Catalytic behavior of MnMCM-48 and WMnMCM-48 ordered mesoporous catalysts in a reductive environment: a study of the conversion of methylcyclopentane
Ioana
Fechete
*a,
Ovidiu
Ersen
b,
Francois
Garin
a,
Liliana
Lazar
c and
Alain
Rach
a
aLaboratoire des Matériaux, Surfaces et Procédés pour la Catalyse, UMR 7515 CNRS, Université de Strasbourg, 25 rue Becquerel, 67087, Strasbourg Cedex 2, France. E-mail: ifechete@unistra.fr; Fax: +33 (0)368852761; Tel: +33 (0)368852737
bInstitut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504 CNRS, 23 rue du Loess BP 43, F-67034, Strasbourg Cedex 2, France
cDepartment of Chemical Engineering/Engineering of Inorganic Products, Gh. Asachi University of Iasi, 73 D.Mangeron Bd., 700050, Iasi, Romania
First published on 25th September 2012
Abstract
Direct hydrothermal synthesis (HT), template ion-exchange (TIE) and molecular dispersion (MD) approaches were used to introduce manganese species into MCM-48-type mesoporous materials. The tungsten was highly dispersed within the channels of mesoporous MnMCM-48 samples by chemical grafting (CG) via a liquid–solid reaction. The prepared manganesosilicate and tungstenomanganesosilicate MCM-48-type mesoporous materials were characterized by physico-chemical techniques, including TEM, N2 sorption, XRD, XPS, UV-Vis and elemental analysis. The physico-chemical characterization revealed that all of the samples retained a high surface area, a regular cubic mesoporosity and that the Mn and W were highly dispersed. The higher unit cell parameter/pore wall thickness values of the MnMCM-48 and WMnMCM-48 samples relative to that of pure MCM-48 indicate the incorporation of Mn and W into the framework/channels of MCM-48. The Mn and W species existed as isolated sites in the framework/extra-framework or as monomolecular species. No bulk manganese or tungsten was observed outside the MCM-48-type mesoporous materials. The coexistence of Mn2+, Mn3+ or/and W6+ was evidenced by XPS and UV-Vis spectroscopic measurements. The catalytic activities of these samples were studied with respect to the conversion of methylcyclopentane (MCP) in reductive media as a function of Mn loading and reaction temperature (200–500 °C). Whatever the manganese content and irrespective of the temperature of reaction, the MnMCM-48 mesoporous samples did not exhibit catalytic activity, which suggests that the electrophilic manganese oxygen species, operative over all the samples, were inactive sites for the conversion of MCP. The addition of tungsten favored catalytic activity. When the WMnMCM-48 was not calcined, only the methane (C1) cracking product was observed to be formed by successive rupture of C–C bonds starting at 400 °C. When the WMnMCM-48 was calcined, ring-opening products formed by the C–C rupture, at substituted and unsubstituted carbon atoms, were observed starting at 400 °C. This behavior is associated with a symbiotic process between the MnMCM-48 support and the W nanoparticles.
1. Introduction
Much attention has been directed toward the conversion of methylcyclopentane (MCP) on supported noble-metal catalysts because these catalysts are able to facilitate the reaction with atom economy.1–4 The conversion of MCP involves the formation of 2-methylpentane (2-MP), 3-methylpentane (3-MP) and n-hexane (n-H) as desired ring-opening products and the formation of cracking and ring-enlargement products as by-products. The competition among these products is governed by the nature of the metal, the nature of the support and the reaction conditions.1–10 Two mechanisms for the ring opening of MCP have been observed: the selective mechanism, which results primarily in 2-MP and 3-MP, and the non-selective mechanism, which forms 2-MP, 3-MP and n-H.11,12 The 2-MP and 3-MP products have been established to form on metal sites, whereas the n-H required both metal and acidic sites. Although supported noble-metal catalysts exhibit high activities and selectivities, they suffer from disadvantages that include sensitivity to poisoning, requiring special conditions for their preparation, high price values and limited availability. These limitations are currently driving renewed interest in the development of new catalytic systems that do not incorporate noble metals.13,14Non-noble metal oxides constitute an outstanding alternative to the precious metals.13 Non-noble metal oxides are used in many catalytic applications, including hydrodesulfurization,15 methanol decomposition to hydrogen, methane and CO,16 the N2O oxidation of benzene to phenol,17,18 the reduction of environmental pollution olefin epoxidation,19 the conversion of aromatic hydrocarbons20,21 and Aldol/Prins condensations22,23 because they are less costly. Among the non-noble metal oxides, the manganese oxides, in particular, have been a key topic of investigation because of their potential catalytic applications24–30 which can be conducted in oxidative environments.
Until now, no studies on the conversion of MCP on manganese oxides under a reductive environment have been reported. However, recent research in the area of catalysis for MCP conversion has concentrated on the development of alternative catalysts, especially supported tungsten, iron and molybdenum catalysts.14,31,32 These catalysts have demonstrated that the catalytic activity of the catalyst is dependent on the density of active sites, the dispersion, and the location and coordination of the metal atoms.14,31,32 The role of the support is dominant, which indicates that it must have a high surface area to disperse a large fraction of the active metal phase. By dispersing the metal in this way, a large total area of metal surface is available for catalytic reactions to occur. A high metal particle dispersion is desirable to achieve a high level of catalytic conversions.14,31,32
The discovery of the MCM-48 siliceous mesostructured material has attracted considerable attention as a catalyst support because this material possesses a high surface area and an ordered cubic structure with a controllable, uniform pore size.33 MCM-48, because of its cubic structure, has been suggested to be a more advantageous system than MCM-41 for catalytic applications because the three-dimensional pore system of MCM-48 may allow faster diffusion through the channels and make this material more resistant to pore blocking.14,34
Nonetheless, no previous studies on MCM-48-supported manganese oxides in a reductive environment have been reported. This knowledge gap provides an opportunity to enrich our understanding of manganese oxides for the conversion of MCP with hydrogen at atmospheric pressure. The purpose of this work is to investigate the oxidation state and the local environment of manganese species, their spatial distribution (framework/inside the pores), and their role in the conversion of MCP. The reactivity of MnMCM-48 catalysts prepared using different strategies, such as hydrothermal synthesis, ion template exchange, and designed metal dispersion, were evaluated for the first time in the conversion of MCP in the temperature range of 200 to 500 °C. The tungsten was dispersed by chemical grafting via a liquid–solid reaction.
2. Experimental part
2.1. Materials and instrumentation
Cetyltrimethylammonium bromide (CTAB), manganese acetylacetonate Mn(acac)3 and tungsten hexachloride (WCl6) (99%) were purchased from Sigma-Aldrich. Manganese acetate tetrahydrate (CH3COO)2Mn·4H2O, potassium permanganate (KMnO4) and sodium hydroxide (NaOH) 98% were purchased from SDS. Tetraethylorthosilicate (TEOS) 98% was purchased from Acros. The methylcyclopentane (MCP) used for the catalytic tests was supplied by Aldrich (puriss >99%). All chemicals were used as received.Transmission electron microscopy (TEM) images were obtained on a TopCon 2100 FCs microscope operated at an accelerating voltage of 200 kV. The samples were dispersed in ethanol in an ultrasonic bath for several minutes and then deposited on a Cu grid and dried at room temperature. Nitrogen sorption measurements were performed at −196 °C on a TriStar instrument. The samples were first outgassed at 250 °C overnight. The linear part of the Brunauer–Emmett–Teller (BET) equation35 was used to determine the specific surface area. The pore size distribution curves were calculated from the desorption branches of the isotherms using the Barett–Joyner–Halenda (BJH) method.36 The mean thickness of the pore wall (w) of the cubic structure in the MCM-48 mesoporous solid was calculated according to the formula w = (a0/3.092) − (DBJH/2), where a0 is the unit cell parameter, 3.092 is a constant that represents the minimal surface area for the MCM-48 space group,37 and DBJH is the pore diameter. The cumulative pore volume (VBJH) of the mesopores was obtained from the amount of nitrogen adsorbed at a relative pressure p/p0 of 0.99,38 assuming complete surface saturation with nitrogen. Powder X-ray diffraction (XRD) patterns were recorded on a Bruker D8 powder diffractometer equipped with a CuKα radiation source (λ = 0.154 nm) operated at 40 kV and 100 mA. The diffractograms were recorded in the 2θ range of 0.5–5° (low angle) and 10–90° (wide angle). The scans were performed with a step size of 0.02° and a step time of 1.2 s. The unit-cell parameter (a0), a0 = d{hkl} (h2 + k2 + l2)1/2 for the Ia3d symmetry group, was evaluated using the interplanar spacing (d) of the (211) XRD peak. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a Multilab 2000 spectrometer equipped with an AlKα X-ray radiation source (1486.6 eV) operated at an anode current of 20 mA and 10 keV. The XPS measurements were performed in a static in situ reaction chamber at room temperature and at a base pressure of 10−9 Pa. The samples were outgassed in a vacuum oven overnight before the XPS measurements. The binding energies of the Mn and W photoelectron peaks were corrected for charge effects based on the C 1s peak at 284.6 eV as reference in the XPS measurements. Peak decomposition was performed using curves that were 85% Gaussian type and a 15% Lorentzian type and using a Shirley non-linear sigmoid-type baseline. The following peaks were used for the quantitative analysis: C 1s, W 4f, and Mn 2p. Based on the XPS analysis, the XPS surface ratio of Si/Mn or/and Si/W is defined as the molar concentration of the Si element (%) divided by the molar concentration of Mn or W (%). The atomic concentrations of all elements were estimated based on comparisons of the integrated peak intensities normalized by the atomic sensitivity factors.39,40 BEs were reproducible to within ±0.2 eV. UV-Vis reflectance absorption spectra were recorded on a PerkinElmer spectrophotometer under computer guidance. Spectra were recorded between 200 and 800 nm and are presented as normalized spectra. The chemical analysis of the solid samples was performed at the Service Central d'Analyse of CNRS-Solaize, France.
In this article we have presented the physico-chemical characterization of the calcined samples. It must be noted that the physico-chemical characterization of the reduced catalysts has been performed but no changes were observed and that only the calcined samples, which are stable under reductive conditions up to 500 °C, have been presented here.
2.2. Catalysts preparation
Manganese was introduced onto the MCM-48 support by hydrothermal synthesis (HT), template ion exchange (TIE) and molecular dispersion (MD) approaches. The tungsten was dispersed on MnMCM-48 (HT) Si/Mn = 45 sample following chemical grafting (CG) by a liquid–solid reaction.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :(0.02–0.1) Mn:0.48 CTAB:0.25 Na2O: 55 H2O. In a typical synthesis, the cetyltrimethylammonium bromide, doubly distilled water and sodium hydroxide were stirred for 0.5 h at room temperature, and the tetraethylorthosilicate was added with vigorous stirring.14 After 0.5 h, the required amount of manganese acetate was added. The gel mixtures were stirred for 3 h at room temperature. The prepared gel mixture was transferred to an autoclave and aged for 72 h at 110 °C. The white precipitate was filtered, washed with distilled water and dried for 24 h at 60 °C. The surfactant was removed using an extraction procedure,14 and the sample was calcined at 500 °C for 5 h in a muffle furnace with a heating rate of 1 °C min−1. The catalysts were denoted MnMCM-48 (HT).
:(0.02–0.1) Mn:0.48 CTAB:0.25 Na2O: 55 H2O. In a typical synthesis, the cetyltrimethylammonium bromide, doubly distilled water and sodium hydroxide were stirred for 0.5 h at room temperature, and the tetraethylorthosilicate was added with vigorous stirring.14 After 0.5 h, the required amount of manganese acetate was added. The gel mixtures were stirred for 3 h at room temperature. The prepared gel mixture was transferred to an autoclave and aged for 72 h at 110 °C. The white precipitate was filtered, washed with distilled water and dried for 24 h at 60 °C. The surfactant was removed using an extraction procedure,14 and the sample was calcined at 500 °C for 5 h in a muffle furnace with a heating rate of 1 °C min−1. The catalysts were denoted MnMCM-48 (HT).
2.3. Catalytic tests
Reaction of MCP was carried out using a pulse method with hydrogen under atmospheric pressure. The mass of catalysts used was 200 mg and the injected volume of methylcyclopentane was 5 μl. The gas flow used for treatments and catalytic tests was fixed at 40 cm3 min−1. The catalytic products were analyzed by gas chromatography using a 50 m (CP-SIL-5CP) column and a flame ionization detector (FID). The catalytic activity and performance of the catalysts were measured in terms of conversion and selectivity which are defined as follows: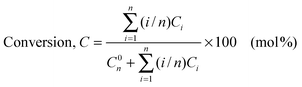
where Ci is the mole percent of product with i carbon atoms and C0n is the mole percent in the feed of MCP (n = 6).
3. Results and discussion
3.1. Catalyst preparation and elemental analysis
The incorporation of manganese at different Si/Mn ratios into the MCM-48 mesoporous matrix was performed using three approaches:| Catalyst | Si/Mea | Si/Me | Unit cell parameter, a0 (nm) | Unit cell volume, V (nm3) | Surface area, BET (m2 g−1) | Pore volume, Vp (cm3 g−1) | Pore diameter, Dp (nm) | Pore wall thickness, w (nm) | |
|---|---|---|---|---|---|---|---|---|---|
| ICP | XPS | ||||||||
| a Me: metal (Mn, W), HT: hydrothermal, CG: chemical grafting, TIE: template ion exchange, MD: molecular dispersion. | |||||||||
| SiMCM-48 | — | — | — | 7.86 | 485.60 | 1265 | 1.21 | 2.34 | 1.37 |
| MnMCM-48 (HT) | 75 | 82 | 89 | 7.92 | 496.80 | 1250 | 1.20 | 2.34 | 1.39 |
| MnMCM-48 (HT) | 45 | 54 | 61 | 8.01 | 513.92 | 1208 | 1.18 | 2.34 | 1.42 |
| WMnMCM-48 (CG) | 45 | 57 W | 64 W | 8.12 | 535.38 | 1130 | 1.03 | 2.36 | 1.44 |
| 49 Mn | 54 Mn | ||||||||
| MnMCM-48 (IE) | 75 | 81 | 87 | 7.98 | 508.16 | 1252 | 1.18 | 2.34 | 1.41 |
| MnMCM-48 (IE) | 45 | 49 | 57 | 8.07 | 525.55 | 1190 | 1.14 | 2.36 | 1.42 |
| MnMCM-48 (DM) | 75 | 86 | 91 | 7.88 | 489.30 | 1230 | 1.18 | 2.34 | 1.38 |
| MnMCM-48 (DM) | 45 | 54 | 57 | 7.97 | 506.26 | 1181 | 1.07 | 2.30 | 1.42 |
3.2 Structural characterization
We performed TEM experiments to obtain information about changes in the mesoporous structure and in the Mn and W species during their incorporation into the MCM-48 mesoporous matrix. Fig. 1 shows TEM images of the purely siliceous MCM-48, the manganese and tungsten manganese MCM-48 calcined samples. The ordered arrangement of lattice dots along the [100] direction indicates that the samples have excellent textural uniformity. These results also indicate that the ordered mesoporous structure of the support was unambiguously unaffected by the manganese and tungsten species; the cubic arrangement is maintained for all the samples, and no large aggregates were observed outside of the mesopores. In this case, the MCM-48 is a good matrix for highly dispersed Mn and W species. Similar observations have been reported in the literature for the Mn-MCM-48, Cr-MCM-48, V-MCM-48 and W-MCM-48.14,28,41 | ||
| Fig. 1 TEM images of calcined SiMCM-48 (a), MnMCM-48 with Si/Mn = 45 prepared by HT (b), TIE (c), MD (d) approaches and WMnMCM-48 (CG) (e and f) samples. | ||
The maintenance of the ordered cubic arrangement of the MCM-48 frameworks upon the introduction of manganese is further supported by the N2 sorption data. N2 sorption isotherms for all the samples are illustrated in Fig. 2. As shown in this figure, at a relative pressure of p/p0 ≤ 0.3, the nitrogen adsorption occurs as a monolayer. The isotherms exhibited sharp inflections characteristic of capillary condensation within mesopores, which occurred at p/p0 = 0.2–0.3. The sharpness of this step indicated a uniform size pore system of the high quality of the MCM-48 samples. These results suggest also that the pore channels are not obstructed. The textural properties of these mesoporous materials are listed in Table 1, which details the BET specific surface area (SBET), the pore volume (Vp), the mesopore diameter (DBJH), which were calculated from the desorption branch of the nitrogen sorption isotherms. Although the incorporation of Mn led to a decrease in the BET specific surface area, high surface areas are observed in MCM-48 with Mn loadings up to a Si/Mn ratio of 45. Similar effects on the BET specific surface area and pore volume have been previously reported.14,41 A high BET specific surface area was observed even for the WMnMCM-48 sample. We therefore concluded that well-organized mesoporous MCM-48 catalysts with a high surface area could be obtained using all of the described procedures. The total pore volume decreased from the pure silica MCM-48 to the MnMCM-48 with a Si/Mn ratio of 45 and finally to the WMnMCM-48 with Si/W and Si/Mn ratios of 45. The value of mesopore diameter (DBJH) does not change drastically from one approach to another. In all samples pore blockage does not appear, and the manganese species are most likely located in the framework (HT approach) or on the inner surface of the pore channels (TIE and DM approaches). The values of the wall thickness are summarized in Table 1. The wall thickness of the MnMCM-48 and WMnMCM-48 samples increased with increasing manganese/tungsten content, and the walls were denser than those of pure silica MCM-48. These results are consistent with the results reported in the literature.14
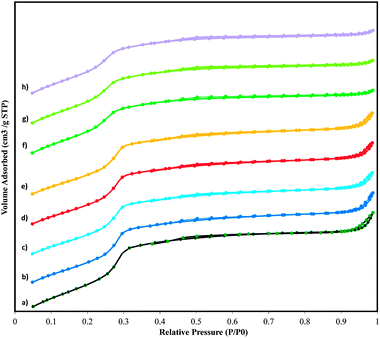 | ||
| Fig. 2 Nitrogen sorption isotherms at −196 °C for SiMCM-48 (a), MnMCM-48 prepared by HD with Si/Mn = 75 (b), Si/Mn = 45 (c), TIE with Si/Mn = 75 (d), Si/Mn = 45 (e), MD with Si/Mn = 75 (f), Si/Mn = 45 (g) and WMnMCM-48 (CG) (h) samples. | ||
Fig. 3 shows the powder X-ray diffraction patterns for calcined samples of the pure silica MCM-48, the MnMCM-48 and of the WMnMCM-48 mesoporous materials with different metal contents in the low-angle range from 0.5° < 2θ < 5°. The XRD patterns exhibit exclusively (211) and (200) planes belonging to the Ia3d cubic symmetry reported for the MCM-48 mesostructure and imply a high degree of order in the mesoporous materials.33 No remarkable shifts in the diffraction peaks to higher or lower 2-theta values were observed with increasing Mn content.
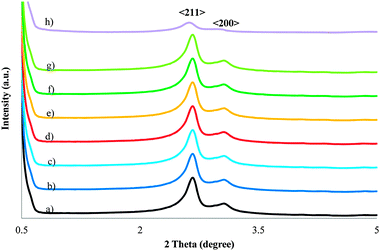 | ||
| Fig. 3 X-ray diffraction pattern for the calcined SiMCM-48 (a), MnMCM-48 prepared by HD with Si/Mn = 75 (b), Si/Mn = 50 (c), TIE with Si/Mn = 75 (d), Si/Mn = 50 (e), MD with Si/Mn = 75 (f), Si/Mn = 50 (g) and WMnMCM-48 (h) samples. | ||
Moreover, the intensity of the d211 peak is gradually attenuated with increasing Mn content in the XRD patterns of the samples. An important decrease in intensity was observed for the WMnMCM-48 sample prepared by chemical grafting. However, the addition of W to MnMCM-48 (HT) of Si/Mn = 45 sample does not significantly alter the degree of order in the mesoporous materials. Logically, the reduced intensity of the reflections may be caused by a degradation of the cubic arrangement of the MCM-48 pores. As evident from the TEM and N2 adsorption data, the cubic arrangement of the MCM-48 framework was retained after Mn and W were incorporated. In this case, the progressive reduced intensity of the d211 reflections is likely due to a dilution of Si with the continuous incorporation of Mn (W) as a consequence of the greater X-ray absorption factor of Mn (W) relative to that of Si. The absorption of X-rays by Mn (W) was indeed significant, as indicated by the strong reduction of the main beam intensity. These results are consistent with those previously reported.14,28,41,44,45 The cubic Ia3d unit-cell parameter (a0) was calculated using Bragg's equation from d211, which was obtained from the (211) peak in the XRD pattern. The values of the unit-cell parameter from the manganese samples increase with increasing manganese/tungsten content for all the samples. This behavior is similar to those observed for the unit cell volume, V. These results are in line with those already reported44 and indicate the incorporation of Mn atoms into the framework and on the inner walls of the MCM-48 matrix. The change in the unit-cell parameter for the Mn and MnW samples can also be rationalized, in terms of bond lengths. The W–O and Mn–O bonds are longer than the Si–O bond, and the crystal radii of W and Mn are larger than that of Si. These results are consistent with results reported in the literature.14,41,44
The high-angle X-ray diffraction patterns (10° < 2θ < 90°) for the MnMCM-48 and WMnMCM-48 samples with various loadings are shown in Fig. 4. The XRD patterns in the high-angle area for all the samples show no distinct reflection of Mn/W oxide phases. This result suggests the presence of very small, well-dispersed species within the silica matrix or on the silica walls. Only a broad maximum centered at 2θ = 23° was observed, which was attributed to amorphous silica; the presence of this maximum suggests a low overall degree of crystallization of siliceous walls. As evidenced by the TEM, BET and XRD results, the ordered mesoporous structures of all the samples were maintained after the Mn and W were incorporated into the MCM-48 structures.
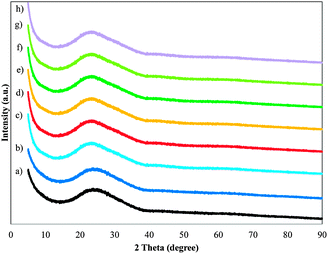 | ||
| Fig. 4 X-ray diffraction pattern for the calcined SiMCM-48 (a), MnMCM-48 prepared by HD with Si/Mn = 75 (b), Si/Mn = 45 (c), TIE with Si/Mn = 75 (d), Si/Mn = 45 (e), MD with Si/Mn = 75 (f), Si/Mn = 45 (g) and WMnMCM-48 (h) samples. | ||
3.3. Spectroscopic characterization
The valence bond of Mn and the oxidation states of the catalyst surface entities were examined by XPS analysis. The Mn 2p XPS spectra for the calcined Mn catalysts prepared by various approaches are shown in Fig. 5. The energy position and the shape of the two photoelectron peaks are approximately the same for all the samples. For the samples with a Si/Mn ratio of 45, the binding energy values were 641.5 eV and 652.9 eV with a spin–orbit splitting of 11.4 eV. These binding-energy values for Mn are consistent with those of manganese oxide and are in agreement with other results reported previously.41,46 A comparison of the spectra of samples with a Si/Mn ratio of 75 with those of samples with a Si/Mn ratio of 45 reveals that the spectral shape remains unchanged; however, the change in peak intensities was observed and is more intense for samples with high concentrations of Mn. However, the assignment of the oxidation state of manganese oxide is difficult because these binding-energy values correspond to several oxidation states, including Mn2+, Mn3+ and Mn4+, which funds the assertion that several oxidation states are present on the surface of MnMCM-48 catalysts, irrespective of the manganese concentration and the preparation approach. According to literature data, the binding-energy values of 640 eV, 641 eV, and 641.7 eV correspond to Mn2+, values of 641–642 eV and 641.60 eV correspond to Mn3+, and 642 eV and 642.65 eV correspond to Mn4+.41,47–49 Based on this analysis, the XPS peak binding energies alone are not sufficient to accurately determine the chemical state of Mn in the manganese oxides.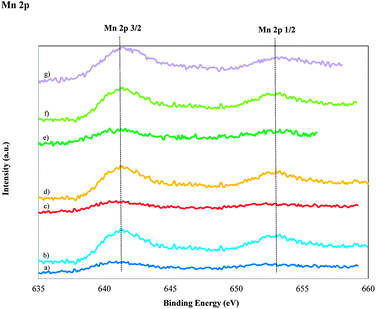 | ||
| Fig. 5 XPS spectra of the Mn 2p XPS region for calcined MnMCM-48 and WMnMCM-48 catalysts; MnMCM-48 prepared by HD with Si/Mn = 75 (a), Si/Mn = 45 (b), TIE with Si/Mn = 75 (c), Si/Mn = 45 (d), MD with Si/Mn = 75 (e), Si/Mn = 45 (f) and WMnMCM-48 (g) samples. | ||
Fig. 6 shows the W4f peak of the WMnMCM-48 catalyst. The major peak W4f7/2 appears at 35.5 eV and assigned to W6+. This binding-energy value for W is consistent with those of tungsten oxide reported in the literature.50
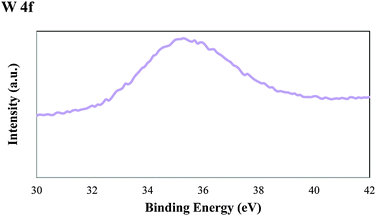 | ||
| Fig. 6 XPS spectra of the W 4f region for the calcined WMnMCM-48 catalyst. | ||
The calculated Si/Mn and Si/W ratios from integral intensities of Si 2p, Mn 2p and W 4f photoelectron spectra are summarized in Table 1. A slight but systematic increase in the Si/Mn and Si/W ratios was observed compared with the chemical analysis.
The identification and characterization of the coordination environment of Mn and W and their locations in the framework/extraframework positions of MCM-48 samples were performed by diffuse-reflectance UV–Vis spectroscopy. Fig. 7 shows the diffuse-reflectance UV–Vis spectra of calcined MnMCM-48 samples with different Mn contents prepared by three methods and also that of the WMnMCM-48 (CG) sample.
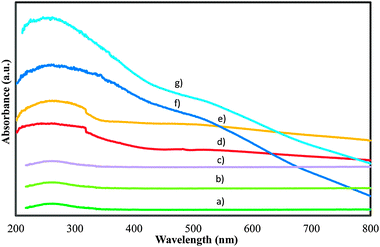 | ||
| Fig. 7 Diffuse reflectance UV-Vis spectra for the calcined WMnMCM-48 CG (a), MnMCM-48 prepared by HT with Si/Mn = 75 (b), Si/Mn = 45 (c), TIE with Si/Mn = 75 (d), Si/Mn = 45 (e), MD with Si/Mn = 75 (f), Si/Mn = 45 (g) samples. | ||
The spectra were acquired over the entire ultraviolet and visible regions (200 to 800 nm). The samples with a Si/Mn ratio of 75, prepared by the HT and TIE methods, showed only one absorption band maximum at 270 nm, while the sample with the same Si/Mn ratio prepared by the MD exhibited two overlapped absorption bands at 270 and 500 nm. The absorption maxima are quasi-identical with those reported in the literature.51,52 When the Mn content was increased (Si/Mn = 45), the peak position of the band shifted to 280 nm, and the absorption edge also shifted to a longer-wavelength position. The less resolved and broad absorption band at a wavelength less than 270 nm is attributed to a ligand-to-metal charge transfer of O2− to Mn3+ in a tetrahedral coordination that involves, in this case, isolated transition-metal sites. The Mn3+ ions are probably coordinated with the Si and located in the framework of MCM-48. These structural features are not rare because they have been observed by other authors.51–53 The broad absorbance at approximately 500 nm, which was observed in the spectra of the samples prepared by MD, is associated with the d–d transition in Mn2+/Mn3+, which suggests that the two sites should coexist in these samples. Similar results have been reported for the other structures.51,52,54 Moreover, this band was attributed to the 6A1g → 4T2g crystal-field transition.52,54 Our results confirmed that both Mn3+ and Mn2+ sites are present in the MnMCM-48 (MD) and also support the XPS analysis that multiple Mn oxidation states are present in these samples.
3.4. Catalytic reactivity toward the conversion of MCP
Oxide catalysts have recently been considered a modern alternative to noble-metal catalysts and represent a scientific, economic and ecological challenge.13 In this work, the well-organized cubic MnMCM-48 and WMnMCM-48 mesoporous solids were investigated as a novel catalyst system for the conversion of MCP. Previously reported results have shown that MnMCM-48 mesoporous catalysts deliver excellent results in oxidative media,24–30 and we applied the same catalysts in reductive media. The chemistry of these catalysts in reductive media was unknown, and, to our knowledge, these results represent the first catalytic tests of MCP on MnMCM-48 and WMnMCM-48 mesoporous oxides. The catalysts with variable Mn contents and with the same W loadings were evaluated as a function of the reaction temperature in the range of 200 to 500 °C. As observed with noble-metal catalysts tested under the same conditions,3,6,9 the possible products formed in the conversion of MCP are 2-methylpentane, 3-methylpentane and n-hexane, which are formed from a ring-opening reaction of MCP. Cyclohexane and benzene can be formed via ring-enlargement reactions, whereas methane, ethane, propane, butane, i-butane and pentane/i-pentane can be formed from cracking reactions.Prior to the catalytic tests, two treatments were applied to better understand the catalytic behavior of the tungstenomanganesosilicate mesoporous catalysts: (i) we tested the catalysts after they were dried at 100 °C and heat-treated under hydrogen for 2 h at 500 °C and (ii) after they were calcined at 500 °C and heat-treated under hydrogen for 2 h at 500 °C.
The catalytic potential of the WMnMCM-48 catalysts is presented in Fig. 8–10 as a function of the reaction temperature.
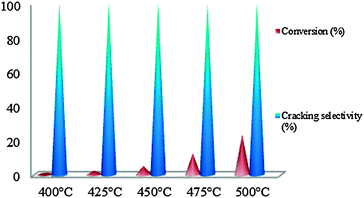 | ||
| Fig. 8 Conversion of MCP on WMnMCM-48 catalysts dried at 100 °C and heat-treated under hydrogen for 2 h at 500 °C. | ||
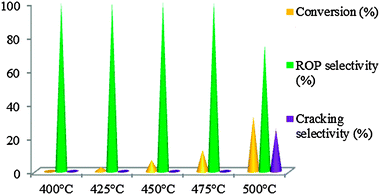 | ||
| Fig. 9 Conversion of MCP on WMnMCM-48 catalysts calcined at 500 °C and heat-treated under hydrogen for 2 h at 500 °C. | ||
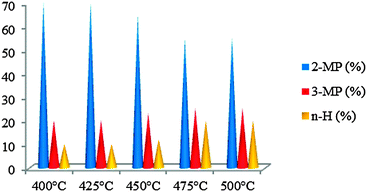 | ||
| Fig. 10 Ring opening products on WMnMCM-48 catalysts calcined at 500 °C and heat-treated under hydrogen for 2 h at 500 °C. | ||
3.4.2.1. WMnMCM-48 dried at 100 °C and heat-treated under hydrogen for 2 h at 500 °C. Although the catalysts were tested between 200 and 500 °C, the W sites were inactive at temperatures less than 400 °C. These sites exhibited catalytic activity only between 400 and 500 °C. The catalytic results show a linear increase in the percent conversion in this temperature range. The values of the conversion varied between 0.69% at 400 °C and 23.40% at 500 °C (Fig. 8).
Olefins were not observed among the products over the studied temperature range (200–500 °C). No products derived from ring-opening reactions were observed, which indicates the difficulty of the endocyclic C–C bond rupture. In addition, no products derived from ring-enlargement reactions were formed. Only the C1 products from the cracking reaction were observed as the major products. The data show that, irrespective of the temperature, the cracking-selectivity values were 100% (Fig. 8). These results reflect the fact that the WMnMCM-48 catalysts, when dried only at 100 °C, are active in extensive cracking reactions that involve successive C–C bond rupture, which underscores the poorly metallic behavior of the catalyst. It is expected that the non-calcined active sites have a low dispersion and the heat treatment under hydrogen leads to a progressive reduction of WO3 (particles size < 5 nm) to highly reduced tungsten oxide phase W3O with poor metallic character. In this case, on the poorly metallic sites (W3O) the rate of desorption of the products from the WMnMCM-48 catalyst surface diminished and the multiple C–C ruptures occurred. The results may indicate strong heat adsorption of the active sites on the dried WMnMCM-48 catalyst.
Consequently, the dried WMnMCM-48 catalysts were active toward MCP conversion but not selective toward ring-opening products.
3.4.2.2. WMn-MCM-48 calcined under air at 500 °C and heat-treated under hydrogen for 2 h at 500 °C. The catalysts were tested between 200 and 500 °C, but the catalytic activity was observed starting at 400 °C (Fig. 9). At 400 and 425 °C, all the catalysts showed discrete catalytic activity, <0.25%, with only traces of ring-opening products. The reactivity of the calcined WMnMCM-48 catalyst to the ring opening of MCP is slow but occurs at three positions with the formation of three products—2-MP, 3-MP and n-H—in different proportions (Fig. 10). This behavior is probably attributed to the high dispersion of W on the inner pore structure framework, thus resulting in a higher number of accessible active sites to MCP molecules. Under these conditions, the main role of calcinations is the redistribution of the WO3 species over the catalyst surface and into the channels/pore of the MnMCM-48 mesoporous solid. The calcination treatments help probably to the formation of WMnMCM-48 catalysts with higher dispersion, improving in this case the accessibility of active sites. Thereafter, under hydrogen at 500 °C, as a result of dissociative chemisorption of hydrogen, the highly dispersed WO3 species became more difficult to reduce to poorly metallic sites (W3O) and are reduced by hydrogen atoms only to the WO3−x/WO2 phase forming Bronsted acid sites. These active acid sites are responsible for the formation of active sites for the ring opening of MCP. Furthermore, the catalytic activity gradually increased with increasing reaction temperature. Only the ring-opening products were observed at 450 and 475 °C and were considered to be the primary products under our conditions (Fig. 9). These results suggest that the formed ring opening products on highly dispersed WMnMCM-48 catalyst are strongly desorbed in the gas phase. At 500 °C, the cracking products appeared; however, the ring-opening reaction remained the major reaction. The decrease in the ring-opening selectivity at high temperatures and the high conversion are associated with cracking of the ring-opening products by the multiple ruptures of C–C bonds. As is evident from the results in Fig. 10, among the ring opening products, 2-MP is the major product. This result indicated that the catalytically active W species predominantly catalyzed the C–C rupture at unsubstituted secondary–secondary carbon atoms. The formation of only ring-opening products in the temperature range of 400 to 475 °C suggests that the catalysts possess the same active sites. The percentage of the ring-opening products, although low, decreased further with increasing temperature at 500 °C.
Taken together, the results of this study revealed that the difference in activities between the non-calcined and calcined catalysts would results not only from the W dispersion but also from the nature of W sites (W3O or WO3−x) present at the surface. Moreover, on the calcined WMnMCM-48 catalyst (WO3−x sites) the ring opening products desorb easily, while on the non-calcined WMnMCM-48 catalyst (W3O site) the ring opening products are hindered to desorb, leading to multiple ruptures of the C–C bonds.
3.4.2.3. Roles of W on catalytic activity. It must be noted that our experimental data cannot rule out the complete structure of the WMnMCM-48 catalyst concerning the possible interaction between W and Mn and implicitly concerning the nature of active sites in the catalytic conversion of MCP. No W–Mn bonds were observed on the WMnMCM-48 catalysts, as evidenced by the spectroscopic studies (XPS). As shown in the foregoing catalytic results on MnMCM-48, the electrophilic Mn–O site was unable to catalyze the C–C bond rupture, but the introduction of W, remarkably increase the catalytic activity. Excepting the high acidity introduced by W, such change in catalytic activity could be explained by assuming that W could change the electronic properties of MnMCM-48. The special behavior of the WMnMCM-48 catalyst could be interpreted by a possible electronic interaction between W and Mn. Theoretically, the electron transfers from the low electronegative elements to high electronegative elements.55 There is an electron transfer from the low electronegative Mn element (1.55) to the high electronegative W element (2.36). In this case, the promotion by W induces changes in the density of d electrons at the Fermi level which leads to a synergistic effect between W–Mn of the WMnMCM-48 catalyst, improving their catalytic activity. It seems that the electronic effect, which transfers electrons between Mn and W and leads to an electron-deficient state of the Mn entities, is essential for the ring opening of MCP by the rupture of secondary–secondary or secondary–tertiary bonds.
4. Conclusions
This study details our investigation of the MnMCM-48 mesoporous catalysts, prepared by three strategies, that contain Mn2+ and Mn3+ cations whose oxidation states do not change after the addition of the tungsten species, as revealed in the UV-Vis/XPS results. The introduction of Mn and W species into MCM-48-type mesoporous materials resulted in increased wall thickness, and all samples were prepared without destruction of the mesoporous cubic structure. The MnMCM-48 and WMnMCM-48 mesoporous catalysts contained either framework manganese cations or extra-framework monomolecular clusters (UV-Vis). No Mn–W bonds were observed to be formed during the tungsten grafting procedure, as evidenced by the spectroscopic studies. However, under these circumstances, the characterization is limited, which resulted in our inability to discern the electronic properties of this WMnMCM-48 mesoporous catalyst. In this situation, catalytic reaction of MCP serves as a probe to obtain information concerning possible interactions between the atoms of the two (W–Mn) components. Notably, manganese alone (electrophilic Mn–O site) was unable to catalyze the C–C bond rupture in reductive media, whereas the Mn–O sites were very active and selective in oxidative media.24–30 Whatever the manganese content and irrespective of the reaction temperature, the MnMCM-48 did not show catalytic activity. The WMnMCM-48 catalyst did not show catalytic activity at low temperatures; however, this catalyst starts converting MCP at approximately 400 °C. The WMnMCM-48 dried at 100 °C contains active W for the successive rupture of C–C bonds by consecutive reactions but ruptures the endocyclic C–C bonds with difficulty. The WMnMCM-48 calcined at 500 °C contains active sites suitable for the endocyclic C–C bond rupture. A symbiotic process between the support and the W nanoparticles likely occurs, and a metal support effect can be envisaged in which the W particles are highly dispersed on the Mn surface of the catalysts as revealed by the catalytic tests. During the MCP conversion, W has three major functions: the acidity effect, the dispersion effect and the electronic effect. Although the conversion of MCP is relatively low, these experimental results in reductive media for WMnMCM-48 mesoporous catalysts are essential for the development and optimization of novel catalysts for the petrochemical industry.Acknowledgements
We are pleased to acknowledge the REALISE network and the IDECAT network of excellence for their support. The generous support of the CNRS France is acknowledged with gratitude by I. Fechete.References
- R. J. Fenoglio, G. M. Nunez and D. E. Resasco, Appl. Catal., 1990, 63, 319 CrossRef CAS.
- P. Samoila, M. Boutzeloit, C. Especel, F. Epron and P. Marecot, Appl. Catal., A, 2009, 369, 104 CrossRef CAS.
- A. Djeddi, I. Fechete and F. Garin, Appl. Catal., A, 2012, 413–414, 340 CrossRef CAS.
- C. Poupin, L. Pirault-Roy, C. La Fontaine, L. Toth, M. Chamam, A. Wootsch and Z. Paal, J. Catal., 2010, 272, 315 CrossRef CAS.
- P. Samoila, M. Boutzeloit, C. Especel, F. Epron and P. Marecot, J. Catal., 2010, 276, 237 CrossRef CAS.
- A. Djeddi, I. Fechete and F. Garin, Catal. Commun., 2012, 17, 173 CrossRef CAS.
- N. Gyorffy, I. Bakos, S. Szabo, L. Toth, U. Wild, R. Schlogl and Z. Paal, J. Catal., 2009, 263, 372 CrossRef CAS.
- Z. Paal, Catal. Today, 1988, 2, 595 CrossRef CAS.
- A. Djeddi, I. Fechete and F. Garin, Top. Catal., 2012, 55, 700 CrossRef CAS.
- M. Chow and G. B. McVicker, J. Catal., 1988, 112, 303 CrossRef CAS.
- G. Maire, G. Plouidy, J. C. Prudhomme and F. G. Gault, J. Catal., 1965, 4, 556 CrossRef CAS.
- M. J. Dees, M. H. B. Bol and V. Ponec, Appl. Catal., 1990, 64, 279 CrossRef CAS.
- I. Fechete, Y. Wang and J. C. Vedrine, Catal. Today, 2012, 189, 2 CrossRef CAS.
- I. Fechete, B. Donnio, O. Ersen, T. Dintzer, A. Djeddi and F. Garin, Appl. Surf. Sci., 2011, 257, 2791 CrossRef CAS.
- G. Shi, D. Fang and J. Shen, Microporous Mesoporous Mater., 2009, 120, 339 CrossRef CAS.
- T. Tsoncheva, J. Rosenholm, M. Linden, F. Kleitz, M. L. Ivanova, M. Dimitrov, D. Paneva, I. Mitov and C. Minchev, Microporous Mesoporous Mater., 2008, 112, 327 CrossRef CAS.
- F. Zhang, X. Chen, J. Zhuang, Q. Xiao, Y. Zhong and W. Zhu, Catal.: Sci. Technol., 2011, 1, 1250 CAS.
- D. Meloni, R. Monaci, E. Rombi, C. Guimon, H. Martinez, I. Fechete and E. Dumitriu, Stud. Surf. Sci. Catal., 2002, 142, 167 CrossRef.
- P. C. Bakala, E. Briot, L. Salles and J.-M. Bregeault, Appl. Catal., A, 2006, 300, 91 CrossRef CAS.
- E. Dumitriu, C. Guimon, V. Hulea, D. Lutic and I. Fechete, Appl. Catal., A, 2002, 237, 211 CrossRef CAS.
- I. Fechete, E. Gautron, E. Dumitriu, D. Lutic, P. Caullet and H. Kessler, Rev. Roum. Chim., 2008, 53, 49 CAS.
- E. Dumitriu, V. Hulea, I. Fechete, C. Catrinescu, A. Auroux, J. F. Lacaze and C. Guimon, Appl. Catal., A, 1999, 181, 15 CrossRef CAS.
- E. Dumitriu, V. Hulea, I. Fechete, A. Auroux, J. F. Lacaze and C. Guimon, Microporous Mesoporous Mater., 2001, 43, 341 CrossRef CAS.
- S. L. Brock, N. Duan, Z. R. Tian, O. Giraldo, H. Zhou and S. L. Suib, Chem. Mater., 1998, 10, 2619 CrossRef CAS.
- Z.-R. Tang, S. A. Kondrat, C. Dickinson, J. K. Bartley, A. F. Carley, S. H. Taylor, T. E. Davies, M. Allix, M. J. Rosseinsky, J. B. Claridge, Z. Xu, S. Romani, M. J. Crudace and G. J. Hutchings, Catal.: Sci. Technol., 2011, 1, 740 Search PubMed.
- G. G. Xia, Y. G. Yin, W. S. Willis, J. Y. Wang and S. L. Suib, J. Catal., 1999, 185, 91 CrossRef CAS.
- S. Singha and K. M. Parida, Catal.: Sci. Technol., 2011, 1, 1496 CAS.
- S. Gomez, L. J. Garces, J. Villegas, R. Ghosh, O. Giraldo and L. S. Suib, J. Catal., 2005, 233, 60 CrossRef CAS.
- K. M. Parida, S. S. Dash and S. Singha, Appl. Catal., A, 2008, 351, 59 CrossRef CAS.
- M. Selvaraj, K. S. Seshadri, A. Pandurangan and T. G. Lee, Microporous Mesoporous Mater., 2005, 79, 261 CrossRef CAS.
- A. Boulaoued, I. Fechete, B. Donnio, M. Bernard, P. Turek and F. Garin, Microporous Mesoporous Mater., 2012, 155, 131 CrossRef CAS.
- S. Haddoum, I. Fechete, B. Donnio, F. Garin, D. Lutic and C. E. Chitour, Catal. Commun., 2012, 27, 141 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- K. Schumacher, P. I. Ravikovitch, A. Du Chesne, A. V. Neimark and K. K. Unger, Langmuir, 2000, 16, 4648 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309 CrossRef CAS.
- E. P. Barret, L. G. Jouner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373 CrossRef.
- M. Hartmann and C. Bischof, J. Phys. Chem. B, 1999, 103, 6230 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. Haul, L. Moscou, R. A. Pirroti, J. Rouquerol and T. Siemoeniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- S. J. Lee, A. Gavriilidis, Q. A. Pankhurst, A. Kyek, F. E. Wagner, P. C. L. Wong and K. L. Yeung, J. Catal., 2001, 200, 298 CrossRef CAS.
- S. Gomez, O. Giraldo, L. J. Garces, J. Villegas and S. L. Suib, Chem. Mater., 2004, 16, 2411 CrossRef CAS.
- P. Van Der Voort, M. Morey, G. D. Stucky, M. Mathieu and E. F. Vansant, J. Phys. Chem. B, 1998, 102, 585 CrossRef CAS.
- D. Zhao and D. Goldfarb, Stud. Surf. Sci. Catal., 1995, 97, 181–188 CrossRef CAS.
- N. N. Tusar, S. Jank and R. Glaser, ChemCatChem, 2011, 3, 254 CrossRef.
- L. Vradman, M. V. Landau, M. Herskowitz, V. Ezersky, M. Talianker, S. Nikitenko, Y. Koltypin and A. Gedanken, J. Catal., 2003, 213, 163 CrossRef CAS.
- J. S. Foord, R. B. Jackman and G. C. Allen, Philos. Mag., 1984, 49, 657 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Perkin Elmer, Eden Praitie, MN, 1992 Search PubMed.
- H. Cao and S. L. Suib, J. Am. Chem. Soc., 1994, 116, 5334 CrossRef CAS.
- B. J. Aronson, C. F. Blanford and A. Stein, J. Phys. Chem. B, 2000, 104, 449 CrossRef CAS.
- G. Lu, X. Li, Z. Qu, Q. Zhao, H. Li, Y. Shen and G. Chen, Chem. Eng. J., 2010, 159, 241 CrossRef.
- G. S. Kumar, M. Palanichamy, M. Hartmann and V. Murugesan, Microporous Mesoporous Mater., 2008, 112, 53 CrossRef CAS.
- Q. Zhang, Y. Wang, S. Itsuki, T. Shishido and K. Takehira, J. Mol. Catal. A: Chem., 2002, 188, 189 CrossRef CAS.
- C. Y. Chen, H. X. Li and M. E. Davis, Microporous Mater., 1993, 2, 17 CrossRef CAS.
- S. Velu, N. Shah, T. M. Jyothi and S. Sivasanker, Microporous Mesoporous Mater., 1999, 33, 61 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1932, 54, 3570 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |