PVP-stabilized mono- and bimetallic Ru nanoparticles for selective ring opening
Jing
Shen
a,
Xing
Yin
a,
Dimitre
Karpuzov
b and
Natalia
Semagina
*a
aDepartment of Chemical and Materials Engineering, University of Alberta, 9107–116 Str. Edmonton, Alberta T6G 2V4, Canada. E-mail: semagina@ualberta.ca; Fax: +1-(780)-492-2881; Tel: +1-(780)-492-2293
bAlberta Center for Surface Engineering and Science, University of Alberta, Edmonton T6G 2V4, Canada
First published on 7th September 2012
Abstract
Selective ring opening of naphthenic molecules in oil upgrading should result in no loss in molecular weight. Benzocyclopentane (indan) ring opening was studied under hydrogen atmospheric pressure at 609 K over poly-(vinylpyrrolidone)-stabilized Ru, Ir and Pd monometallic and Ru–Ir and Ru–Pd bimetallic nanocatalysts prepared by simultaneous reductions. The particle size (mostly within 2 nm) and their monodispersity were confirmed by transmission electron microscopy, while X-ray photoelectron spectroscopy indicated the bimetallics' alloy structure. Pd catalysts displayed the lowest activity in the ring opening; Ru showed the highest formation of undesired o-xylene and lights. Monometallic Ir displayed the highest activity and selectivity toward 2-ethyltoluene and n-propylbenzene. In bimetallic structures, higher Ir content led to improved catalytic performance. Next to the monometallic Ir catalyst, the newly developed Ru1Ir4/γ-Al2O3 catalyst (with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio of Ru to Ir) displayed superior single cleavage selectivity as well as lower cracking activity compared to industrial Pt–Ir catalysts at a comparable indan conversion. The study can pave the way in the development of Pt-free Ru-containing catalysts with narrow size distributions for selective ring opening, especially taking into consideration a possibility of their higher S-resistance as compared to the Pt catalysts.
:4 molar ratio of Ru to Ir) displayed superior single cleavage selectivity as well as lower cracking activity compared to industrial Pt–Ir catalysts at a comparable indan conversion. The study can pave the way in the development of Pt-free Ru-containing catalysts with narrow size distributions for selective ring opening, especially taking into consideration a possibility of their higher S-resistance as compared to the Pt catalysts.
1. Introduction
Exhaust gas emissions from vehicles and engines contribute to decreased air quality, and lead to negative environmental and health effects. In Canada, since May 2006 the sulfur content of diesel fuel produced or imported has been required to be less than 15 ppm. At the same time, the quality of crude oil has decreased, which poses significant challenges for the refining industry. A catalytic ring opening (RO) of naphthenes, serving as one of the upgrading steps during hydroprocessing of heavy crude oils, is a preferred reaction for improving the cetane number of fuels. During the selective ring opening (SRO), the naphthenic ring is only cleaved once, maintaining the same number of carbon atoms, as contrary to hydrogenolysis and cracking.Platinum group metals are known to selectively catalyze the ring opening of naphthenes. However, the most significant drawback of the Pt catalysts for fuel hydrotreating applications is their very low sulfur tolerance.1 Many studies have focused on improving sulfur tolerance by alloying the active component with another metal. In the 1970s, Exxon introduced Pt–Ir catalysts in reforming units, which were several-fold more active and stable than Pt.2 Ir ensures high RO activity and low coke formation; the addition of Pt tempers the undesirable excessive cracking by Ir, and increases its sulfur tolerance and resistance to agglomeration.3 The conventional Pt–Ir catalysts are prepared using traditional methods via support impregnation/co-precipitation with the Pt and Ir metal precursors. Such methods typically result in polydispersed metal nanoparticles without control over their size or structure, which often consume expensive metals in catalytically unfavorable size and structure modes.
Recent advances in nanotechnology and colloidal chemistry techniques allow the production of metal nanoparticles with controlled size and structures. The use of various stabilizing agents, such as surfactants, polymers, and electrostatic stabilizers, to prepare metallic nanoparticles has been actively exploited in hundreds of research papers and patents. Although the studies have involved the same basic principle of controlled metal nanoparticle formation from molecular precursors in the presence of steric and/or electrostatic protective agents, the final nanoparticle structure and properties are known to depend strongly on the synthesis conditions and metal nature. Typically, the polymer-protected nanoparticles are highly stable for years.3 The preparation of noble metal nanoparticles using “magic polymer” poly-(vinylpyrrolidone) (PVP) is widely described for various nanoparticles with a controlled size.
The importance of size control of metal nanoparticles in catalysis is far from the traditional concept that the decrease of nanoparticle size leading to a higher surface-to-volume ratio leads to the reaction rate increase. On the contrary, there are numerous reactions that occur on quite large nanoparticles (above 3 nm) with much higher rates and selectivities than on smaller particles.4 The reason is in the surface configuration: the proportion of atoms on edges and vertices decreases with the particle size increase, while the proportion of terrace atoms increases.5 Such atoms possess different electronic and geometric properties, affecting the chemisorption strength and mode of reaction substrates. For example, alkyne adsorbs too strongly on the edge atoms of Pd nanoparticles and does not react, while chemisorption on the terrace atoms favors alkyne hydrogenation, so the reaction rates on larger particles with higher proportions of terrace atoms are higher.4 With the increase in size, the reactions demanding large ensembles of atoms as active sites are affected most. The concept of structure-sensitive reactions was introduced in 1960s.6,7 Since then, they have attracted considerable attention, both in fundamental and applied catalytic science. For example, an increase of Pd nanoparticle size from 1.7 to 4.2 nm leads to a 15 fold increase of the turnover frequency in 1-butyne hydrogenation;8 8 nm Pd nanoparticles show 14% higher selectivity to alkenes in alkyne hydrogenations than 2 nm nanoparticles.9 Meanwhile, 2.0 nm Pt particles exhibit 90% selectivity to the RO product n-butylamine vs. 70% selectivity of 1.5 nm particles in the ring opening of pyrrole.10 In the ring opening of methylcyclopentane on Pt, for the particles below 2 nm n-hexane, 2-methylpentane, and 3-methylpentane are formed as 2:2:1, while on larger particles no n-hexane formation took place.11 The brutal rupture corresponds to a cubooctahedron with four atoms on the edge, the smallest structure allowing the formation of a reactive species diadsorbed on two contiguous-edge.12 A study on the methylcyclohexane ring opening over Ir showed that RO selectivity is 5% for 1 nm Ir nanoparticles and 40% for larger particles.13 Different crystal surfaces of Ir show activities in the cyclopropane ring opening that differ by a ratio of up to ten.14 As seen, the fine-tuning of active metal nanoparticles size (i.e., its surface structure) can lead to dramatic changes in the catalytic behavior of the very same metal.
Special attention in the preparation of size-controlled particles is paid to bimetallic systems.15 Toshima et al. have made a great contribution to the synthesis of bimetallic nanoparticles with precisely controlled composition and size using PVP.3,16,17 Theoretically, bimetallic particles may form a random alloy, cluster-in-cluster, core–shell, and inverted core–shell structures with hetero- and homo-bonds.3 Electronic and geometric properties of the surface atoms will be strongly affected by the structure mode and nanoparticle size. Such properties underlie the chemisorption strength and mode of catalytic reaction substrates and deposits (sulfur, coke); thus, by changing precisely the atom position and its surroundings, one can control their catalytic properties on an atomic level.
