 g−1) as compared to other SZ (150 μmol min−1 g−1) and sugar catalyst (478 μmol min−1 g−1) for methyl oleate formation at 80 °C.38 The conversion rates were also observed in a similar range for esterification of lauric acid with 2-ethyl hexanol by the SZ (SSZr-1) catalyst, studied at 170 °C with 10 wt% of catalyst.8 The authors reported similar results for esterification of lauric and myristic acids with iso-propanol. TOF values of the reported reactions may be higher. A high reaction rate for esterification of myristic acid with methanol was observed over SZ18 (75–85% conversion in 5 min) at 22 bar in a Parr reactor at 120–170 °C. The high temperature and pressure are important factors to significantly enhance the reaction rates besides the catalyst properties. Therefore, a proper comparison of catalytic activity, though normalized with acid site concentration, can only be viable when the reactions have been performed at comparable reaction parameters such as temperature, catalyst amount and acid to alcohol ratio9,18,40,41 along with the carbon chain length of alcohol8,20,41 and carboxylic33/fatty acid.19,20
2.7 Structure–activity co-relation
The co-relation between acidic features in terms of total surface acidity, Brönsted acidity, Brönsted acid site concentration, B/L ratio and the activity of all three SZ catalysts reveals that esterification of myristic acid with short chain alcohols is mainly Brönsted acid catalyzed reaction. The activity decreases with a decrease in Brönsted acidity in either terms. Furthermore, a SZ catalyst with lower number of weak and moderate acid sites is also able to demonstrate the esterification activity similar to a SZ catalyst having comparatively higher number of strong acid sites; however, the latter is requisite to retain the original activity of re-used SZ catalysts along with a higher B/L ratio. The difference in the activity of re-used catalysts clearly revealed that re-usability of SZ catalysts strongly depends on the acidity of the catalysts.3. Experimental
3.1 Materials
Zirconium n-propoxide (70 wt% in n-propanol) was procured from Sigma-Aldrich, Germany; n-propanol (99.5%), n-butanol (99.5%), aqueous ammonia solution (25%) and concentrated sulfuric acid (98%) were procured from s.d. Fine chemicals, India. Methanol (99%) was purchased from Ranbaxy, India and Merck, India. Myristic acid (99.5%) and ethanol (99.9%) were procured from Spectrochem Pvt. Ltd., India.3.2 Catalyst synthesis and characterization
A sulfated zirconia catalyst was prepared using a two-step sol-gel technique as described previously.26a In a typical synthesis procedure, Zr(OPr)4 was hydrolyzed by aqueous ammonia and the obtained gel was treated with H2SO4 after oven drying at 120 °C for 12 h. The sulfated powder was calcined at 600–700 °C for 4 h and designated SZ-T, where T represents the calcination temperature.PXRD patterns were obtained using a Philips X'pert diffractometer. The crystallite size was determined from the characteristic peak of tetragonal phase (2θ = 30.22) using the Scherrer equation,42 crystallite size = Kλ/W cosθ, where K is the Scherrer constant (0.9), λ = 1.5406 Å (CuKα radiation), W = Wb − Ws; Wb is the broadened profile width of the experimental sample and Ws is the standard profile width of the reference silicon sample and θ is the angle of diffraction. TEM micrographs were obtained using a JEOL JEM 2100 transmission electron microscope by dispersing the catalyst sample in ethanol by sonication and deposited on a Cu grid coated with carbon film. IR spectra were recorded on a Perkin Elmer GX spectrophotometer. The bulk sulfur present before and after calcination at different temperatures was analyzed by ICP emission spectroscopy using Perkin-Elmer, Optima 2000 DV. BET surface area, pore volume and pore diameter were calculated from nitrogen sorption isotherms after pre-activation at 120 °C for 4 h at −196 °C on ASAP 2010, Micromeritics.
NH3-TPD was used to estimate the total surface acidity, i.e. strength and number of acid sites present in the catalysts using Micromeritics Pulse Chemisorb 2720 as described earlier.26a Vapor phase cyclohexanol dehydration in a fixed bed reactor was used as a model reaction to assess the Brönsted acidity of the catalysts.26b Diffuse Reflectance FT-IR spectroscopy (DRIFT) was used to differentiate between Brönsted and Lewis acidity of pyridine adsorbed SZ samples using Perkin Elmer GX equipped with DRIFT Selector accessory (Graseby Specac, P/N 19900 series).25e The spectra were recorded at room temperature (∼27 °C) to 450 °C after holding at each temperature for 10 min, thus allowing sufficient time for pyridine desorption.
3.3 Catalytic activity
Esterification of myristic acid was carried out in a liquid phase batch reactor.26b In a typical reaction procedure, required amounts of acid, alcohol, and catalyst were taken in a round bottom flask and the suspension was magnetically stirred (600 rpm) in an oil bath maintained at constant temperature (±1 °C) in the temperature range of 40–60 °C. The reactions were performed in duplicate with ±2% variations in conversion. The samples were withdrawn at regular intervals and the reaction product was analyzed using a gas chromatograph (HP 6890) having a DB-225 capillary column (20 m length, 100 μm diameter and 0.10 μm film thickness) and a FID detector.4. Conclusions
The acidity and activity of a SZ catalyst for esterification of myristic acid with methanol and other short chain alcohols are strongly influenced by its calcination temperature along with the reaction parameters mainly acid to alcohol ratio, catalyst concentration and reaction temperature. Among the structural, textural and acidic properties of the catalyst, the acidic features play a major role in its activity and re-usability behaviour for the studied reaction.The present study reveals a very remarkable result that the SZ-catalyst having higher number of Brönsted acid sites could be re-used for five reaction cycles without significant loss in activity for myristic acid esterification, whereas the SZ-catalyst having less number of acid sites showed decrease in activity after five reaction cycles. This explicitly confirms the requirement of more number of Brönsted acid sites for better re-usability of SZ catalysts for esterification of myristic acid.
The present study concludes that the requirement of a very small concentration of the catalyst yielding maximum conversion and selective formation of the ester at lower temperature within reasonable reaction time makes SZ an appealing catalyst for the synthesis of FAAEs. In addition, eco-friendly, recyclable SZ catalysts may find wide applications in reactions where conc. H2SO4 is currently being used as a catalyst.
Acknowledgements
The authors are thankful to CSIR Network Programme on Inorganic Materials for Diverse Application and to Analytical Science discipline for catalyst characterization analysis. The authors are also thankful to Dr Ram S. Shukla for helping in initial reaction rate determination.Notes and references
- J. Beare-Rogers, A. Dieffenbacher and J. V. Holm, Lexicon of lipid nutrition (IUPAC Technical Report), Pure Appl. Chem., 2001, 73, 685–744 CrossRef CAS.
- T. A. Hughes, M. Heimberg, X. Wang, H. Wilcox, S. M. Hughes, E. A. Tolley, D. M. Desiderio and J. T. Dalton, Metabolism, 1996, 45, 1108–1118 CrossRef CAS.
- R. W. Johnson and E. Fritz, in Fatty Acids in Industry: Processes, Properties, Derivatives, Applications, ed. Marcel Dekker, New York, 1988 Search PubMed.
- Commonwealth of Australia Gazette no. APVMA 37, March 2006, p. 13.
- S. Hartwig, V. Auwärter and F. Pragst, Alcohol Alcohol., 2003, 38, 163–167 CAS.
- E. Lotero, Y. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin Jr., Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 111, 285–1308 Search PubMed.
- M. L. Grecea, A. C. Dimian, S. Tanase, V. Subbiah and G. Rothenberg, Catal. Sci. Technol., 2012, 2, 1500–1506 CAS.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Adv. Synth. Catal., 2006, 348, 75–81 CrossRef CAS.
- K. Suwannakarn, E. Lotero, J. G. Goodwin Jr. and C. Lu, J. Catal., 2008, 255, 279–286 CrossRef CAS.
- D. E. López, J. G. Goodwin Jr., D. A. Bruce and S. Furuta, Appl. Catal., A, 2008, 339, 76–83 CrossRef.
- (a) S. Furuta, H. Matsuhashi and K. Arata, Biomass Bioenergy, 2006, 30, 870–873 CrossRef CAS; (b) S. Furuta, H. Matsuhashi and K. Arata, Catal. Commun., 2004, 5, 721–723 CrossRef CAS.
- K. F. Yee, J. C. S. Wu and K. T. Lee, Biomass Bioenergy, 2011, 35, 1739–1746 CrossRef CAS.
- M. Hino, S. Kobayashi and K. Arata, J. Am. Chem. Soc., 1979, 101, 6439–6441 CrossRef CAS.
- B. M. Reddy and M. K. Patil, Chem. Rev., 2009, 109, 2185–2208 CrossRef CAS.
- G. D. Yadav and J. J. Nair, Microporous Mesoporous Mater., 1999, 33, 1–48 CrossRef CAS.
- X. Song and A. Sayari, Catal. Rev. Sci. Eng., 1996, 38, 329–412 CAS.
- D. Rattanaphra, A. P. Harvey, A. Thanapimmetha and P. Srinophakun, Renewable Energy, 2011, 36, 2679–2686 CrossRef CAS.
- X. Hu, Z. Zhou, D. Sun, Y. Wang and Z. Zhang, Catal. Lett., 2009, 133, 90–96 CrossRef CAS.
- K. Srilatha, N. Lingaiah, P. S. S. Prasad, B. L. A. P. Devi, R. B. N. Prasad and S. Venkateswar, Ind. Eng. Chem. Res., 2009, 48, 10816–10819 CrossRef CAS.
- J. C. Juan, J. Zhang and M. A. Yarmo, Catal. Lett., 2008, 126, 319–324 CrossRef CAS.
- T. Yalçinyuva, H. Deligöz, I. Boz and M. A. Gürkaynak, Int. J. Chem. Kinet., 2008, 40, 136–144 CrossRef.
- A. Sakthivel, K. Komura and Y. Sugi, Ind. Eng. Chem. Res., 2008, 47, 2538–2544 CrossRef CAS.
- K. Mantri, K. Komura and Y. Sugi, Green Chem., 2005, 7, 677–682 RSC.
- (a) R. V. Jasra, B. Tyagi and M. K. Mishra, US Pat., 2009 7, 544 831 B2, Aug 2009 Search PubMed; (b) B. Tyagi, M. K. Mishra and R. V. Jasra, J. Mol. Catal. A: Chem., 2010, 317, 41–45 CrossRef CAS; (c) B. Tyagi, M. K. Mishra and R. V. Jasra, J. Mol. Catal. A: Chem., 2009, 301, 67–78 CrossRef CAS; (d) B. Tyagi, M. K. Mishra and R. V. Jasra, Catal. Commun., 2006, 7, 52–57 CrossRef CAS; (e) B. Tyagi, M. K. Mishra and R. V. Jasra, J. Mol. Catal. A: Chem., 2008, 286, 41–46 CrossRef CAS; (f) B. Tyagi, M. K. Mishra and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 276, 47–56 CrossRef CAS.
- (a) K. Saravanan, B. Tyagi and H. C. Bajaj, J. Sol-Gel Sci. Technol., 2012, 61, 275–280 CrossRef CAS; (b) K. Saravanan, B. Tyagi and H. C. Bajaj, J. Sol-Gel Sci. Technol., 2012, 62, 13–17 CrossRef CAS.
- S. J. Gregg and K. S. W. Sing, Adsorption, Surface Area and Porosity, Academic Press, New York, 2nd edn, 1982 Search PubMed.
- T. Yamaguchi, T. Jin and K. Tanabe, J. Phys. Chem., 1986, 90, 3148–3152 CrossRef CAS.
- B. T. Loveless, A. Gyanani and D. S. Muggli, Appl. Catal., B, 2008, 84, 591–597 CrossRef CAS.
- A. L. Cardoso, R. Augusti and M. J. D. Silva, J. Am. Oil Chem. Soc., 2008, 85, 555–560 CrossRef CAS.
- M. Hino, S. Takasaki, S. Furuta, H. Matsuhashi and K. Arata, Catal. Commun., 2006, 7, 162–165 CrossRef CAS.
- S. Ramu, N. Lingaiaha, B. L. A. P. Devi, R. B. N. Prasad, I. Suryanarayanaa and P. S. S. Prasad, Appl. Catal., A, 2004, 276, 163–168 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin Jr., J. Catal., 2006, 243, 221–228 CrossRef CAS.
- D. Kusdiana and S. Saka, Bioresour. Technol., 2004, 91, 289–295 CrossRef CAS.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Energy Fuels, 2008, 22, 598–604 CrossRef CAS.
- F. Omata, A. C. Dimian and A. Bliek, Chem. Eng. Sci., 2003, 58, 3175–3185 CrossRef.
- X. B. Li, K. Nagaoka and J. A. Lercher, J. Catal., 2004, 227, 130–137 CrossRef CAS.
- M. H. Zong, Z. Q. Duan, W. Y. Lou, T. J. Smith and H. Wu, Green Chem., 2007, 9, 434–437 RSC.
- X. R. Chen, Y. H. Ju and C. Y. Mou, J. Phys. Chem. C, 2007, 111, 18731–18737 CAS.
- A. A. Kiss, F. Omota, A. C. Dimian and G. Rothenberg, Top. Catal., 2006, 40, 141–150 CrossRef CAS.
- M. C. de Jong, R. Feijt, E. Zondervan, T. A. Nijhuis and A. B. de Haan, Appl. Catal., A, 2009, 365, 141–147 CrossRef CAS.
- B. D. Cullity and S. R. Stock, Elements of X-ray Diffraction, Prentice Hall: Upper Saddle River, NJ, 3rd edn, 2001, p. 388 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD pattern, TG/DTA, nitrogen sorption isotherms, IR spectra, DRIFT spectra from RT to 450 °C, pH vs. time after stirring the SZ-600 catalyst in water and methanol and comparison with other acid catalysts. See DOI: 10.1039/c2cy20462b |
| This journal is © The Royal Society of Chemistry 2012 |