Integration of heterogeneous catalysts into complex synthetic routes: sequential vs. one-pot reactions in a (Knoevenagel + Mukaiyama–Michael + hydrogenation + transesterification) sequence†
José M.
Fraile
*,
Nuria
García
,
Clara I.
Herrerías
and
José A.
Mayoral
Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) and Instituto Universitario de Catálisis Homogénea (IUCH), C.S.I.C. – Universidad de Zaragoza, E-50009 Zaragoza, Spain. E-mail: jmfraile@unizar.es
First published on 19th September 2012
Abstract
Heterogeneous catalysts of different nature can be combined in one-pot tandem reactions, but the compatibility issues of catalysts with reagents, products, and by-products make more convenient the application of a sequential procedure, in which the catalyst is filtered after each reaction and the crude is used in the following reaction without purification. In this way the recovery and reuse of each catalyst can be optimized to obtain the maximum productivity. The nature of the reactions, and the catalysts involved in them, conditions the maximum number of successive steps in the sequence. In the case of the (Knoevenagel + Mukaiyama–Michael + hydrogenation + transesterification) process tested in this work, the optimal results are obtained when the sequential method is applied to only three reactions, whereas the four reactions sequence shows limitations in the yield of the last transesterification step.
1. Introduction
The substitution of conventional stoichiometric methodologies by catalytic processes, together with the possibility of process intensification by combining several catalytic steps into a one-pot catalytic system,1,2 provides a means to improve the economical and environmental aspects of the chemical processes by minimizing the use of chemicals, the waste production and the processing time.Although it is possible to carry out tandem reactions with a single catalyst, as in the case of tandem coupling reactions with Pd catalysts,3,4 in most cases several different catalysts are needed. Soluble chemical catalysts have been employed in some tandem catalysis processes,5,6 but the site isolation provided by heterogeneous catalysis should avoid the interaction between incompatible sites.7–10 As an additional advantage, heterogeneous catalysts can be easily separated and reused, with the subsequent increase in productivity and operational simplicity. Thus tandem heterogeneous catalysis should constitute a powerful tool in applied chemistry, allowing extremely complex chemical transformations to take place in cleaner and more efficient processes.
In some cases multifunctional catalysts have been prepared and applied in tandem reactions,11,12 but in general the combination of two or more single-site catalysts is simpler. In spite of the interest in this strategy, the examples in the literature are rather scarce, and include one-pot combinations of solid acids and bases,13–17 various types of heterogeneous catalysts,18–21 and biocatalysts with chemical catalysts.22–24 Even if compatibility between catalysts is not an issue, their compatibility with the components of the other reaction (solvent, reagents, concomitant products) may condition the applicability of this methodology. Furthermore, the need for compatibility would be even more difficult in the synthesis of highly functionalized molecules.
In a previous work, we studied the combination of Mukaiyama–Michael addition and hydrogenation reaction with heterogeneous catalysts, a copper-exchanged laponite and Pd/alumina respectively.25 It was shown that both reactions were compatible for a one-pot process, but by-products generated in the hydrogenation reaction were detrimental for recovery of the mixture of solids. In contrast, a sequential procedure that uses the crude of the first reaction in the second one allows the optimization of each catalyst used individually whereas the separation and purification steps are minimized. In our current work, we intend to use heterogeneous catalysts in combinations of more than two reactions that include, in addition to Mukaiyama–Michael and hydrogenation, both the synthesis of the α,β-unsaturated substrate of the Mukaiyama–Michael addition, and the transformation of the lactone products by transesterification with methanol (Scheme 1).
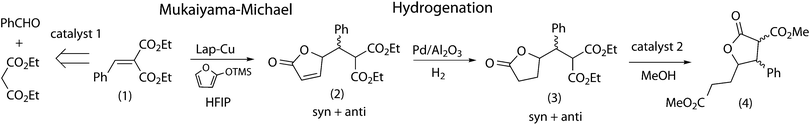 | ||
| Scheme 1 Insertion of the (Mukaiyama–Michael + Hydrogenation) tandem reaction in a longer synthetic pathway with heterogeneous catalysts. | ||
2. Experimental section
1,5,7-Triazabicyclo[4.4.0]dec-5-ene supported on silica (TBD-SiO2) and on poly(styrene-divinylbenzene) (TBD-PS) and Pd/alumina were purchased from Aldrich. Laponite was a generous gift from Rockwood additives and it was dried at 120 °C for at least 12 h before use. Lap-Cu was prepared by cationic exchange of laponite with Cu(OTf)2 in methanol.25 All reagents were also purchased from Aldrich and used without further purification, except benzaldehydes that were distilled under vacuum prior to use. Solvents were dried following conventional procedures.2.1 Preparation of Lap-Mn(salen)
2.2 Catalytic tests
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent. Isolated yield was always over 90%.
:1) as an eluent. Isolated yield was always over 90%.
2.3 Combinations of catalytic reactions
3. Results and discussion
3.1 Heterogeneous catalysis for diethyl benzylidenemalonate synthesis by Knoevenagel condensation
The synthesis of diethyl benzylidenemalonate (1) can be envisaged through Knoevenagel condensation of diethyl malonate with benzaldehyde (Scheme 2), a reaction typically catalyzed by bases. Several heterogeneous catalysts have been described for model Knoevenagel reactions, for example mixed oxides derived from hydrotalcites,26,27 alumina,28 and KF–alumina,29 or for Michael additions in the case of guanidine-like organic bases, such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) supported on mesoporous materials.30 Mixed oxides had been tested in the condensation of cyanoacetate or malononitrile with benzaldehyde, but in our hands Mg/Al31 and Mg/La32 mixed oxides were totally ineffective for condensation of diethyl malonate. Three types of alumina (basic, acidic and neutral) were also tested, with only 16% yield at most in the case of basic alumina at 120 °C, in spite of the excellent results described in the literature with the same substrate.28 These results were not improved by microwaves activation. KF–alumina was also ineffective, even at temperatures as high as 140 °C. With respect to TBD, the two commercially available supported catalysts, on silica and on a polystyrene–divinylbenzene resin, were completely unsuccessful. Ni(NO3)2/SiO2 has recently been described as an efficient catalyst for Knoevenagel reactions,33 but again in our hands very low yields were obtained with diethyl malonate (up to 5%), and in addition an important amount of the dicondensation product was observed (Scheme 2).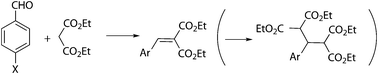 | ||
| Scheme 2 Knoevenagel condensation of diethyl malonate with benzaldehydes. | ||
One of the best catalysts described for this reaction is the MnIII(salen)OAc complex34 (Scheme 3). Our tests using this catalyst with diethyl malonate showed total conversion to the expected diethyl benzylidenemalonate in 24 h. As the homogeneous nature of this catalyst precluded its use in a multistep sequence, it was immobilized onto laponite clay.35 A simple cation exchange in methanol (Scheme 3) led to Lap-Mn(salen) with a functionalization of 0.49 mmol Mn per gram. This catalyst was also efficient in the Knoevenagel condensation but less active (57% yield after 24 h) than the homogeneous complex and it required longer reaction times (3 days) to reach quantitative yield. The efficiency was similar with different para substituted benzaldehydes (Table 1). The catalyst was also recoverable once with similar performance, but the activity was significantly reduced in the third use (Table 1). The IR analysis of the used catalyst (ESI†) shows the presence of additional bands, probably due to the adsorption of reagents or products, and even a chemical modification of the salen ligand cannot be discarded.
| ArCHO | Yield (%) |
|---|---|
| a Reaction conditions: 1 mmol of aldehyde, 1.2 mmol of diethyl malonate, 135 mg of Lap-Mn(salen) (0.066 mmol Mn), 5 mL of toluene under reflux, 72 h. | |
| PhCHO | 100 |
| p-Cl-PhCHO | 100 |
| p-NO2-PhCHO | 98 |
| p-tBu-PhCHO | 95 |
| p-Ph-PhCHO | 90 |
| PhCHO (2nd use) | 92 |
| PhCHO (3rd use) | 54 |
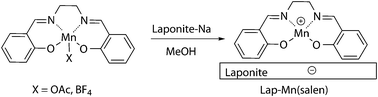 | ||
| Scheme 3 Synthesis of Lap-Mn(salen) from homogeneous (salen)MnX complexes. | ||
One possible explanation for the loss in catalytic activity upon immobilization by cationic exchange may be the change of counterion from acetate to laponite. In order to check this hypothesis the analogous homogeneous complexes with BF4− and PF6− anions were prepared by reaction of MnIII(salen)(OAc) with KBF4 and KPF6 respectively.36 These complexes were poorly active in the Knoevenagel reaction, leading up to only 10% yield of diethyl benzylidenemalonate, as well as MnIII(salen)Cl. On the contrary, when the immobilized catalyst was prepared by exchange of MnIII(salen)(BF4), the solid was nearly as active as that prepared from acetate, showing that the species on laponite is the same irrespective of the anion of the starting complex. The important role of the additional anion/ligand is then evident, which explains the loss of activity upon immobilization. However the acetate itself is not responsible for the catalysis, as different acetate species demonstrated to be almost completely inactive for this reaction.37
The proposed mechanism for this reaction is the abstraction of a proton from malonate by one of the phenoxide groups of the salen ligand. In such a case, any complex of salen with a trivalent metal should be suitable at the same time for immobilization by cationic exchange and catalysis of the Knoevenagel condensation. Complexes of salen with Sc(OTf)3, Cr(NO3)3, and V(acac)3 were prepared and tested in Knoevenagel condensation, but very low yield of diethyl benzylidenemalonate was obtained in all cases. The same happened with MnII(salen), demonstrating that the combination of MnIII, salen and acetate (or laponite) is mandatory to obtain good results.
3.2 Heterogeneous catalysis for transesterification
As in the case of Knoevenagel condensation, many different basic heterogeneous catalysts had been described for transesterification reactions, mainly related with biodiesel production.38 In our case Mg–La mixed oxide and KF–alumina were unable to promote the transesterification under reflux of methanol. Given that supported 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) had been successfully used as a transesterification catalyst in a multistep process,17 silica-supported and polystyrene-divinylbenzene (PS)-supported TBD were tested and both of them showed to be active in the transesterification of the hydrogenated product 3 (Scheme 4). In this reaction all ester and lactone groups underwent transesterification and the final product (4) was a new more substituted lactone obtained by cyclization between the hydroxyl group and one of the starting ester groups with nearly quantitative yield after 24 h. Two diastereomeric products were obtained. The major isomer was prepared by transesterification of the pure major syn-3, whose stereochemistry controls the relative cis position of β and γ substituents, and the relative stereochemistry of α and β substituents was determined to be trans by NMR. It was not possible to determine the relative configuration of the minor product. The behavior of both supported catalysts upon recovery was completely different. TBD-PS was fully recoverable once and deactivated for the third cycle (yields: 100–99–20%), whereas TBD-SiO2 was deactivated after the first reaction. Hence TBD-PS was chosen for compatibility studies and combined reactions.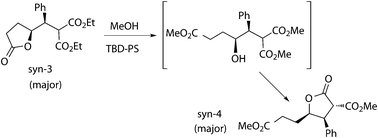 | ||
| Scheme 4 Transesterification of the hydrogenated product (3). | ||
3.3 Combination of Knoevenagel condensation + Mukaiyama–Michael addition
Before combining Mukaiyama–Michael and Knoevenagel condensation, several compatibility studies between both reactions were carried out. Firstly it was checked if the manganese catalyst was able to promote the Mukaiyama–Michael reaction. It was found that this solid did not catalyze the Mukaiyama–Michael addition and thus, Lap-Cu was required to complete the sequence. Another point to be checked was the need for heterogeneous catalysts for this combination of reactions. In fact a mixture of Mn(salen)(OAc) and Cu(OTf)2 was poorly active in Knoevenagel (26% yield) and Mukaiyama–Michael (10% yield), in contrast with the isolated catalysts that are able to reach total conversions. The presence of Cu(OTf)2 also deactivated Lap-Mn(salen) (40% Knoevenagel yield after 4 days) and Mn(salen)(OAc) was able to deactivate Lap-Cu (only 20% Mukaiyama–Michael yield after 24 h).In the compatibility study with the heterogeneous catalysts, a Lap-Mn(salen) catalyzed Knoevenagel condensation was carried out in the presence of Lap-Cu, and the reaction proceeded in the same way as in the absence of the heterogeneous copper catalyst (Scheme 5A). When the Lap-Cu catalyzed Mukaiyama–Michael addition was tested in the presence of Lap-Mn(salen) (Scheme 5B), no negative effect was observed, demonstrating the total compatibility between both heterogeneous catalysts.
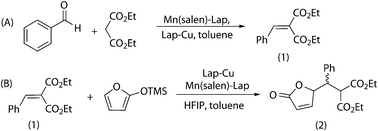 | ||
| Scheme 5 Compatibility between Knoevenagel and Mukaiyama–Michael catalysts. | ||
Apart from the compatibility between catalysts, it was also necessary to check the compatibility between the Mukaiyama–Michael reaction and the reagents of Knoevenagel condensation, since diethyl malonate is generally used in a slight excess over benzaldehyde. When the Mukaiyama–Michael reaction was carried out in the presence of diethyl malonate the yield decreased to 80%, probably due to coordination of this reagent to the copper catalyst (Lewis acid). Therefore, the possibility of carrying out Knoevenagel condensation with only the stoichiometric amount of diethyl malonate was studied and under those conditions the reaction also proceeded with quantitative yield in 72 h.
On the other hand, it is well known that benzaldehyde in the presence of an enol silane (as 2-(trimethylsilyloxy)furan) and a Lewis acid may result in the Mukaiyama aldol reaction39 (Scheme 6) consuming the corresponding amount of furan and decreasing the yield of the Mukaiyama–Michael addition. In fact catalysts similar to Lap-Cu have shown to be efficient in Mukaiyama aldol reactions.40 However, the reaction with benzaldehyde proceeds more slowly (10% yield at 24 h) than the Mukaiyama–Michael addition under the same conditions, and the impact of a small amount of benzaldehyde would not be very important.
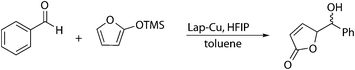 | ||
| Scheme 6 Mukaiyama aldol reaction of benzaldehyde with 2-(trimethylsilyloxy)furan. | ||
The combined process was tried by adding Lap-Cu to the Knoevenagel condensation, carried out under reflux, and subsequent addition of HFIP and 2-(trimethylsilyloxy)furan at room temperature once the first reaction had finished. Although the compatibility studies had been positive, the global yield of the combination of both reactions (Scheme 7) dropped to 53%, due to a yield decrease in the Mukaiyama–Michael addition (Table 2). This deactivation of Lap-Cu is even stronger when the mixture of catalysts is recovered under the same conditions. The difference in behavior between fresh and aged Lap-Mn(salen) was checked by stirring this catalyst in toluene under reflux and then addition of all the components of the Mukaiyama–Michael reaction. The significant reduction in yield (only 20%, Table 2) demonstrated the role of aging of Lap-Mn(salen). Leaching of Mn, favored by the long stirring period under reflux, was considered as the potential origin of this effect. Two consecutive washings of Lap-Mn(salen) with toluene under reaction conditions (and hot filtration) demonstrated the existence of leaching: the activity of the resulting solid was significantly reduced (45% yield, Table 2) whereas the two toluene extracts were active in the Knoevenagel condensation (Table 2). Surprisingly, Lap-Cu catalyzed Mukaiyama–Michael addition proceeded with total conversion when the components were added to the toluene extracts from Lap-Mn(salen), showing that the origin of Lap-Cu deactivation is not (only) Mn leaching.
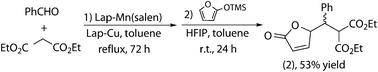 | ||
| Scheme 7 (Knoevenagel + Mukaiyama–Michael) combination. | ||
| Mn catalyst | Yield (%) | |
|---|---|---|
| Individual Knoevenagel | Mukaiyama–Michael in the presence of Mn cat. | |
| a Knoevenagel reaction in the presence of Lap-Cu and addition of HFIP and TMSO-furan at the end of the first reaction. b Stirred in toluene under reflux for 72 h. c Lap-Mn(salen) washed twice with hot toluene. d Extracts of each hot washing. | ||
| Lap-Mn(salen) fresh | 99 | 100 |
| Combined reactionsa | 99 | 53 |
| (2nd run) | 99 | 15 |
| Lap-Mn(salen) agedb | – | 20 |
| Hot toluene washing twicec | 45 | – |
| Toluene extract 1d | 38 | 100 |
| Toluene extract 2d | 37 | 100 |
Finally, in view of the incompatibilities between the catalysts, a sequential process was envisaged. The catalysts were separated after each reaction by filtration, and the reaction crude was used in the following reaction without further purification. Under such conditions, when the manganese catalyst was filtered off after Knoevenagel condensation, the yield of the global process was almost quantitative (Table 3). However, as conversion of the second Knoevenagel reaction is not complete (92% yield), the presence of unreacted diethyl malonate and benzaldehyde causes a drop in the yield of the Mukaiyama–Michael addition (60%, Table 3) due to the competition for coordination to the copper catalyst. This fact was even more evident in the third cycle, when the Knoevenagel reaction proceeded with lower yield (54%).
| Cycle | Yield (%) | ||
|---|---|---|---|
| Knoevenagel | Mukaiyama–Michael | Overall | |
| a Knoevenagel reaction conditions as in Table 1. Lap-Mn(salen) filtered off after 72 h. Reagents and catalyst for Mukaiyama–Michael (ref. 20) added to the crude. b Lap-Cu washed with THF in a Soxhlet extractor after the second cycle. | |||
| 1 | 99 | 90 | 99 |
| 2 | 92 | 60 | 54 |
| 3 | 54 | 10b | 5 |
Therefore, to carry out the combination of both reactions, the best results are obtained in a sequential tandem process, including filtration of the manganese catalyst and use of the reaction crude in the next reaction without further purification. Furthermore, it is necessary to obtain diethyl benzylidenemalonate with 100% yield, because the presence of small amounts of unreacted reagents competes with the catalyst of the second reaction.
3.4 Combination of hydrogenation + transesterification
As transesterification is carried out in a different solvent (methanol), it was necessary to check the effect of the presence of toluene. It was shown that toluene was not detrimental up to a 50/50 (v/v) methanol/toluene ratio.The hydrogenation was carried out in the presence of the transesterification catalyst (TBD-PS) that did not have any negative effect on the hydrogenation yield (Scheme 8A). On the contrary, the presence of Pd/Al2O3 in the transesterification inhibited the formation of 4 (Scheme 8B). By increasing the reaction temperature to 85 °C, 67% yield of 4 was obtained, but this value could not be improved by increasing the amount of TBD-PS or the reaction time.
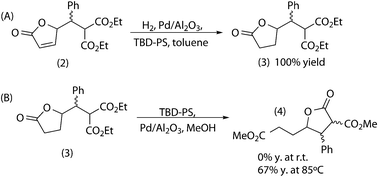 | ||
| Scheme 8 Compatibility between hydrogenation and transesterification catalysts. | ||
Due to these problems found in the compatibility studies, the combination of reactions was carried out in a sequential way, that is, with filtration of the first catalyst and using the crude in the second reaction. Under such conditions, the final product (4) was obtained at rt with high yield (97%) (Scheme 9).
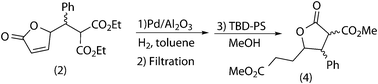 | ||
| Scheme 9 Sequential (hydrogenation + transesterification) tandem process. | ||
Although the palladium catalyst could be reused for 4 cycles of the reaction, the transesterification catalyst was deactivated after the first cycle and it could not be regenerated by continuous extraction with toluene in a Soxhlet apparatus. Thus, a new batch of catalyst must be added in each reaction cycle.
3.5 Knoevenagel + Mukaiyama–Michael + hydrogenation + transesterification sequential process
Due to previously observed incompatibilities between some catalysts and taking advantage of the heterogeneous nature of the catalysts, a sequential process was envisaged (Scheme 10). Thus, the catalysts must be separated after each reaction by filtration and reused in subsequent reaction cycles.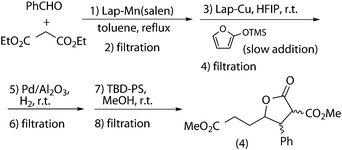 | ||
| Scheme 10 Sequential (Knoevenagel + Mukaiyama–Michael + hydrogenation + transesterification) tandem process. | ||
When the crude after three sequential reactions was used, the transesterification yield dropped to 48% (Table 4). Moreover, the catalyst of this reaction was fully deactivated, as it happened in the combination of hydrogenation–transesterification (Table 4, cycle 2). A new batch of Lap-Mn(salen) was used in the third cycle, to prevent the drop in yield of Mukaiyama–Michael, as a consequence of the lower Knoevenagel conversion. The use also of a new batch of TBD-PS allowed recovering the initial results (Table 4, cycle 3). In view of the problems associated with the combination of the four reactions, the two possible combinations of three reactions were studied.
3.6 Sequential processes involving three reactions
Firstly, the combination of the last three reactions was studied (Scheme 11). When purified diethyl benzylidenemalonate was used as the starting material, the overall yield of the three-reaction sequential process was close to 90% (Table 5, cycle 1). Thereby, the components of the Knoevenagel reaction proved to be responsible for the yield drop of the transesterification reaction. Unfortunately, TBD-PS was deactivated in each cycle of the reaction, as observed in the combined (hydrogenation + transesterification) system, so a new batch of catalyst had to be used in every reaction. Characterization by 13C CP-MAS-NMR (ESI†) shows that the TBD moiety is modified under reaction conditions, and it has been shown that in particular the combination of 2-(trimethylsilyloxy)furan and HFIP is highly detrimental for activity. This question is currently under study. As previously described25 Lap-Cu could be reused at least ten times, provided that it is Soxhlet extracted every two cycles, and Pd/alumina could be reused 4 times but it could not be regenerated (Table 5, cycles 2–10).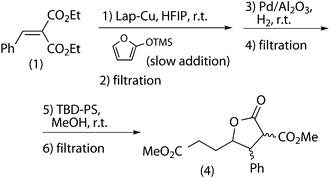 | ||
| Scheme 11 Sequential (Mukaiyama–Michael + hydrogenation + transesterification) tandem process. | ||
| Cycle | Yielda (%) | |||
|---|---|---|---|---|
| Mukaiy. | Hydrogen. | Transesterif. | Overall | |
| a Determined by NMR in the two first reactions. Isolated in the case of transesterification and overall yields. b Lap-Cu washed with THF in a Soxhlet extractor every two cycles. c A new batch of Pd/alumina was used every four cycles. d A new batch of TBD-PS was used every cycle. | ||||
| 1 | 100 | 100 | 89 | 89 |
| 2 | 99 | 100 | 0 | 0 |
| 3 | 100b | 100 | 90d | 90 |
| 4 | 100 | 100 | 88d | 88 |
| 5 | 99b | 100c | 89d | 88 |
| 6 | 95 | 100 | 89d | 85 |
| 7 | 97b | 100 | 91d | 88 |
| 8 | 94 | 100 | 88d | 83 |
| 9 | 98b | 100c | 87d | 85 |
| 10 | 95 | 100 | 91d | 86 |
The second tested combination was Knoevenagel + Mukaiyama–Michael + hydrogenation (Scheme 12), whose results are gathered in Table 6. Given the problems encountered with recovery of the Lap-Mn(salen) catalyst, a new batch of the catalyst was used in each cycle, whereas Lap-Cu and Pd/alumina were recovered as described above without any loss of performance. Under such conditions, overall yields in the range of 93–99% were obtained in ten consecutive runs, with no purification intermediate steps, except the simple catalyst filtration.
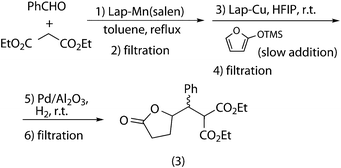 | ||
| Scheme 12 Sequential (Knoevenagel + Mukaiyama–Michael + hydrogenation) tandem process. | ||
| Cycle | Yielda (%) | |||
|---|---|---|---|---|
| Knoeven. | Mukaiy. | Hydrogen. | Overall | |
| a Determined by NMR except in the case of isolated overall yield. b A new batch of Lap-Mn(salen) was used every cycle. c Lap-Cu washed with THF in a Soxhlet extractor every two cycles. d A new batch of Pd/alumina was used every four cycles. | ||||
| 1 | 99 | 100 | 100 | 97 |
| 2 | 99b | 99 | 100 | 96 |
| 3 | 99b | 100c | 100 | 97 |
| 4 | 98b | 98 | 100 | 94 |
| 5 | 98b | 99c | 100d | 95 |
| 6 | 99b | 96 | 100 | 93 |
| 7 | 97b | 96c | 100 | 91 |
| 8 | 98b | 95 | 100 | 91 |
| 9 | 99b | 97c | 100d | 94 |
| 10 | 99b | 95 | 100 | 92 |
This is the most suitable three-reaction sequential process when compared with the “classical” approach. In such a case the isolated yields are 88% for Knoevenagel, 96% for Mukaiyama–Michael and 100% for hydrogenation, with an overall yield of 84% after three filtrations and evaporations, and two chromatography columns on silica. On the contrary, the sequential process allows an overall isolated yield of 97% with only three filtrations and one evaporation and silica column chromatography. In addition to the higher overall yield, an additional advantage of the Knoevenagel + Mukaiyama–Michael + hydrogenation is that purification of the final product 3 should allow the transesterification catalyst to be recovered once, doubling in this way its productivity.
4. Conclusions
One-pot fine chemistry catalytic processes present some limitations due to incompatibilities between the different components of the reactions. On the contrary the sequential methodology minimizes the incompatibility issues, while optimizing at the same time the recovery and reuse of each catalyst, and hence its productivity. The elimination of intermediate purification steps is an additional practical advantage of this approach.The full sequence of four reactions shows inherent limitations that drastically reduce the overall yield by deactivation of the last reaction catalyst. On the contrary, different sequences reactions, either two (Knoevenagel + Mukaiyama–Michael; Mukaiyama–Michael + hydrogenation; hydrogenation + transesterification) or three (Mukaiyama–Michael + hydrogenation + transesterification; Knoevenagel + Mukaiyama–Michael + hydrogenation) have led to very high yields, with optimal recovery of the catalysts in subsequent cycles of the reaction.
Acknowledgements
Financial support from the Spanish Ministerio de Economía y Competitividad (Project CTQ2011-28124) and the Diputación General de Aragón (E11 Group co-financed by the European Regional Development Funds) is gratefully acknowledged.Notes and references
- A. Bruggink, R. Schoevaart and T. Kieboom, Org. Process Res. Dev., 2003, 7, 622 CrossRef CAS.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS.
- M. Gruber, S. Chouzier, K. Koehler and L. Djakovitch, Appl. Catal., A, 2004, 265, 161 CrossRef CAS.
- L. Joucla, G. Cusati, C. Pinel and L. Djakovitch, Adv. Synth. Catal., 2010, 352, 1993 CrossRef CAS.
- H. Lebel and V. Paquet, J. Am. Chem. Soc., 2004, 126, 11152 CrossRef CAS.
- H. Lebel, C. Ladjel and L. Bréthous, J. Am. Chem. Soc., 2007, 129, 13321 CrossRef CAS.
- S. J. Broadwater, S. L. Roth, K. E. Price, M. Kobašlija and D. T. McQuade, Org. Biomol. Chem., 2005, 3, 2899 CAS.
- S. L. Poe, M. Kobašlija and D. T. McQuade, J. Am. Chem. Soc., 2006, 128, 15586 CrossRef CAS.
- F.-X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072 CrossRef CAS.
- B. M. Choudary, N. S. Chowdari, S. Madhi and M. L. Kantam, Angew. Chem., Int. Ed., 2001, 40, 4620 Search PubMed.
- B. M. Choudary, N. S. Chowdari, S. Madhi and M. L. Kantam, J. Org. Chem., 2003, 68, 1736 CrossRef CAS.
- K. Motokura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2005, 127, 9674 CrossRef CAS.
- N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209 CrossRef CAS.
- K. K. Akagawa, S. Sakamoto and K. Kudo, Tetrahedron Lett., 2007, 48, 985 CrossRef CAS.
- A. W. Pilling, J. Boehmer and D. J. Dixon, Angew. Chem., Int. Ed., 2007, 46, 5428 CrossRef CAS.
- J. M. Fraile, R. Mallada, J. A. Mayoral, M. Menéndez and L. Roldán, Chem.–Eur. J., 2010, 16, 3296 CrossRef CAS.
- F. Gelman, J. Blum and D. Avnir, J. Am. Chem. Soc., 2000, 122, 11999 CrossRef CAS.
- R. Abu-Reziq, D. Wang, M. Post and H. Alper, Chem. Mater., 2008, 20, 2544 CrossRef CAS.
- J. M. Fraile, N. García, C. I. Herrerías and J. A. Mayoral, Catal. Today, 2011, 173, 15 CrossRef CAS.
- M. Genelot, V. Dufaud and L. Djakovitch, Tetrahedron, 2011, 67, 976 CrossRef CAS.
- L. Veum and U. Hanefeld, Chem. Commun., 2006, 825 RSC.
- P. Mäki-Arvela, S. Sahin, N. Kumar, J.-P. Mikkola, K. Eränen, T. Salmi and D. Y. Murzin, React. Kinet. Catal. Lett., 2008, 94, 281 CrossRef.
- A. Caiazzo, P. M. L. Garcia, R. Wever, J. C. M. van Hest, A. E. Rowan and J. N. H. Reek, Org. Biomol. Chem., 2009, 7, 2926 CAS.
- J. M. Fraile, N. García, C. I. Herrerías, M. Martín and J. A. Mayoral, ACS Catal., 2012, 2, 56 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra, K. Epping and A. Velty, J. Catal., 2004, 225, 316 CrossRef CAS.
- U. Costantino, M. Curini, F. Montanari, M. Nocchetti and O. Rosati, Microporous Mesoporous Mater., 2008, 107, 16 CrossRef CAS.
- Y. Moussaoui and R. Ben Salem, C. R. Chim., 2007, 10, 1162 CrossRef CAS.
- B. E. Blass, Tetrahedron, 2002, 58, 9301 CrossRef CAS.
- Y. V. Subba Rao, D. E. De Vos and P. A. Jacobs, Angew. Chem., Int. Ed. Engl., 1997, 36, 2661 CrossRef.
- J. M. Fraile, N. García, J. A. Mayoral, E. Pires and L. Roldán, Appl. Catal., A, 2009, 364, 87 CrossRef CAS.
- J. M. Fraile, N. García, J. A. Mayoral, E. Pires and L. Roldán, Appl. Catal., A, 2010, 387, 67 CrossRef CAS.
- V. S. R. R. Pullabhotla, A. Rahman and S. B. Jonnalagadda, Catal. Commun., 2009, 10, 365 CrossRef CAS.
- M. L. Kantam and B. Bharathi, Catal. Lett., 1998, 55, 235 CrossRef CAS.
- J. M. Fraile, J. I. García, J. Masam and J. A. Mayoral, J. Mol. Catal. A: Chem., 1998, 136, 47 CrossRef CAS.
- A. Miyafuji and X. Katsuki, Tetrahedron, 1998, 54, 10339 CrossRef CAS.
- Knoevenagel condensation was tested under the same conditions using as catalysts NaOAc, (18-crown-6)NaOAc, Mn(OAc)3, (18-crown-6)Mn(OAc)3, and laponite (Na form). Yields in the range of 5–15% were obtained in all cases.
- M. Di Serio, R. Tesser, L. Pengmei and E. Santacesaria, Energy Fuels, 2008, 22, 207 CrossRef CAS.
- M. Szlosek and B. Figadere, Angew. Chem., Int. Ed., 2000, 39, 1799 CrossRef CAS.
- J. M. Fraile, I. Pérez and J. A. Mayoral, J. Catal., 2007, 252, 303 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra of freshly prepared and used Lap-Mn(salen), and 13C CP-MAS-NMR spectra of TBD-PS unused and treated with HFIP and HFIP + 2-(trimethylsilyloxy)furan. See DOI: 10.1039/c2cy20442h |
| This journal is © The Royal Society of Chemistry 2013 |