PVP-stabilized bimetallic nanoparticles as catalysts have recently received special attention for various catalytic reactions. The addition of the second metal may result in improved catalytic activity, selectivity, and stability (sulfur tolerance). Electron and geometric effects as well as the occurrence of mixed sites are claimed to be responsible for synergism between two metallic components.18 As with monometallic particles, the described methods for the nanoparticle preparation originate from well-known colloidal chemistry techniques, but each catalytic application requires its own most optimal combination of metals and nanoparticle structure modes. There are numerous recent reports on improved catalytic activity of bimetallic catalysts synthesized in a controlled environment.3 For example, the enhancement in activity was observed for Au–Pd particles stabilized by PVP as compared to those prepared by impregnation for hydrogenation of toluene and naphthalene in the presence of dibenzothiophene.19 PVP-stabilized Pt–Au nanoparticles exhibited higher activity and were more stable than the monometallic particles in naphthalene hydrogenation.20 Pd–Pt and Pd–Au bimetallic nanoparticles with surfaces fully covered by Pd atoms showed the highest activity, which was greater than Pd nanoparticles themselves.21 It is very important that the addition of the second metal does not simply “add” its catalytic activity but often leads to synergism between the two metals. For example, in selective aerobic oxidation of crotyl alcohol, PVP-stabilized Au–Pd bimetallic nanoparticles with precisely controlled structure, i.e., Pd-rich surfaces, showed significantly enhanced activities and higher selectivity than co-reduced Au–Pd and their monometallic forms.22
Catalytic selective ring opening in heavy oil upgrading has been studied using both single-ring compound models like methylcyclopentane (MCP) and methylcyclohexane, and multiple-ring compound models such as indan, decalin, tetralin, naphthalene, phenanthrene, etc. These compounds were found in diesel after different levels of hydrogenation.23 Among them, the ring opening of MCP over supported metal catalysts has been most extensively studied. In the study on the conversion of MCP over Pt, Ir and Pt–Ir catalysts, researchers have found that Ir/γ-Al2O3 is the most active catalyst, while MoO2 support provides the highest selectivity.24,25 Some noble metal free catalysts, such as Mo, Fe, and molecular sieves, were also found to be active in the MCP conversion.26,27 As shown by Gilson et al., monofunctional acid and metal catalysts were applied in the conversion of methylcyclohexane (MCH), another simple mono-naphthenic molecule and it was found that the monofunctional metal (Ir) catalysts are better suited than acid solids for LCO upgrading in terms of CN improvement.28 The metal–acid balance is the key parameter for optimum performance in the conversion of MCH. This bifunctional catalyst may lead to more effective Ir based catalysts for the RO of other model compounds, such as decalin, tetralin, and alkyl-naphthalenes.29,30
Although many studies used catalytic RO of MCP as the model compound, its structure is not close enough to real industrial feed molecules; thus, its use as a probe molecule to study the heavy oil upgrading can be questioned.28 A multiple-ring compound model, indan (benzocyclopentane), has a structure and properties that are closer to the petroleum real feed molecules than the commonly used MCP.31 The desired ring opening products are 2-ethyltoluene and n-propylbenzene, where the naphthenic ring has been cleaved only once, leaving the molecular weight unchanged. Among them, the formation of n-propylbenzene is preferable from the viewpoint of the cetane number improvement. Due to the consecutive dealkylation, further stripping of hydrocarbon fragments occurs irrevocably and products such as o-xylene, ethylbenzene, toluene, benzene, and lights are formed.31 The ring opening of indan over bimetallic Pt–Ir catalysts at 325 °C and atmospheric pressure was previously studied by Boutonnet et al. Pt–Ir bimetallic catalysts were synthesized from a microemulsion system. Their catalysts showed superior activities for Ir and Pt–Ir bimetallic catalysts as compared to Pt catalyst. Although Pt–Ir catalysts showed fast deactivation at atmospheric pressure, a better operating stability with no deactivation at high pressure (40 bar) was obtained. The indan conversion increased as the Ir content increased in the bimetallic catalysts.32 Later, Boutonnet et al. also studied the RO of indan over Pt–Ir bimetallic catalysts supported on seven different materials, and found that 2 wt% Pt5Ir95/CeO2 is the best catalyst for selective RO of indan.31 However, no size or structure control was achieved, as the catalysts were prepared via traditional impregnation of the supports with metal precursors followed by reduction.
RO reactions of model compounds with two fused rings, such as decalin, have been mostly reported for acid catalysts, mainly zeolites, and follow the mechanism comprising protolytic dehydrogenation, skeletal isomerization, β-scission, and hydride transfer. The resultant products over acid catalysts are branched and low in molecular weight because of excessive cracking. Thus, these are not potential catalysts for selective ring opening and cetane number improvement.28,33,34 Murzin et al. also summarized the drawbacks of monofunctional catalysts. High Brønsted acidity favored excessively cracking reactions regardless of the zeolite structure.35 The zeolites were modified with noble metals like Pt and Ir; the interactions between Pt and Brønsted acid sites reduced the strength of the Brønsted acid and less cracking products were formed.36 Later, they reported that the high selectivity to RO products could be obtained only on mildly acidic Ir-modified zeolite catalyst. The importance of the metal–acid balance was also stressed by Daage et al. in the study of a variety of model compounds ring opening over metal catalysts deposited on the supports with different acidity. Ir was also reported as the most active and selective catalyst to cleave unsubstituted bonds, while Pt was able to break substituted C–C ring bonds.37
In terms of the cetane number CN improvement for diesel, only selective RO at substituted C–C bonds will lead to the CN increase. When unsubstituted bonds are cleaved, highly branched molecules are produced, which increase octane number ON.38 As seen, six-member rings of single- or multiple-ring compounds are more difficult to rupture than a five-member ring. Acid function is required to isomerise the six-member structures to five-member rings (ring shrinkage) before opening; and then either acid or metal catalyst can open the five-member rings. However, acid catalysts always favor excessive cracking reactions and are not good options for selective ring opening.38,39 In the 1980's Gault has summarized that RO reactions could be proceeded over metal catalysts via three different mechanisms: dicarbene, π-adsorbed olefin, and metallocyclobutane reaction paths. Dicarbene reaction path results in the cleavage of unsubstituted secondary–secondary C–C bonds; thus, producing highly branched isoparaffins with low CNs (high ONs). Both π-adsorbed olefin and metallacyclobutane reaction pathways result in C–C cleavage at substituted positions; therefore, the latter two paths are desirable for enhancing CNs, as the main consequence of cleaving a substituted C–C bond is the elimination of molecular branching.11 Metal nature, nanoparticle size and support could affect the ring opening path. For example Gault et al. have reported that highly dispersed Pt catalysts favored C–C bond cleavage via a π-adsorbed olefin, whereas Pt catalysts of low dispersion follow the dicarbene path.11 π-adsorbed olefin mode requires flat adsorption of three neighboring atoms, while dicarbene mode requires metal–carbon bonding on two contiguous metal atoms, with the molecule adsorbed perpendicular to the surface.23 Ir catalyst did not show the same size effect as Pt catalyst did, but strong support effect has been reported by Resasco et al. When Ir was supported on SiO2, the reaction followed the dicarbene mode and resulted in low CN; whereas on Ir/Al2O3 catalysts, the preferred reaction path was metallocyclobutane mode that cleaves C–C bonds at substituted positions for products of high CN.38 Moreover, the addition of a second metal could also lead to different RO mechanisms than the existing metal. Resasco's group has found that both Ni and K are potential promoters to inhibit the secondary hydrogenolysis on Ir/Al2O3 catalysts.33 A variety of factors affect the ring opening positions, i.e., substituted C–C bonds (high CNs) or unsubstituted C–C bonds (high ONs), such as the type of catalyst used (acid or metal or bifunctional), metal particle size, type of support, as well as the model compound that has been studied.28,38
A variety of transition metals were evaluated in metal-catalyzed RO reactions. As an example, one of the previous studies used 11 different metals to test the ring opening of MCP. Among the monometallic catalysts tested, Ir was the most active and selective catalyst.40 In order to obtain bimetallic catalysts leading to selective RO, Marécot et al. studied the RO of MCP over several Pt-based bimetallic catalysts. Although Pt was the least active catalyst for the RO of MCP as compared to other monometallic catalysts, Pt–Rh bimetallic catalyst allowed increasing RO activity and selectivity, which are similar to those of Ir catalysts.41 In another example, Ru–Pt catalyst with Ru excess on the surface resulted in a synergism factor of 4 in a ring opening reaction.42 Moreover, catalytic activity also depends on the metal composition of the bimetallic nanoparticles. It has been found that Pt(core)Pd(shell) bimetallic catalyst with a Pt/Pd molar ratio of 1/4 was the most active catalyst for the hydrogenation of 1,3-cyclooctadiene to cyclooctene.16 From the sulfur resistance point of view, greater resistance of Pt–Pd bimetallic catalyst (as compared to their monometallic counterparts) to poisoning by sulfur compounds was confirmed by the study involving the hydrogenation of toluene and naphthalene in the presence of 1200 ppm dibenzothiophene.43
This study is focused on the development of Ru-containing catalysts without expensive and unstable Pt component found in industrial catalysts for SRO. The special interest in ruthenium stems from the fact that RuS2 is the most active hydrodesulfurization catalyst44 (an order-of-magnitude more active than conventional sulfided Mo), so its high activity in the sulfur feed is anticipated. The current study is a preliminary investigation of Ru ring opening activity and selectivity in hydrogenolysis of model compound indan in a sulfur-free feed aiming to get insight into Ru SRO performance, as well as to find some bimetallic compositions with improved catalytic properties. The main distinctive feature of our work as compared to other studies on Ru performance in ring opening reactions42 is the precise control over the nanoparticles' size using a PVP stabilizer. Contrary to the polydispersed samples with mean sizes, as reported in the literature, the ability to produce and evaluate monodispersed nanoparticles may provide unique information on the performance of Ru-containing nanoparticles of a definite size, which is especially important for structure-sensitive reactions. As follows from the literature review above, particle size no less than 1 nm is preferable to ensure high selectivity. We also evaluated Ir and Pd as co-metals in bimetallic compositions with Ru, as Ir is known to have high RO activity and Pd is an excellent hydrogenation catalyst. The majority of the reported catalysts are within a 2 nm range with narrow size distributions; thus, their catalytic behavior can be directly correlated to the metal nature without any size effects. The study may pave the way to the development of Pt-free Ru-based catalysts with high stability in sulfur feed for ultra-deep hydrodesulphurization.
2. Experimental section
2.1. Materials
Ruthenium(III) nitrosylnitrate (Ru(NO)(NO3)3, Alfa Aesar), hydrogen hexachloroiridate(IV) hydrate (H2IrCl6, 99.98%, Sigma-Aldrich), palladium(II) chloride solution (PdCl2, 5% w/v, Acros), poly-(vinylpyrrolidone) (PVP) (MW: 10000 and 40000, Sigma-Aldrich), ethylene glycol (EG, 99.8%, Sigma-Aldrich), gamma aluminum oxide (γ-Al2O3, 150 mesh 58 Å pore size, Sigma-Aldrich), reagent alcohol (ethanol, 95 vol.%, Fisher Scientific), and acetone (99.7%, Fisher Scientific) were used as received. Argon and hydrogen of ultra-high purity 5.0 (99.999%) were purchased from Praxair. MilliQ water was used throughout the work. Pt–Ir/Al2O3 is an industrial catalyst; due to data confidentiality, no physico-chemical characteristics are presented for this catalyst, apart from its catalytic activity and selectivity.
2.2. Preparation of catalysts
000) were dissolved in 100 mL EG in a 250 mL 3-neck flask under stirring. The concentration of metal ions was 1.12 mM, and the molar ratio of PVP/metal was 10/1. The mixture was stirred and refluxed under air. The reduction temperature was increased from room temperature to the reflux point of ethylene glycol (198 °C), and then maintained at 198 °C for 3 hours. After the reactions, transparent dark-brown colloidal dispersions of Ru nanoparticles were obtained without any precipitates.
PVP-stabilized Pd nanoparticles were synthesized by Miyake's one-step alcohol (ethanol/water system) reducing method.46 To synthesize PVP-stabilized Pd nanoparticles, 50 mL of 2.0 mM PdCl2 aqueous solution was prepared by diluting PdCl2 solution (5% w/v) with milliQ water (resistivity 18.2 MΩ cm). A mixture of 50 mL 2.0 mM PdCl2 solution, 167 mL ethanol/water solution ([ethanol] = 40 vol.%), and 0.111 g of PVP (MW 40000) (molar ratio of PVP/Pd = 10) was stirred and refluxed in a 250 mL 3-neck round bottom flask for 3 hours under air. Transparent dark-brown colloidal dispersions of Pd metal nanoparticles were obtained without any precipitate.
The size control of PVP-stabilized Ir nanoparticles has been barely studied. Therefore, Miyake's alcohol reduction method for Pd nanoparticles (as described above) was applied to synthesize PVP-stabilized Ir nanoparticles.46 In order to get a better understanding of the size control of bimetallic Ru–Ir and Ru–Pd nanoparticles, monometallic Ir and Pd nanoparticles were also prepared by Li's EG reduction method used for Ru nanoparticles as described above, which would be applied for the syntheses of Ru-based bimetallic nanoparticles later.
2.3. Colloids and catalyst characterization
2.4. Low-pressure ring opening of indan
600) was used to avoid coke formation. The 99.999% pure Ar and H2 flows were controlled by calibrated mass flow controllers (Sierra Instruments). The catalytic reactions were carried out at an internal temperature of 336 °C and atmospheric pressure. Selective ring opening was studied at low pressure, as low pressure promotes the RO route vs. the hydrogenation route at high pressure (40 bar).31 The reactor up- and down- streamlines were heated to 220 °C to preheat reactants and avoid product condensation. The outgoing stream was analyzed online with a Varian 430-GC-FID every 30 minutes after the reaction was started. The GC capillary column is a WCOT fused silica column, 50 m length × 0.32 mm inside diameter × 1.2 μm thickness. Initially, the oven temperature was stabilized at 40 °C for 2.5 min; and then it started to increase at a rate of 30 °C min−1 until the temperature reached 110 °C and maintained at 110 °C for 20 min. FID and injector temperature were 280 °C. The split ratio was 1. Helium flow rate was constant at 25 mL min−1.
The reactant (indan and impurities), major reaction products, and by-products were confirmed by GC-MS. GC-MS was performed with an Agilent Technologies 7890 GC coupled with 5975C MSD. The GC column used is a ZB-50 (Phenomenex) column, 30 m length ×25 mm i.d. ×25 μm thickness. Oven temperature was stabilized at 40 °C for 0.5 min, then increased to 110 °C at 30 °C min−1, and then increased to 280 °C at 50 °C min−1.
For the online product analysis, after steady state was achieved (at 150 min time on stream), no more than 5% deviation in the mass balance was observed (typically, within 2%) as compared to the mass flow of incoming indan. Raw GC results were corrected for indan impurities, and all calculations for indan conversion and product selectivities were based on the corrected GC results. Indan purified by distillation contains 0.09% benzene, 0.15% n-propylbenzene, 0.02% 2-ethyltoluene, 0.02% lights, and 2.91% other.
Selectivities are reported on a mass basis as molar selectivity can give a distorted picture of indan utilization because up to 9 moles of methane may be produced per mole of indan. The FID detector is a mass-sensitive analyzer that responds to the number of carbon atoms entering per unit of time; thus, for each RO opening product, the selectivity was determined as the GC area of each product dividing by the difference between total GC area and the GC area of indan. For most of the catalysts, 8 (or 6) data points were obtained at 150, 180, 210 (and 240) minutes of time on stream with a duplicate experiment (unless stated otherwise). One standard deviation never exceeded 10% and was typically within 3%. Within the indicated times on stream the catalysts did not show noticeable deactivation.
3. Results and discussions
3.1. Catalyst synthesis and characterization
Monometallic Ru, Ir, Pd and bimetallic Ru–Ir and Ru–Pd nanoparticles were synthesized by the ethylene glycol reduction method at the same reduction conditions in the presence of PVP. Ir and Pd nanoparticles were also prepared by the alcohol reduction method. Nanoparticles sizes and size distributions were determined by TEM; the formation of metal clusters was confirmed by UV-visible spectroscopy; the formation of the Ru–Ir and Ru–Pd bimetallic structures was confirmed by XPS, while with ISS the presence of both metals was confirmed at the bimetallic nanoparticle surface; and the loadings of supported nanocatalysts were determined by either AAS or NAA.Fig. 1 shows TEM images of monodispersed PVP-stabilized Ru, Ir and Pd nanoparticles with mean diameters of 2.3 nm, 1.6 nm, and 2.3 nm, respectively. With the applied synthesis methods, all three kinds of monometallic nanoparticles show narrow size distributions. TEM images of supported nanocatalysts also show that Ru, Ir, and Pd nanoparticles were dispersed evenly on γ-Al2O3 (Fig. 2(a)–(c)). Fresh and used Ru and Ir catalysts (after RO reaction for 4 hours on stream at 336 °C) showed no significant sintering, but sintering could be observed on portions of the used Pd catalysts (Fig. 2(d)–(f)).
 | ||
| Fig. 1 TEM images of PVP-stabilized monometallic Ru, Ir and Pd colloids and corresponding size distribution histograms: (a) Ru (EG), (b) Ir (ethanol/water) and (c) Pd (ethanol/water). | ||
 | ||
| Fig. 2 TEM images of PVP-stabilized (a) Ru/γ-Al2O3, (b) Ir/γ-Al2O3, (c) Pd/γ-Al2O3 catalysts; and (d) Ru/γ-Al2O3 after RO reaction, (e) Ir/γ-Al2O3 after RO reaction, and (f) Pd/γ-Al2O3 after RO reaction (336 °C, 6 hours on stream). | ||
Fig. 3 shows TEM images of PVP-stabilized Ru–Ir and Ru–Pd bimetallic nanoparticles and corresponding size distribution histograms. With the same synthesis conditions, an average mean diameter of 2.0 nm was obtained among all Ru–Ir bimetallic nanoparticles (Fig. 3 (a)–(c)). This result is consistent with the sizes of monometallic Ru and Ir nanoparticles, which range from 1.6 nm to 2.3 nm. For Ru–Pd bimetallic nanoparticles, the particle size was affected significantly by the molar ratio of the two metals, i.e., with the same synthesis conditions (except Ru1Pd2 synthesized at 160 °C) the more the Pd in the bimetallic structure, the larger the mean diameter of nanoparticles (Fig. 3 (d) and (e)). Fig. 3 also shows that the mean diameter of Ru1Pd2 is 5.3 nm; such a large particle size could be due to both a higher Pd ratio in the bimetallic structure and a lower reduction temperature (160 °C). Unlike Ru, monometallic Pd nanoparticles synthesized under the same experimental conditions as Ru–Pd bimetallic nanoparticles (EG reduction) have a relatively large particle size of 7.1 nm. Particles sizes of PVP-stabilized Ru–Pd bimetallic nanoparticles show consistency with their monoforms, i.e., all bimetallic nanoparticles are in the range of 2.3 nm (Ru) and 7.1 nm (Pd).
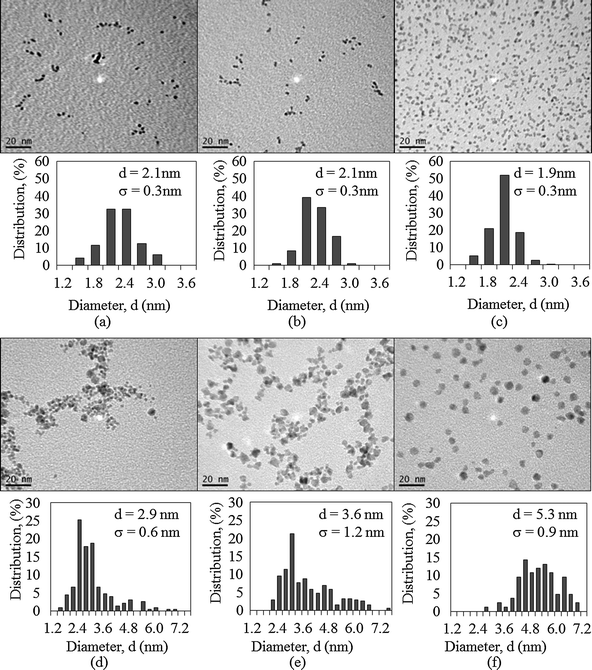 | ||
| Fig. 3 TEM images of PVP-stabilized bimetallic Ru–Ir and Ru–Pd colloids (the scale bar is 20 nm) and corresponding size distribution histograms: (a) Ru4Ir1, (b) Ru2Ir1, (c) Ru1Ir4, (d) Ru4Pd1, (e) Ru1Pd1, and (f) Ru1Pd2. | ||
The formation of Ru–Ir and Ru–Pd nanoparticles was confirmed by UV-vis spectroscopy. The solutions containing metal ions turned from yellow to dark brown after reflux was achieved. After a 3 hour reduction, the absorbance from ultraviolet to visible region increases and the peaks corresponding to metal ions disappear, as shown in Fig. 4. All these are evidence of the formation of PVP-stabilized Ru–Ir and Ru–Pd nanoparticles.46,48
 | ||
| Fig. 4 UV-vis spectroscopy of PVP-stabilized Ru–Ir and Ru–Pd nanoparticles and the precursors, Ru(NO)(NO3)3, H2IrCl6 and PdCl2. | ||
Metal loadings for the synthesized supported catalysts determined by either AAS or NAA are shown in Table 1 (NAA was applied as Ir cannot be dissolved in aqua regia). The target loading was 0.3 wt%. Lower than desired loadings are most likely due to the incomplete reduction of metal precursors that were removed during the catalyst washing procedure.
Prior to the catalytic reactions, the active sites of nanoparticles must be made available for chemisorption, so the catalysts were subjected to oxidation–hydrogenation pretreatment to remove PVP. According to the XPS results of the pretreated catalysts (Table 2), the mass ratios of metal-to-nitrogen show 93–99% removal of PVP as compared to the ratio used in the synthetic procedures.
| Ru | Ir | Pd | Ru4Ir1 | Ru2Ir1 | Ru1Ir4 | Ru4Pd1 | Ru1Pd1 | Ru1Pd2 | |
|---|---|---|---|---|---|---|---|---|---|
| Ru | 5.9 | — | — | 10.3 | 10.8 | 1.2 | 13.6 | 16.7 | 5.4 |
| Ir | — | 13.5 | — | 6.4 | 7.7 | 17.6 | — | — | — |
| Pd | — | — | 16.6 | — | — | — | 2.2 | 8.3 | 9.3 |
| Al | 40.9 | 36.0 | 30.3 | 35.2 | 34.2 | 34.5 | 35.3 | 29.1 | 33.9 |
| O | 43.7 | 38.5 | 37.0 | 38.0 | 36.2 | 36.7 | 35.5 | 32.0 | 36.2 |
| C | 9.3 | 10.8 | 14.0 | 8.9 | 10.0 | 8.7 | 11.5 | 12.2 | 14.5 |
| Cl | 0.1 | 0.4 | 0.6 | 0.5 | 0.0 | 0.8 | 0.3 | 0.3 | 0.4 |
| N | 0.2 | 0.7 | 1.5 | 0.3 | 0.3 | 0.3 | 0.3 | 0.5 | 0.3 |
| Na | — | — | — | 0.5 | 0.4 | 0.3 | 0.4 | 0.4 | 0.2 |
| F | — | — | — | — | 0.5 | — | 0.8 | 0.6 | 0 |
The binding energies of Ru (3d5/2), Ir (4f7/2), and Pd (3d5/2) for the metals in monometallic colloids were 280.2 eV, 61.1 eV, and 335.4 eV, respectively (Table 3, Fig. 5), and were in good agreement with those for the corresponding pure metals.49 Two other Ru chemical states, RuO2 and RuO3, can be identified from the Ru (3d5/2) binding energy at 281.2 and 283.2 eV (for the monometallic Ru sample), respectively; PdO can be identified from the Pd (3d5/2) binding energy at 336.5 eV (for the monometallic Pd sample). The binding energies of oxidized Ru and Pd show good agreement with literature data as well.50–52 It should be noted that the XPS analyses were performed on the samples exposed to air (room temperature) for about 2 weeks; hence Pd and Ru surface oxidation was possible. Previous study on PVP-Pd nanoparticles showed that Pd0 content decreased from 82% to 25% after 3 months' exposure to the air at room temperature.53
| Catalysts | Binding energies (eV) | Zero-oxidation state | ||||
|---|---|---|---|---|---|---|
| Ru0 (3d5/2) | Ir0 (4f7/2) | Pd0 (3d5/2) | Ru | Ir | Pd | |
| Mono-Ru | 280.2 | 31.5% | ||||
| Mono-Ir | 61.1 | 100% | ||||
| Mono-Pd | 335.4 | 58.6% | ||||
| Ru4Ir1 | 279.8 | 60.5 | 49.7% | 100% | ||
| Ru2Ir1 | 279.9 | 60.8 | 63.1% | 100% | ||
| Ru1Ir4 | 280.0 | 60.7 | 56.0% | 100% | ||
| Ru4Pd1 | 279.8 | 335.3 | 60.1% | 100% | ||
| Ru1Pd1 | 279.7 | 335.1 | 65.3% | 100% | ||
| Ru1Pd2 | 279.8 | 335.1 | 68.9% | 100% | ||
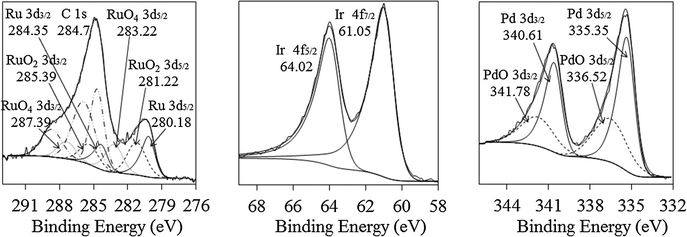 | ||
| Fig. 5 X-ray photoelectron spectra of monometallic Ru, Ir, and Pd nanoparticles supported on γ-A2O3. | ||
In Ru–Ir bimetallic systems the binding energy of Ir0 (4f7/2) shifts to lower values, suggesting electron transfer from Ru to Ir, which correlates with the electron affinity values of both metals: the electron affinity of Ir (112 kJ mol−1) is higher than that of Ru (101 kJ mol−1), indicating electron-accepting properties of Ir. Besides, a higher proportion of Ru is in its zero-oxidation state as compared to mono-Ru of the same ∼2 nm size probably because less Ru is located on the surface of nanoparticles; thus, lower Ru portion is oxidized (Fig. 6 and 7, Table 3).
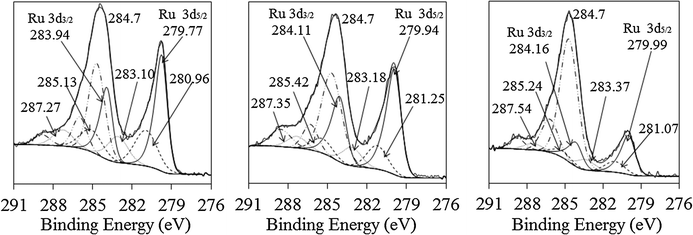 | ||
| Fig. 6 X-ray photoelectron spectra of the Ru 3d peak for three different Ru–Ir bimetallic nanoparticles supported on γ-A2O3 (from left to right: Ru4Ir1, Ru2Ir1, and Ru1Ir4). | ||
 | ||
| Fig. 7 X-ray photoelectron spectra of the Ir 4f peak for three different Ru–Ir bimetallic nanoparticles supported on γ-A2O3 (from left to right: Ru4Ir1, Ru2Ir1, and Ru1Ir4). | ||
When Pd was co-reduced with Ru for the Ru–Pd catalyst preparation, no oxidized Pd was found, as in the case of monometallic Pd nanoparticles, indicating the absence of monometallic nanoparticles in the bimetallic Ru–Pd structures (Fig. 8 and 9 and Table 3). The relative amount of unoxidized Ru increased twice, which could be in part due the larger size of Ru1Pd1 and Ru1Pd2 colloids (3.6 and 5.3 nm, respectively) as compared to monometallic Ru particles (2.3 nm). The electron affinities of Ru and Pd are 101 and 54 kJ mol−1, respectively, indicating the electron transfer from Pd to Ru, which correlates with the XPS data: Ru0 BE is shifted toward lower values when alloyed with Pd. These findings confirm the formation of bimetallic Ru–Ir or Ru–Pd nanoparticles instead of a physical mixture of monometallic Ru and Ir (or Pd) nanoparticles.
 | ||
| Fig. 8 X-ray photoelectron spectra of the Pd 3d peak for three different Ru–Pd bimetallic nanoparticles supported on γ-A2O3 (from left to right: Ru4Pd1, Ru1Pd1, Ru1Pd2). | ||
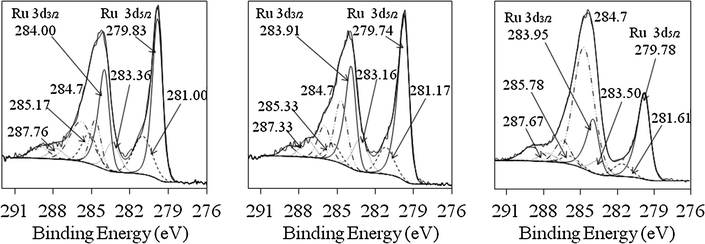 | ||
| Fig. 9 X-ray photoelectron spectra of the Ru 3d peak for three different Ru–Pd bimetallic nanoparticles supported on γ-A2O3 (from left to right: Ru4Pd1, Ru1Pd1, Ru1Pd2). | ||
Ion scattering spectroscopy was performed on the monometallic and bimetallic catalysts and confirmed the presence of both metals in the top surface layer of the bimetallic formulations (Fig. 10). The results quantification is limited by the uncertainty of the inelastic losses and the neutralization rate depending on ion trajectories; however, we estimated the ratios of peak areas under the deconvoluted peaks to roughly assess the abundance of each metal on the nanoparticle surface. For the Ru4Ir1, Ru2Ir1 and Ru1Ir4 catalysts the area ratios were 3/1, 2/1 and 1/1, respectively, indicating the abundance of Ru in the nanoparticle shell, which correlates well with the observed selectivities discussed below. For Ru4Pd1, Ru1Pd1 and Ru1Pd2 catalysts the area ratios were 1/2, 1/1 and 1/1, respectively. These ratios are even less accurate than for Ru–Ir catalysts due to Ru and Pd peaks overlapping, but they still correlate with the observed catalytic activity, when the behavior of Ru4Pd1 catalyst was closer to the Pd catalyst behavior than to that of the monoRu, regardless of the high bulk Ru content: according to the ISS results, the Ru–Pd catalysts are characterized by the Pd abundance in the nanoparticle shell. This is also in line with the XPS results, when the proportion of unoxidized Ru in the bimetallic catalysts decreased as compared to the monometallic Ru.
 | ||
| Fig. 10 Ion scattering spectra of Ru–Ir and Ru–Pd bimetallic, as well as mono-Ru, mono-Pd and mono-Ir nanoparticles supported on γ-A2O3. | ||
3.2. Catalytic behaviour in the ring opening of indan
The comparison of activities and conversions among synthesized catalysts and industrial Pt–Ir catalyst is shown in Fig. 11; Table 4 shows the product selectivities. The desired products are 2-ethyltoluene and n-propylbenzene, when the C5-ring of indan is cleaved only once. In terms of cetane number improvement, n-propylbenzene formation is required. | ||
| Fig. 11 Catalytic activities and conversions vs. time on stream for monometallic and bimetallic catalysts in indan ring opening (336 °C; 1 atm; 1.2 mg of total active metal loading). | ||
| Catalysts | Particle size/nm | Conversion/% | Selectivities/% | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2-Ethyl-toluene | n-Propyl-benzene | Ethyl-benzene | o-Xylene | Benzene | Toluene | Lights | n-Propylbenzene yield, % | |||
| Ru | 2.3 | 51 | 8 | 1 | 3 | 38 | 4 | 20 | 26 | 0.4 |
| Ir | 1.6 | 50 | 63 | 11 | 2 | 13 | 1 | 5 | 5 | 5.7 |
| Pd | 2.3 | 4 | 68 | 11 | 2 | 2 | 1 | 2 | 14 | 0.4 |
| Ru4Ir1 | 2.1 | 7 | 31 | 2 | 3 | 30 | 2 | 12 | 20 | 0.1 |
| Ru2Ir1 | 2.1 | 18 | 26 | 2 | 3 | 39 | 1 | 12 | 17 | 0.3 |
| Ru1Ir4 | 1.9 | 55 | 57 | 5 | 2 | 24 | 0 | 5 | 7 | 2.8 |
| Ru4Pd1 | 2.9 | 9 | 6 | 1 | 2 | 10 | 3 | 7 | 71 | 0.1 |
| Industrial Pt–Ir | n.a. | 62 | 37 | 6 | 4 | 22 | 2 | 16 | 12 | 4 |
Monometallic Ir nanocatalyst with a mean diameter of 1.6 nm is the most active catalyst, it also showed 1.4-fold higher activity than the industrial Pt-containing catalyst. The monometallic Pd catalyst showed almost negligible activity, while Ru took the intermediate position between Ir and Pd. In this study, palladium nanoparticles sintered under the reaction conditions (Fig. 2(f)), which also could be the reason for low catalytic activity. Dieguez et al. reported that deactivation occurred for Pd/Al2O3 catalysts with different palladium contents (thus, nanoparticle sizes) in the hydrogenolysis of MCP.54
The most selective catalysts towards 2-ethyltoluene and n-propylbenzene among the monometallic catalysts are 2.3 nm Pd and 1.6 nm Ir. It should be noted that the high selectivity of Pd is observed at low conversions (Table 4), contrary to ∼50% conversion for the Ir and Ru catalysts; however, other catalysts (Ru4Ir1, Ru4Pd1) showed low selectivities at low conversions. In terms of the light production, Pd takes an intermediate position between Ir and Ru catalysts. Hydrocracking products were also found in the hydrogenolysis of MCP over 1 wt% Pd/Al2O3.55 The iridium catalyst displays low cracking activity, which may be seen by the relatively low selectivities to o-xylene and lights. Boutonnet et al. have reported high selectivity in indan ring opening over 2 wt% Ir/boehmite at steady state.32 Another work has showed that Ir is the most active and selective catalyst in the ring opening of MCP.41 Also, in a study in the ring opening of cyclohexane, 4.6 nm Ir catalyst showed high conversion, high selectivity to desired n-hexane, and low selectivity toward undesired benzene and lights.55
The Ru catalyst promotes dealkylation reactions as evidenced by high selectivity to o-xylene and lights. Previous studies on the hydrogenolysis of cyclohexane over the Ru/Al2O3 catalyst show that Ru was less selective to desired n-hexane formation than Rh, and had a high cracking selectivity.56 Kustov et al. have also reported that the hydrogenolysis of cyclohexane was unselective over 1 wt% Ru/Al2O3 as the reaction temperature reached 280 °C, which led to a 100 wt% yield of light products (C1–C3).57 Moreover, the ring opening of MCP shows that C1–C5 cracking products were favored in the presence of 1.5 wt% Ru/SiO2.41
Although both mono-Ru and mono-Ir resulted in high catalytic activities in the ring opening of indan, the combinations of these two metals are less active (Fig. 10) than their monoforms unless a very high Ir content is present (Ru1Ir4: Ru/Ir molar ratio = 1/4). The higher the Ir content in the alloy structure, the higher the activity of the bimetallic Ru–Ir catalysts. A similar trend is noticed in terms of selectivity (Table 4): the Ru–Ir alloys showed significantly higher selectivity to 2-ethyltoluene and n-propylbenzene than that of the mono-Ru. This improvement is due to the presence of Ir metal in the bimetallic structure, as it tempers the undesirable excessive cracking of Ru. As Ir content decreases in the bimetallic structure, Ru2Ir1 and Ru4Ir1 become less selective to the desired RO products and lead to excessive cracking similar to the monometallic Ru nanocatalyst. Relatively high o-xylene formation by the Ru1Ir4 catalyst is most likely due to the Ru abundance on the nanoparticle surface as detected by ISS. It is important that the activities and selectivities of the Ru–Ir bimetallic catalysts are compared for the same particle size with a narrow size distribution, i.e., ∼2 nm.
Alloying Ru with Pd resulted in lower activities as compared to the monoform of Ru which correlated with low activity of mono-Pd. The activity of the Ru4Pd1 catalyst was closer to the Pd catalyst activity than to that of the monoRu, regardless of the high bulk Ru content: according to the ISS results, the Ru–Pd catalysts are characterized by the Pd abundance in the nanoparticle shell. However, the presence of Ru significantly reduced the selectivity: at similar conversions and particle sizes the Ru4Pd1 catalyst displayed higher selectivity to lights as compared to the monometallic Pd. A similar result has been reported by Marecot's group for Ru–Pt catalysts. In their study, the ring opening of MCP favored the production of cracking products, as Ru added onto a parent Pt/Al2O3 catalyst by a surface redox reaction.41 Notably, in our study, bimetallic Ru–Pd catalysts displayed lower activities and single cleavage selectivities than monometallic Ru and Pd, indicating that the electron transfer between Pd and Ru evidenced by XPS leads to antagonism between these two metals.
In terms of single cleavage selectivity for CN improvement, the formation of n-propylbenzene is favorable, which occurs at substituted positions via π-adsorbed olefin. At similar conversions (50–60%), only monometallic Ir allowed relatively high n-propylbenzene yield (Table 4), as well as Ru–Ir bimetallic particle with the highest Ir content. Ru in its mono and bimetallic forms showed excessive hydrogenolysis to o-xylene and toluene.
Fig. 11 and Table 4 also contain results on the industrial Pt–Ir catalyst. The developed Ir catalyst shows both higher activity and single cleavage selectivity (sum of selectivities to 2-ethyltoluene and n-propylbenzene), as well as higher yield of n-propylbenzene. The bimetallic Ru1Ir4 catalyst exhibits higher single cleavage selectivity at a comparable conversion as the industrial catalyst. Both catalysts do not contain an expensive platinum component, and the Ru1Ir4 catalyst is believed to be a promising alternative to the industrial catalyst, as Ru forms sulfided active sites in the presence of sulfur in the feed,44 contrary to the sulfur-poisoned platinum. The catalyst performance should also be evaluated at high hydrogen pressure, which promotes hydrogenation route31 affecting the resulting cetane number. This study is currently underway.
Based on the catalytic test results, the activity of the synthesized catalysts in indan ring opening (calculated per mole of total active metals) is in the order of Ir > Ru1Ir4 > Ru > Ru2Ir1 > Ru4Pd1 > Ru4Ir1 > Pd > Ru1Pd1 > Ru1Pd2. Comparing only monodispersed catalysts with the sizes of 1.9–2.3 nm and omitting Pd due to its sintering, the trend is Ru1Ir4 > Ru > Ru2Ir1 > Ru4Ir1. The selectivity to single cleavage products within the latter trend is Ru1Ir4 > Ru2Ir1 ≈ Ru4Ir1 ≫ Ru, with the selectivity to lights being the lowest for Ru1Ir4. The importance of these findings is that the activities and selectivities are compared for the same particles size with narrow size distribution, contrary to the polydispersed catalysts typically prepared via the traditional impregnation technique followed by calcination-reduction at various temperatures.
4. Conclusions
Monometallic and bimetallic Ru, Ir, Pd, and Ru–Pd, Ru–Ir nanoparticles were synthesized in the presence of PVP and deposited on γ-Al2O3 for a two-phase indan ring opening at atmospheric H2 pressure and 336 °C. TEM images show monodispersed particles ranging from 2 to 5 nm with narrow size distributions. XPS analysis of mono- and bimetallic structures suggested that bimetallic particles of Ru–Pd and Ru–Ir were formed instead of a physical mixture of monometallic particles; binding energy shifts correlate with the electron affinity of the corresponding metals. ISS analysis confirmed the presence of both metals on the nanoparticle surface in bimetallic formulations. Pd nanoparticles sintered under the reaction conditions, while other catalysts were resistant to agglomeration. High Ir content in both monometallic and bimetallic catalysts was responsible for high activity and selectivity to 2-ethyltoluene and n-propylbenzene, with Ru exhibiting higher selectivity to o-xylene and lights. Pd showed the lowest activity, which was improved by the addition of Ru. The study compares catalytic properties of the monodispersed mono- and bimetallic nanoparticles with similar ∼2 nm sizes. Next to the monometallic Ir, the Ru1Ir4/γ-Al2O3 catalyst has superior indan ring opening selectivity at a lower cost as compared to the industrial Pt–Ir catalyst at a comparable activity. The study can pave the way in the development of Pt-free Ru-containing catalysts for selective ring opening, especially taking into consideration a possibility of their higher S-resistance as compared to the Pt catalysts.Acknowledgements
Financial support was provided by the Centre for Oil Sands Innovation at the University of Alberta.References
- C. Song and A. D. Schmitz, Energy Fuels, 1997, 11, 656 CrossRef CAS.
- J. L. Carter, G. B. McVicker, W. Weissman, W. S. Kmak and J. H. Sinfelt, Appl. Catal., 1982, 3, 327 CrossRef CAS.
- B. Corain, G. Schmid and N. Toshima, Metal nanoclusters in catalysis and materials science: the issue of size control, Elsevier, 2007 Search PubMed.
- A. Molnar, A. Sarkany and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185 CrossRef CAS.
- R. van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189 CrossRef CAS.
- M. Boudart, A. Aldag, J. E. Benson, N. A. Dougharty and C. G. Harkins, J. Catal., 1966, 6, 92 CrossRef CAS.
- M. Che and C. O. Bennett, Adv. Catal., 1989, 36, 55 CrossRef CAS.
- J. P. Boitiaux, J. Cosyns and S. Vasudevan, Appl. Catal., 1983, 6, 41 CrossRef CAS.
- G. D. Zakumbaeva, N. A. Zakarina, V. A. Naidin, A. M. Dostiyarov, N. F. Toktabaeva and E. N. Litvyakova, Kinet. Catal., 1983, 24, 379 Search PubMed.
- J. N. Kuhn, W. Huang, C.-K. Tsung, Y. Zhang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 14026 CrossRef CAS.
- F. G. Gault, Adv. Catal., 1981, 30, 1 CrossRef CAS.
- F. Luck, J. L. Schmitt and G. Maire, React. Kinet. Catal. Lett., 1982, 21, 219 CrossRef CAS.
- C. G. Walter, B. Coq, F. Figueras and M. Boulet, Appl. Catal., A, 1995, 133, 95 CrossRef CAS.
- J. A. Rodriguez and D. W. Goodman, Surf. Sci. Rep., 1991, 14, 1 CrossRef CAS.
- N. Semagina and L. Kiwi-Minsker, Catal. Rev., 2009, 51, 147 CAS.
- N. Toshima, T. Yonezawa and K. Kushihashi, J. Chem. Soc., Faraday Trans., 1993, 89, 2537 RSC.
- P. Lu, T. Teranishi, K. Asakura, M. Miyake and N. Toshima, J. Phys. Chem. B, 1999, 103, 9673 CrossRef CAS.
- B. Coq and F. Figueras, J. Mol. Catal. A: Chem., 2001, 173, 117 CrossRef CAS.
- B. Pawelec, A. Venezia, V. Laparola, E. Canoserrano, J. Camposmartin and J. Fierro, Appl. Surf. Sci., 2005, 242, 380 CrossRef CAS.
- B. Pawelec, A. M. Venezia, V. La Parola, S. Thomas and J. L. G. Fierro, Appl. Catal., A, 2005, 283, 165 CrossRef CAS.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179 RSC.
- T. Balcha, J. R. Strobl, C. Fowler, P. Dash and R. W. J. Scott, ACS Catal., 2011, 1, 425 CrossRef CAS.
- R. C. Santana, P. T. Do, M. Santikunaporn, W. E. Alvarez, J. D. Taylor, E. L. Sughrue and D. E. Resasco, Fuel, 2006, 85, 643–656 CrossRef CAS.
- A. Boulaoued, I. Fechete, B. Donnio, M. Bernard and P. Turek, Microporous Mesoporous Mater., 2012, 155, 131 CrossRef CAS.
- A. Djeddi, I. Fechete and F. Garin, Appl. Catal., A, 2012, 413–414, 340 CrossRef CAS.
- A. Djeddi, I. Fechete and F. Garin, Catal. Commun., 2012, 17, 173 CrossRef CAS.
- I. Fechete, B. Donnio, O. Ersen, T. Dintzer, A. Djeddi and F. Garin, Appl. Surf. Sci., 2012, 257, 2791 CrossRef.
- R. Moraes, K. Thomas, S. Thomas, S. van Donk, G. Grasso, J. P. Gilson and M. Houalla, J. Catal., 2012, 286, 62 CrossRef CAS.
- J. W. Park, K. Thomas, J. van Gestel, J. P. Gilson and C. Collet, Appl. Catal., A, 2010, 388, 37 CrossRef CAS.
- S. Lecarpentier, J. v. G. Gestel, K. Thomas, J. P. Gilson and M. Houalla, J. Catal., 2008, 254, 49 CrossRef CAS.
- U. Nylén, L. Sassu, S. Melis, S. Järås and M. Boutonnet, Appl. Catal., A, 2006, 299, 1 CrossRef.
- U. Nylén, J. F. Delgado, S. Järås and M. Boutonnet, Appl. Catal., A, 2004, 262, 189 CrossRef.
- S. Dokjampa, T. Rirksomboon, D. T. M. Phuong and D. E. Resasco, J. Mol. Catal. A: Chem., 2007, 274, 231 CrossRef CAS.
- D. G. Blackmond, J. G. Goodwin and J. E. Lester, J. Catal., 1982, 78, 34 CrossRef CAS.
- D. Kubicka, M. Kangas, N. Kumar, M. Tiitta, M. Lindblad and D. Y. Murzin, Top. Catal., 2010, 53, 1438 CrossRef CAS.
- D. Kubicka, N. Kumar, P. Maki-Arvela, M. Tiitta, V. Niemi, H. Karhu, T. Salmi and D. Y. Murzin, J. Catal., 2004, 227, 313 CAS.
- G. B. McVicker, M. Daage, M. S. Touvelle, C. W. Hudson, D. P. Klein, W. C. Baird, Jr., B. R. Cook, J. G. Chen, S. Hantzer, D. E. W. Vaughan, E. S. Ellis and O. C. Feeley, J. Catal., 2002, 210, 137 CrossRef CAS.
- P. T. Do, W. E. Alvarez and D. E. Resasco, J. Catal., 2006, 238, 477 CrossRef CAS.
- M. Santikunaporn, J. E. Herrera, S. Jongpatiwut and D. E. Resasco, J. Catal., 2004, 228, 100 CrossRef CAS.
- Z. Paal and P. Tetenyi, Nature, 1977, 267, 19 CrossRef.
- P. Samoila, M. Boutzeloit, C. Especel, F. Epron and P. Marécot, Appl. Catal., A, 2009, 369, 104 CrossRef CAS.
- F. Xu, L. J. Bauer, R. D. Gillespie, M. L. Bricker and S. A. Bradley, US Pat., WO 2007041605, 2007 Search PubMed.
- R. M. Navarro, B. Pawelec, J. M. Trejo, R. Mariscal and J. L. G. Fierro, J. Catal., 2000, 189, 184 CrossRef CAS.
- P. Castillo-Villalon, J. Ramirez and F. Mauge, J. Catal., 2008, 260, 65 CrossRef CAS.
- Y. Chen, K. Y. Liew and J. Li, Mater. Lett., 2008, 62, 1018 CrossRef CAS.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594 CrossRef CAS.
- R. M. Rioux, H. Song, M. Grass, S. Habas, K. Niesz, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, Top. Catal., 2006, 39, 167 CrossRef CAS.
- M. Zawadzki and J. Okal, Mater. Res. Bull., 2008, 43, 3111 CrossRef CAS.
- J. Hrbek, J. Vac. Sci. Technol., A, 1986, A4, 86 Search PubMed.
- C. S. Huang, M. Houalla, D. M. Hercules, C. L. Kibby and L. Petrakis, J. Phys. Chem., 1989, 93, 4540 CrossRef CAS.
- K. S. Kim, A. F. Gossmann and N. Winograd, Anal. Chem., 1974, 46, 197 CrossRef CAS.
- K. S. Kim and N. Winograd, J. Catal., 1974, 35, 66 CrossRef CAS.
- A. Gniewek, A. M. Trzeciak, J. J. Ziolkowski, L. Kepinski, J. Wrzyszcz and W. Tylus, J. Catal., 2005, 229, 332 CrossRef CAS.
- A. B. Gaspar and L. C. Dieguez, Appl. Catal., A, 2000, 201, 241 CrossRef CAS.
- G. R. Gattorno, L. O. A. Vázquez, X. A. Franco, J. L. C. Domínguez and R. V. Ibarra, Energy Fuels, 2007, 21, 1122 CrossRef.
- O. V. Masloboishchikova, T. V. Vasina, E. G. K. Sergeeva, L. M. Kustov and P. Zeuthen, Russ. Chem. Bull., 2002, 51, 249 CrossRef CAS.
- O. V. Masloboishchikova, E. G. K. Sergeeva, V. I. Bogdan, T. V. Vasina and L. M. Kustov, Russ. Chem. Bull., 2006, 55, 656 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |