A sol–gel auto-combustion method to prepare Cu/ZnO catalysts for low-temperature methanol synthesis†
Lei
Shi
ab,
Chunyang
Zeng
a,
Yuzhou
Jin
a,
Tiejun
Wang
c and
Noritatsu
Tsubaki
*a
aDepartment of Applied Chemistry, School of Engineering, University of Toyama, Gofuku 3190, Toyama, 930-8555, Japan. E-mail: tsubaki@eng.u-toyama.ac.jp; Fax: +81-76-445-6846; Tel: +81-76-445-6846
bDepartment of Applied Chemistry, Shenyang University of Chemical Technology, Shenyang, 110142, PR China
cGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangdong, 510640, PR China
First published on 17th August 2012
Abstract
Metallic Cu/ZnO catalysts were prepared by a sol–gel auto-combustion method using metal nitrates and citric acid. The activity of the burnt catalysts was investigated for low-temperature methanol synthesis from CO2-containing syngas using ethanol as solvent and a promoter. The activity and the methanol selectivity of the burnt Cu/ZnO catalysts were closely related to metallic Cu0 surface areas and Cu crystalline sizes, and were greatly influenced by the reaction temperature. The effects of M/CA mole ratios on the properties of the burnt catalysts and catalytic activity were systematically studied by Thermogravimetry, Differential Thermal Analysis, Fourier Transform-Infrared, Raman spectrum, X-Ray Diffraction, Scanning Electron Microscope-Energy Dispersive Spectroscopy, Temperature-Programmed Reduction, Brunauer–Emmett–Teller, and N2O chemisorption techniques. The fierceness degree and the temperature of the whole catalyst preparation process were closely related to the balance between the combustion and the pyrolysis processes respectively. Upon continually increasing the citric acid content in the initial precursors, the combustion process became much milder, but excess citric acid in the chelated compound could lead to more violent amorphous carbon oxidation and much more severe pyrolysis of organic residues.
1. Introduction
Methanol is widely used as feedstock in chemical and energy industries, and also as one of the most important liquid fuels that can be supplied to fuel cells via either reforming to hydrogen or the methodology of direct methanol fuel cells (DMFC).1–3 Conventional methods, such as co-precipitation, impregnation, and sol–gel, have been employed to prepare Cu/ZnO catalysts used in high-temperature methanol synthesis (ICI process).Particle size, surface area, metallic Cu0 surface area, and composition of the catalysts are important factors that affect catalytic activity and methanol selectivity. These factors are influenced by different catalyst-preparation methods. Very recently, the sol–gel auto-combustion technique based on the principles of propellant chemistry4 has emerged as an attractive method for the production of high-purity, homogeneous, and crystalline oxide powders5–10 using inexpensive raw materials. This technique proceeds via a combination of chemical sol–gel and subsequent combustion processes. An aqueous solution containing the desired metal salts and organic fuel forms a gel through a sol–gel process. Thereafter, the gel is ignited to combust, yielding a voluminous and fluffy product with large surface area. In this process, citric acid is used as a complexant to form a homogeneous precursor xerogel.11,12
Few reports are available on combustion methods using urea or glycine as a complexant to prepare CuO–ZnO–ZrO catalysts for CO2 hydrogenation13,14 and on the usage of acetylacetonates of copper, zinc, and aluminum for CO hydrogenation15 for methanol synthesis at higher reaction temperatures from 493 to 513 K. However, the efficiency of methanol synthesis at high temperature is severely limited by thermodynamics because of an extremely exothermic reaction. Therefore, developing a low-temperature process for methanol synthesis would result in high one-pass CO conversion and greatly reduce the manufacturing cost. A new method for low-temperature methanol synthesis from CO/CO2/H2 on Cu/ZnO-based catalysts using alcohol as solvent and a promoter is proposed by the present authors; the methanol is produced at 443 K and 5.0 MPa.16–22 A small amount of CO2 (5%) in the syngas could promote the conversion of CO. The used alcohol solvent contains a small amount of water, so this new low-temperature process could directly use low-grade syngas containing CO2 and H2O without purification.
In the present work, the effect of citric acid content on the activity and methanol selectivity of the burnt catalysts prepared by a sol–gel auto-combustion method is systemically studied by multifold characterization techniques. The optimal reaction temperature for the burnt Cu/ZnO catalysts in this low-temperature methanol synthesis is obtained.
2. Experimental
2.1 Preparation of catalysts
A schematic flow chart of the preparation of the catalysts by a sol–gel auto-combustion method is presented in Fig. 1. Analytical-grade Cu(NO3)2·3H2O, Zn(NO3)2·6H2O and citric acid were used as raw materials. The metal nitrates with a mole ratio of Cu/Zn = 1/1 (denoted M, M = Cu2+ + Zn2+) and citric acid (denoted CA) were first dissolved in distilled water according to different M/CA mole ratios of 1/0.5, 1/0.7, 1/0.8, 1/1.0, 1/1.2, 1/1.3, 1/1.4, 1/1.5, and 1/1.8, denoted C0.5, C0.7, C0.8, C1, C1.2, C1.3, C1.4, C1.5, and C1.8. The solutions were adjusted by using 28% (wt%) ammonia to reach a pH value of 7.0. Stirring and refluxing at 353 K for 4 h ensured that the citric acid completely chelated with the metal ions. Subsequently, the neutralized solution was evaporated at 343 K on a hot plate by continuously stirring until dark blue viscous xerogels were formed. Thereafter, the gels were dried at 393 K for 40 h and loaded into a muffle oven. The temperature was increased to about 493 K in air at a heating rate of 2 K min−1, leading to the ignition of the gels. According to the principle of propellant chemistry,4 the gels were burnt in a self-propagating combustion manner until all were burnt out, and formed a loose product. Subsequently, the temperature was continuously increased to about 723 K at a heating rate of 2 K min−1 and maintained for 3 h. The burnt catalysts were reduced by a flow of 5% hydrogen in nitrogen at 523 K for 10 h and successively passivated by 1% oxygen diluted by nitrogen.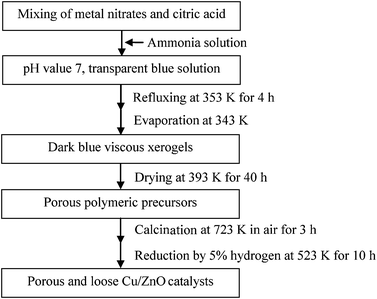 | ||
| Fig. 1 Schematic flow chart of the preparation of the catalysts by a sol–gel auto-combustion method. | ||
2.2 Characterization techniques
The thermal decomposition behaviour of the xerogels was characterized by differential thermogravimetric analysis (DTA/TGA 60, Shimadzu) at a heating rate of 10 K min−1 from 293 to 1173 K in air.XRD patterns were obtained on a Rigaku RINT 2400 X-ray diffractometer using monochromatic Cu Kα radiation, scanning 2θ from 20° to 80°, and operated at 40 kV and 40 mA. The copper crystalline average size was calculated by Scherrer's formula: D = Kλ/(β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ), where D is the average dimension of crystallites, K is Scherrer's constant 0.89, λ is the wavelength of X-ray (Cu Kα = 0.154 nm), and β is the width of the peak at half height.
cosθ), where D is the average dimension of crystallites, K is Scherrer's constant 0.89, λ is the wavelength of X-ray (Cu Kα = 0.154 nm), and β is the width of the peak at half height.
The infrared spectrum was recorded in air using a Shimadzu Fourier Transform Infrared Spectrophotometer (FTIR-8400).
TPR experiments were carried out by using a catalyst analyzer (BELCAT-B, BEL, Japan Co., Ltd.) using 0.035 g burnt catalysts with 5% hydrogen diluted by argon at atmospheric pressure. Before reduction, the catalysts were heated at 323 K in flowing argon for 2 h. Then, 5% hydrogen (50 ml min−1) was passed over the catalyst sample, and the temperature was linearly raised from 298 to 723 K at a heating rate of 5 K min−1. The water formed during the reduction was trapped by 3 Å molecular sieves. The effluent gas was analyzed by using a thermal conductivity detector (TCD) with argon as a reference.
The physical structures of the catalysts were investigated and compositional analysis and surface morphology observation were performed by scanning electron microscopy (SEM) (JEOL, JSM-6360 LV) with energy-diffusive X-ray spectroscopy (EDX) (JEOL, JED-2300). The samples for this analysis were first coated with a platinum layer on the surface by using an auto fine coater (JEOL, JFC-1600).
The copper (Cu0) surface area (SCu) was determined by a nitrous oxide (N2O) pulse method as described elsewhere.23
The surface area of the catalysts was determined by N2 physical desorption at 77 K using a Micromeritics NOVA 1000 SP surface area and porosimetry analyzer. The sample was degassed at 473 K prior to acquiring the adsorption isotherm. The surface area was calculated according to the BET method using 0.164 nm2 as the cross-sectional area of the nitrogen molecule.
2.3 Catalytic activity tests
A batch reactor with 85 ml inner volume and a stirrer were used to study the catalytic activity. Approximately 1 g catalyst and 40 ml ethanol were first poured in the autoclave. The air inside was first purged by the reactant gas, Ar/CO/CO2/H2 = 3.13/33/5.23/58.64, then the pressure in the reactor was raised to 5.0 MPa at 298 K. The reaction took place at 443, 453, 463, or 473 K separately for 8 h. The stirring speed was fixed at 2000 rpm to prevent the diffusion-controlled regime. A continuous low-temperature methanol synthesis was conducted at 453 K and 5.0 MPa for 40 h using a flow-type batch reactor21 with 85 ml inner volume, 3 g catalyst, and 40 ml ethanol. The flow rate of the syngas was fixed at 20 ml min−1. All products were determined on GC-MS (Shimadzu GCMS 1600). A TCD (Shimadzu GC-320) and a flame ionization detector (Shimadzu GC-8A) were used to analyze the gaseous and the liquid products, respectively. The CO, CO2, total carbon conversions, and the selectivity of the liquid products were calculated as follows:| CO conv. = 100 × [(CO/Ar in feed − CO/Ar in effluent)]/(CO/Ar in feed) | (1) |
| CO2 conv. = 100 × [(CO2/Ar in feed − CO2/Ar in effluent)]/(CO2/Ar in feed) | (2) |
| Total carbon conv. = CO conv. × a/(a + b) + CO2 conv. × b/(a + b) | (3) |
The selectivity of the liquid products was calculated as follows:
| π selectivity = 100 × π yield/∑π yield | (4) |
3. Results and discussion
3.1 Studies on the auto-combustion process by TG-DTA analysis in air
The thermal behavior of the precursors with different mole ratios of metal nitrates (denoted M, M = Cu2+ + Zn2+) to citric acid (denoted CA) was investigated by TG-DTA measurements in an air atmosphere and is compared in Fig. 2. During the combustion process, all precursors were ignited at about 493 K and exhibited rapid self-propagating combustion until all were burnt out to form loose and porous powders. | ||
| Fig. 2 TG-DTA curves of (a) pure citric acid and the precursors of the burnt catalysts (b) C0.5, (c) C1, (d) C1.4, (e) C1.5, and (f) C1.8. | ||
As displayed in Fig. 2a, the DTA plot of pure citric acid presented two endothermic and one exothermic peaks. The first endothermic peak at about 430 K could be assigned to the melting of citric acid, without any weight loss. Another endothermic peak at about 473 K came from the decomposition of citric acid,24 releasing H2O and CO2, and resulting in a large weight loss. The exothermic peak at higher temperature (683 K) arose from the energy combination of amorphous carbon oxidation and pyrolysis of organic residues in air. As observed from the TG plot in Fig. 2a, no mass was left.
The DTA plot in Fig. 2b for the precursor with M/CA = 1/0.5 has only one extremely sharp exothermic peak at about 493 K in a very narrow temperature range. Simultaneously, the TG curve displayed an abrupt weight loss, indicating that the combustion process of the precursor (C0.5) occurred as a severe redox reaction, wherein citric acid acted as a reductant. On the other hand, nitrate ions and O2 (in air) acted as oxidants. In agreement with the literature studies,6,25 during this combustion process, a large amount of gases, such as H2O, CO2, N2, and NO2, were liberated. Upon increasing the citric acid content in the initial precursors, the DTA curves in Fig. 2c and d (the precursors of C1 and C1.4) exhibited one sharp exothermic peak with a shoulder peak at lower than 493 K, where the shoulder peak was derived from the overlapped accumulation of the energy absorbed by the decomposition of citric acid (as compared in Fig. 2a at about 473 K) and the energy released from the combustion process. Upon continually increasing the citric acid content in the precursors, the DTA curves in Fig. 2e and f (the precursors of C1.5 and C2.0) exhibited one broad exothermic peak at about 493 K, indicating that the combustion process was significantly smoother. The greater reductant content in the redox reaction made the redox process much milder, and citric acid was just the reductant taking part in the redox reaction. Another reason was that the decomposition of citric acid was an endothermic process accompanied by release of H2O and CO2, which improved heat diffusion and heat adsorption.
The second exothermic peak at about 590 K in Fig. 2c and d arose from the oxidation of amorphous carbon, which was supported by the experimental phenomena reported by Liu et al.26 and Pranda et al.27 The third exothermic peak at about 653 K was derived from the pyrolysis of organic residues. The area of exothermic peak at about 653 K in Fig. 2d was obviously much larger than that in Fig. 2c, indicating that more organic residues remained in the burnt catalyst C1.4 than that in C1. Upon continually increasing the citric acid content, the second exothermic peak at about 683 K in Fig. 2e and f became larger and broader, which corresponded to the energy combination of the amorphous carbon oxidation and the pyrolysis of the organic residues. It was deduced that more content of organic residues (which was further proven by FT-IR, Raman spectrum, and EDS analysis) remained in the burnt catalysts with the increased content of citric acid in the initial precursors. As displayed in Fig. 2b, no exothermic peak was attributed to the pyrolysis of organic residues. With a small content of citric acid (reductant), the combustion was more severe and the flame temperature was much higher, which resulted in the complete decomposition of organic residues.
The fierceness degree and the temperature of the whole catalyst-preparation process, which were strongly influenced by the citric acid content, were closely related to the balance of combustion and pyrolysis processes. During the combustion process which was already proven to be a redox process, more reductants, such as citric acid, could make the combustion process much milder. However, excess citric acid could lead to much more organic residues that might cause a more severe pyrolysis process (as compared in Fig. 2e and f). Therefore, there was a balanced point between the citric acid content and metal nitrates, through which the whole process took place in a less severe way. The DTA curve (the precursor of C1.4) in Fig. 2d displayed an equal exothermic peak area at 493 and 653 K, which suggested the desirable match of the combustion and the pyrolysis processes.
3.2 FT-IR analysis of the precursors and the burnt catalysts with different M/CA mole ratios
In metal ions–citric acid system, the formation of citric acid–metal ion chelated complexes could prevent the hydrolysis of metal ions and yield homogeneous products. The dissociation of carboxylic acid groups was controlled by the pH value of the solution. All metal ions (Cu2+ and Zn2+) were completely chelated at a pH value of 7.0.The FT-IR spectra of the precursors with different M/CA mole ratios are compared in Fig. 3. A transmission band at around 3180 cm−1 indicated the presence of adsorbed moisture.28 The band at 2376 cm−1 was attributed to the adsorption of CO2.28,29 The bands at 820 and 1384 cm−1 were assigned to the NO3−.8,28,29 The significantly broad peaks at around 3000–3100 cm−1 were assigned to molecular H2O with a high extent of hydrogen bonding interaction. Two bands occurred near 1620 and 1400 cm−1, representing the asymmetric and symmetric stretching vibrations for carboxyl ions (COO−), whereas the bands at 1720 and 1770 cm−1 were from the free carboxylic group.8,30 Citric acid had two types of carboxyl groups, one inner carboxyl (1770 cm−1) and two terminal free carboxyl groups (1720 cm−1). Two bands at 1720 and 1770 cm−1 are displayed in Fig. 3b and c. However, no band was found at 1720 cm−1 in Fig. 3a, indicating that two terminal-free carboxyl groups were completely chelated in the precursors with M/CA = 1/0.5.
 | ||
| Fig. 3 FT-IR spectra of the precursors of the burnt catalysts (a) C0.5, (b) C1.4, and (c) C1.8. | ||
Fig. 4 compares the FT-IR spectra (in transmittance mode) of the burnt catalysts with different M/CA mole ratios. It is clear that no NO3− (820 and 1384 cm−1) and carboxyl bands were found for all the burnt catalysts. These findings clarified that all NO3− and carboxyl took part in the combustion process. NO3− provided an in situ oxidizing environment for the decomposition of organic components.
 | ||
| Fig. 4 FT-IR spectra of the burnt catalysts (a) C0.5, (b) C1.4, and (c) C1.8. | ||
Almost no bands were compared in Fig. 4a, indicating that little carbonic residue was left in the burnt catalyst C0.5, which was supported by the result of the TG-DTA analysis in Fig. 2b. Broad bands at about 1400 to 1600 cm−1 are compared in Fig. 4b and c. The bands at 1428, 1534, 1551, and 1582 cm−1 were attributed to the carbonates species (COOO).19,31 The bands at 1352 and 1580 cm−1 were attributed to the symmetric and asymmetric stretches of carboxylate species (OCO).19,31–33 With the increased content of citric acid in the initial precursors, the FT-IR band area of the burnt catalysts C1.4 and C1.8 at about 1352, 1428, 1534, 1551, and 1582 cm−1 increased gradually, suggesting that more carbonate (COOO) and carboxylate species (OCO) were left in the burnt catalysts.
3.3 XRD analysis of the burnt catalysts after reduction with different M/CA mole ratios
The XRD patterns of the burnt catalysts with different M/CA mole ratios and reduced by a flow of 5% hydrogen in nitrogen at 523 K for 10 h are compared in Fig. 5. All diffraction peaks of the burnt catalysts after reduction with different M/CA mole ratios were indexed to Cu and ZnO phases. The Cu crystalline sizes in Fig. 5 calculated by Scherrer's formula are compared in Table 2. As compared in Fig. 5 and Table 2, upon increasing the citric acid content, the Cu crystalline sizes decreased at the outset and were smallest for C1.4 with M/CA = 1/1.4, after which it increased. The Cu crystalline sizes were closely related to the citric acid content in the initial precursors, which has an important effect on the temperature during the combustion and pyrolysis processes. Decreasing the reductant content (citric acid) could lead to the increase in the flame temperature and make the combustion process more severe. However, increasing the citric acid content in the initial precursors caused much more amorphous carbon and organic residues to be left, which made the carbon oxidation and the pyrolysis process more severe, resulting in higher pyrolysis temperature. Higher temperature resulted in larger Cu crystalline sizes. The burnt catalyst C1.4 in Fig. 5c displayed the smallest Cu crystalline size, which suggested the desirable match of the combustion and the pyrolysis processes. This result was in accordance with TG-DTA analysis in Fig. 2. | ||
| Fig. 5 XRD patterns of the burnt catalysts after reduction (a) C0.5, (b) C1, (c) C1.4, (d) C1.5, and (e) C1.8 (☆, Cu; ■, ZnO). | ||
3.4 SEM-EDS analysis of the burnt catalysts with different M/CA mole ratios
The SEM micrographs of the burnt catalysts after reduction with different M/CA mole ratios are compared in Fig. 6. All the burnt catalysts exhibited highly porous microstructures. However, the morphologies were different. The burnt catalyst C0.5 in Fig. 6a displayed a macroporous structure, which was derived from higher flame temperature and abundant gases released in a split second. For the burnt catalyst C1.4 in Fig. 6b, the sample had a large amount of small pores, and the distribution of the small pores was uniform. In contrast to C1.4, the catalyst C1.8 in Fig. 6c presented polymerization on the surface, which might be caused by excess citric acid. The unchelated citric acid changed to the quasi-solid state at about 430 K, then, surrounded the xerogels. Finally, the organic residues derived from the pyrolysis of the excess citric acid were obtained and coated on the surface of the catalyst. | ||
| Fig. 6 SEM micrographs of the burnt catalysts (a) C0.5, (b) C1.4, and (c) C1.8. | ||
The surface element distribution in the defined red line in Fig. 6b was analyzed by an EDS technique and the results are displayed in Fig. 7 and Table 1. For the burnt catalyst C1.4, only Cu, Zn, O, and C elements were detected. The Cu/Zn atomic ratio in the red line was about 1/1.004, almost the same as that in the initial ratio of the solution, and the atomic mole ratio of Cu/Zn along the red line was steady. Hence, the sol–gel auto-combustion method could be a reliable way to prepare heterogeneous metallic catalysts which yielded precise and desired compositions.
 | ||
| Fig. 7 EDS line (a red line in Fig. 6b) scanning: element distributions. Red Cu: 25.1 atom%, blue Zn: 25.0 atom%, black C: 12.0 atom%. | ||
| Surface element contenta (atomic ratio %) | Catalysts | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C0.5 | C0.7 | C0.8 | C1 | C1.2 | C1.3 | C1.4 | C1.5 | C1.8 | |
| a Determined by elemental analysis. | |||||||||
| C | 5.2 | 8.0 | 8.8 | 9.7 | 10.4 | 11.1 | 12.0 | 14.6 | 16.2 |
| O | 31.2 | 32.0 | 32.0 | 33.1 | 34.0 | 35.6 | 37.9 | 38.3 | 42.4 |
| Cu | 32.1 | 29.8 | 29.6 | 28.4 | 27.4 | 26.4 | 25.1 | 23.6 | 21.1 |
| Zn | 31.5 | 30.2 | 29.6 | 28.8 | 28.2 | 26.9 | 25.0 | 23.5 | 20.3 |
The EDS analyses of the burnt catalysts after reduction and with different M/CA mole ratios are listed in Table 1. The SEM images (magnification: 1000 times) for EDS analysis were duplicated five times to enhance the accuracy by accumulation. As listed in Table 1, a part of the carbonic residues stayed on the surface of the burnt catalysts. The amount of carbonic residues was associated with the citric acid content in the initial precursors. Upon increasing the citric acid content, more carbonic residues were left on the surface of the catalysts. Simultaneously, the atomic ratio of Cu and Zn on the surface gradually decreased. These results were consistent with those proven by TG-DTA, FT-IR, and Raman spectrum which are discussed in the ESI† and in Fig. S1.
3.5 TPR analysis of the burnt catalysts with different M/CA mole ratios
The TCD signal in terms of hydrogen consumption from H2-TPR of the burnt catalysts is compared in Fig. 8. All the burnt catalysts have only one peak that corresponded to the reduction of CuO to metallic Cu0 in one step, implying that the CuO phase dispersed homogeneously in the burnt catalyst, which was consistent with the line EDS analysis result in Fig. 7. | ||
| Fig. 8 TPR profiles of the burnt catalyst (a) C0.5, (b) C1.4, and (c) C1.8. | ||
As compared in Fig. 8, the freshly burnt catalyst C0.5 has a surprisingly high temperature, and the main reduction peak was located at about 541 K. The burnt catalyst C1.4 displayed the reduction peak at about 518 K, 23 K lower than that of C0.5. The catalyst C1.8 has a reduction peak at about 531 K. Pure CuO bulk was reduced at a considerably higher temperature of 573 K,34–36 indicating that the presence of ZnO could facilitate the reduction of CuO. According to the results from the XRD analysis and metallic Cu0 surface area determined by N2O chemisorption, the reduction peak at lower temperature represented the well-dispersed CuO species with smaller Cu crystalline sizes, whereas the reduction peak at higher temperature was attributed to the reduction of CuO species with larger Cu crystalline sizes. This conclusion was in accordance with the literature studies37–39 that the reduction of smaller CuO crystalline was easy, and the TPR profiles preferred at lower temperature.
As compared in Fig. 8a–c, all the TPR profiles of the burnt catalysts were almost symmetrical, whereas their shapes were different. The burnt catalyst C0.5 exhibited a narrow and symmetrical TPR profile which indicated a narrow particle size distribution. However, the TPR profiles of the burnt catalysts C1.4 and C1.8 were relatively broader, indicating that CuO particle size distribution was wider.40,41 These phenomena were better explained by the TG-DTA results during the combustion and pyrolysis processes. For the burnt catalyst C0.5, the combustion process was severe because of an insufficient reductant in the precursor, which possibly led to higher flame temperature. However, the combustion process proceeded for only a few seconds, as displayed in Fig. 2b. As the time duration of combustion was short, the burnt catalyst C0.5 had a narrow particle size distribution. For the burnt catalyst C1.4, the combustion process was less severe than that for C0.5 because of more reductant in the precursor. However, a part of amorphous carbon and organic residues remained after the combustion process. The oxidation of amorphous carbon and the pyrolysis of organic residues were also a sustained heat releasing process, as shown in Fig. 2d. Therefore, the catalyst C1.4 had a wide particle size distribution. Likewise, the burnt catalyst C1.8 had a wider particle size distribution, and the TPR profiles were broader than those of C1.4 because much more amorphous carbon and organic residues derived from the decomposition of citric acid remained.
3.6 Activity of the burnt catalysts
The activity of the burnt catalysts was investigated in low-temperature methanol synthesis from CO2-containing syngas using ethanol as solvent and a promoter at 443 K and 5.0 MPa for 8 h. The synthetic route was a new method of low-temperature methanol synthesis.16,17 This new method allowed the use of solid catalysts and syngas containing CO2 and H2O directly from an industrial reformer or biomass gasifier without deep purification and could be performed at significantly low temperature.The characterization results of Cu/ZnO catalysts and batch reaction records for low-temperature methanol synthesis are compared in Table 2. When the sol–gel combustion method was used, the BET surface areas and the copper (Cu0) surface areas of the burnt catalysts increased upon increasing the citric acid content at the outset, reaching a maximum for the burnt catalysts C1.4, and then decreasing further. The variation trend of the BET surface areas and the copper (Cu0) surface areas was similar to that of the Cu crystalline size changes. Upon increasing the citric acid content, more gases were released, which led to the porous structure. However, the excess citric acid resulted in surplus of the carbonic residues, which partly coated on the burnt catalysts and caused the decrease in the surface areas. It was clarified that the BET surface areas and the copper (Cu0) surface areas of the burnt catalysts were influenced by Cu crystalline sizes and the amount of carbonic residues. There was a balanced point between the content of carbonic residues and Cu crystalline sizes, at which the burnt catalysts had the largest BET surface areas and largest Cu0 surface areas.
| Catalysts | S BET a (m2 g−1) | S Cu b (m2 g−1) | Cuc Dia. (nm) | Conversion (%) | TOFd (10−3 s−1) | Selectivity (%) | Yield (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| Ctotal | CH3OH | HCOORe | CH3OH | HCOOR | |||||
| Reaction conditions: T = 443 K, P = 5.0 MPa, catalyst weight: 1 g, ethanol solvent: 40 ml, stirring speed: 2000 rpm, reaction time: 8 h, syngas: Ar/CO/CO2/H2 = 3.13/33/5.23/58.64.a Determined by N2 physical adsorption–desorption at 77 K.b Determined from N2O pulse chemisorption.c Calculated using the Scherrer formula.d TOF is the total carbon turnover frequency.e HCOOR is ethyl formate. | |||||||||
| C0.5 | 5.0 | 1.5 | 68.4 | 25.2 | 9.4 | 31.1 | 68.9 | 7.8 | 17.4 |
| C0.7 | 12.9 | 3.8 | 60.2 | 34.3 | 5.0 | 39.0 | 61.0 | 13.4 | 20.9 |
| C0.8 | 16.3 | 4.7 | 46.8 | 40.3 | 4.8 | 42.1 | 57.9 | 17.0 | 23.3 |
| C1 | 17.5 | 4.9 | 43.1 | 45.2 | 5.1 | 50.3 | 49.7 | 22.7 | 22.5 |
| C1.2 | 22.0 | 5.9 | 41.5 | 46.6 | 4.4 | 64.5 | 35.5 | 30.0 | 16.5 |
| C1.3 | 23.4 | 6.2 | 40.6 | 50.5 | 4.5 | 68.3 | 31.7 | 34.5 | 16.0 |
| C1.4 | 28.9 | 7.7 | 34.0 | 55.3 | 4.0 | 77.1 | 22.9 | 42.6 | 12.7 |
| C1.5 | 17.2 | 4.2 | 42.8 | 43.2 | 5.7 | 56.6 | 43.4 | 24.5 | 18.7 |
| C1.8 | 15.8 | 3.4 | 52.3 | 38.3 | 6.3 | 41.5 | 58.5 | 15.9 | 22.4 |
Only CO and CO2 were the carbon-containing gas-phase products, and only methanol and ethyl formate were the liquid-phase products. As compared in Table 2, the total carbon conversion increased upon increasing the citric acid content at the outset, which reached a maximum for the burnt catalyst C1.4, and decreased further. The variation trend was closely related to that of the Cu0 surface areas. This result was consistent with the literature data,42–44 because higher Cu surface areas provided more active sites, and Cu0 was the active site for the rate-determining step in this low-temperature methanol synthesis.
As compared in Table 2, the selectivity of methanol remarkably increased from 31.1 to 77.1% when the Cu crystalline sizes decreased from 68.4 (C0.5) to 34 nm (C1.4). The proposed mechanism17–19 in our previous work for low-temperature methanol synthesis from syngas (CO/CO2/H2) using ethanol as a promoter (B was the as-burnt catalyst Cu/ZnO) is displayed in Fig. 9. Cu+ and ZnO were the active sites for the formation of formate and ethyl formate. However, only metallic Cu0 was active for the hydrogenation of ethyl formate. The methanol selectivity was possibly associated with the crystalline sizes of Cu0. The smaller crystalline size of Cu0 was beneficial in methanol formation, exhibiting higher hydrogenation capability. As clarified by our previous research on this new low-temperature synthesis method,16–23 metallic Cu0 was the active site for the hydrogenation of formic ester in forming methanol. From the TPR findings, the burnt catalyst with finely dispersed Cu oxide was easier to be reduced, as shown by its behavior in C0.5, C1.4, and C1.8. Consequently, these catalysts exhibited higher activity and methanol selectivity than those with larger Cu crystalline sizes and lower Cu dispersion.
 | ||
| Fig. 9 Mechanism for low-temperature methanol synthesis from syngas (CO/CO2/H2) using ethanol as solvent and a promoter (B was the burnt catalyst Cu/ZnO). | ||
As compared with the other burnt catalysts, the burnt catalyst C1.4 has larger Cu0 surface area and the highest catalytic activity. Therefore, C1.4 was selected for further investigation. The effects of reaction temperature on low-temperature methanol synthesis are investigated in Table 3. In our former work,16–23 443 K was found to be the best temperature when co-precipitation and impregnation method were used to prepare Cu/ZnO catalysts with a series of different alcohols as solvent and promoters. When the sol–gel combustion method was used, the effect of reaction temperature on the activity and methanol selectivity of the burnt catalysts was investigated. At 443 K, the prepared catalyst C1.4 exhibited the total carbon conversion of 55.3% and a methanol selectivity of 77.1%, which indicated that the hydrogenation activity was not high enough at this temperature. When the temperature rose to 453 K, the total carbon conversion of the catalyst C1.4 increased to 58.3% and the methanol selectivity reached 89.5%. Upon further increasing the temperature to 463 K, the methanol selectivity and the conversion decreased. When the temperature was 473 K, the selectivity and the conversion remarkably decreased. These results indicated that temperature had an enormous effect on the low-temperature methanol synthesis reaction. High temperature was unsuitable for increasing the total carbon conversion and the selectivity of methanol synthesis. The efficiency of methanol synthesis was severely limited by thermodynamics because the methanol synthesis was an extremely exothermic reaction. The optimum reaction temperature for the burnt Cu/ZnO catalysts under the present conditions is 453 K.
| Catalysts | Reaction temperature (K) | Conversion (%) | TOFa | Selectivity (%) | Yield (%) | ||
|---|---|---|---|---|---|---|---|
| Ctotal | (10−3 s−1) | CH3OH | HCOOR | CH3OH | HCOOR | ||
| Reaction conditions: P = 5.0 MPa, catalyst weight: 1 g, solvent: ethanol 40 ml, stirring speed: 2000 rpm, reaction time: 8 h, syngas: Ar/CO/CO2/H2 = 3.13/33/5.23/58.64.a TOF is the total carbon turnover frequency. | |||||||
| C1.4 | 443 | 55.3 | 4.0 | 77.1 | 22.9 | 42.6 | 12.7 |
| 453 | 58.3 | 4.2 | 89.5 | 10.1 | 52.2 | 5.9 | |
| 463 | 53.2 | 3.9 | 70.4 | 29.6 | 37.5 | 15.7 | |
| 473 | 48.9 | 3.5 | 54.6 | 45.4 | 26.7 | 22.2 | |
The continuous reaction results for low-temperature methanol synthesis at 453 K and 5.0 MPa for 40 h using 3 g of the burnt catalyst C1.4 are presented in Table 4 and Fig. 10. Fig. 10 shows the time-on-stream activity change of the reaction during 40 h. The analysis of the effluent gas after the trap manifested that only CO, CO2, and H2 existed. No alcohol or ester appeared. At the initial stage of the reaction, 45 ml dead volume of the reactor and 18 ml cold trap must be filled by the pressurized feed gas. Thus, the apparent conversions were low but increased gradually. After 12 h, the conversions were stable. The CO conversion was about 40%, whereas the CO2 conversion was about −2%. The total carbon conversion was about 34%. At the initial 12 h, CO2 conversion dropped to a minimum of −15%, and then increased again to about −2%. The negative CO2 conversion proved that CO was first converted to CO2 through the water-shift reaction, and then CO2 was hydrogenated to methanol. The balance between CO2 and formate formation was reached after 12 h. After 40 h reaction, the methanol selectivity calculated by analyzing the liquid products (the total of those in the cold trap and in the autoclave) was 92.4%. The total carbon TOF calculated by N2O pulse method was about 3.6 × 10−3 s−1.
| Catalyst | Conversion (%) | TOFa | Selectivity (%) | Yield (%) | ||
|---|---|---|---|---|---|---|
| Ctotal | (10−3 s−1) | CH3OH | HCOOR | CH3OH | HCOOR | |
| Reaction conditions: T = 453 K, P = 5.0 MPa, catalyst weight: 3 g, solvent: ethanol 40 ml, stirring speed: 2000 rpm, reaction time: 40 h, syngas: Ar/CO/CO2/H2 = 3.13/33/5.23/58.64, 20 ml min−1.a Based on the N2O pulse method. TOF is the total carbon turnover frequency. | ||||||
| C1.4 | 34.6 | 3.6 | 92.4 | 7.6 | 32.0 | 2.6 |
 | ||
| Fig. 10 Variations of CO, CO2, and total carbon conversions with reaction time for continuous synthesis of methanol (reaction conditions: T = 453 K, P = 5.0 MPa, catalyst weight: 3 g, solvent: ethanol 40 ml, stirring speed: 2000 rpm, reaction time: 40 h, syngas: Ar/CO/CO2/H2 = 3.13/33/5.23/58.64, 20 ml min−1). | ||
4. Conclusion
The effects of citric acid content on the properties of the burnt catalysts and catalytic activity were systematically studied. Upon increasing the citric acid content in the initial precursors, the combustion process became much milder and the flame temperature became much lower. However, excess citric acid led to much more amorphous carbon and organic residues, which caused more severe carbon oxidation and pyrolysis. The variation trend of Cu crystalline sizes, BET areas, and Cu0 surface areas was closely related to the fierceness degree and the temperature of the whole catalyst-preparation process. The burnt catalyst C1.4 suggested the desirable match of the combustion process and the pyrolysis of organic residues. The effects of reaction temperature on low-temperature methanol synthesis were investigated from 443 to 473 K. The activity and the methanol selectivity were highest at 453 K. The continuous reaction was conducted at 453 K and 5.0 MPa for 40 h using the burnt catalyst C1.4. The total carbon conversion was about 34.6%, and it was stable during 40 h reaction. The methanol selectivity was about 92.4%.In the present study, metallic catalysts (Cu/ZnO) were prepared by a sol–gel combustion method with inexpensive raw materials and under uncomplicated operation conditions. This proposed combustion method could be used to prepare metallic and supported metallic catalysts with homogeneously distributed composition.
Acknowledgements
JST-NSFC joint research fund for biomass conversion and C1 chemistry is greatly appreciated.References
- H. S. Liu, C. J. Song, L. Zhang, J. J. Zhang, H. J. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95 CrossRef CAS.
- J. P. Breen and J. R. H. Ross, Catal. Today, 1999, 51, 521 CrossRef CAS.
- M. S. Wainwright and D. L. Trimm, Catal. Today, 1995, 23, 29 CrossRef CAS.
- S. R. Jain, K. C. Adiga and V. R. Pai Verneker, Combust. Flame, 1981, 40, 71 CrossRef CAS.
- Q. Xiao, Z. C. Si, Z. M. Yu and G. Z. Qiu, Mater. Sci. Eng., B, 2007, 137, 189 CrossRef CAS.
- Y. W. Jiang, S. G. Yang, Z. H. Hua and H. B. Huang, Angew. Chem., Int. Ed., 2009, 48, 8529 CrossRef CAS PubMed.
- A. S. Mukasyan and P. Dinka, Adv. Eng. Mater., 2007, 9, 653 CrossRef CAS.
- Y. Y. Li, L. H. Xue, L. F. Fan and Y. W. Yan, J. Alloys Compd., 2009, 478, 493 CrossRef CAS.
- J. Chandradass, M. H. Kim and D. S. Bae, J. Alloys Compd., 2009, 470, L9 CrossRef CAS.
- J. L. Liu, W. Zhang, C. J. Guo and Y. W. Zeng, J. Alloys Compd., 2009, 479, 863 CrossRef CAS.
- Y. J. Hao, Q. Y. Lai, D. Q. Liu, Z. Xu and X. Y. Ji, Mater. Chem. Phys., 2005, 94, 382 CrossRef CAS.
- S. Vivekanandhan, M. Venkateswarlu and N. Satyanarayana, J. Alloys Compd., 2008, 462, 328 CrossRef CAS.
- X. M. Guo, D. S. Mao, S. Wang, G. S. Wu and G. Z. Lu, Catal. Commun., 2009, 10, 1661 CrossRef CAS.
- X. M. Guo, D. S. Mao, G. Z. Lu, S. Wang and G. S. Wu, J. Catal., 2010, 271, 178 CrossRef CAS.
- J. R. Jensen, T. Johannessen, S. Wedel and H. Livbjerg, J. Catal., 2003, 218, 67 CrossRef CAS.
- L. Fan, Y. Sakaiya and K. Fujimoto, Appl. Catal., A, 1999, 180, L11 CrossRef CAS.
- N. Tsubaki, M. Ito and K. Fujimoto, J. Catal., 2001, 197, 224 CrossRef CAS.
- N. Tsubaki, J. Q. Zeng, Y. Yoneyama and K. Fujimoto, Catal. Commun., 2001, 2, 213 CrossRef CAS.
- R. Q. Yang, Y. L. Fu, Y. Zhang and N. Tsubaki, J. Catal., 2004, 228, 23 CrossRef CAS.
- R. Q. Yang, Y. Zhang and N. Tsubaki, Catal. Commun., 2007, 8, 1839 Search PubMed.
- B. L. Xu, R. Q. Yang, F. Z. Meng, P. Reubroycharoen, T. Vitidsant, Y. Zhang, Y. Yoneyama and N. Tsubaki, Catal. Surv. Asia, 2009, 13, 147 CrossRef CAS.
- T. S. Zhao, K. Zhang, X. R. Chen, Q. X. Ma and N. Tsubaki, Catal. Today, 2010, 149, 98 CrossRef CAS.
- R. Q. Yang, X. C. Yu, Y. Zhang, W. Z. Li and N. Tsubaki, Fuel, 2008, 87, 443 CrossRef CAS.
- L. C. Pathak, T. B. Singh, S. Das, A. K. Verma and P. Ramachandrarao, Mater. Lett., 2002, 57, 380 CrossRef CAS.
- J. G. Huang, H. R. Zhuang and W. L. Li, Mater. Res. Bull., 2003, 38, 149 CrossRef CAS.
- J. H. Zhao, Z. Y. Liu and D. K. Sun, J. Catal., 2004, 227, 297 CrossRef CAS.
- P. Pranda, K. Prandová and V. Hlavacek, Fuel Process. Technol., 1999, 61, 211 CrossRef CAS.
- J. Chandradass and K. H. Kim, Adv. Powder Technol., 2010, 21, 100 CrossRef CAS.
- Z. X. Wei, L. Wei, L. Gong, Y. Wang and C. W. Hu, J. Hazard. Mater., 2010, 177, 554 CrossRef CAS PubMed.
- W. D. Li, J. Z. Li and J. K. Guo, J. Eur. Ceram. Soc., 2003, 23, 2289 CrossRef CAS.
- R. Srivastava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1 CrossRef CAS.
- A. Yee, S. J. Morrison and H. Idriss, J. Catal., 1999, 186, 279 CrossRef CAS.
- A. Yee, S. J. Morrison and H. Idriss, J. Catal., 2000, 191, 30 CrossRef CAS.
- R. Kama, C. Selomulya, R. Amal and J. Scott, J. Catal., 2010, 273, 73 CrossRef.
- P. Kurr, I. Kasatkin, F. Girgsdies, A. Trunschke, R. Schlögl and T. Ressler, Appl. Catal., A, 2008, 348, 153 CrossRef CAS.
- M. Turco, G. Bagnasco, U. Costantino, F. Marmottini, T. Montanari, G. Ramis and G. Busca, J. Catal., 2004, 228, 43 CAS.
- L. C. Wang, Y. M. Liu, M. Chen, Y. Cao, H. Y. He, G. S. Wu, W. L. Dai and K. N. Fan, J. Catal., 2007, 246, 193 CrossRef CAS.
- P. Djinović, J. Batista and A. Pintar, Appl. Catal., A, 2008, 347, 23 CrossRef.
- X. B. Zhang, L. Zhong, Q. H. Guo, H. Fan, H. Y. Zheng and K. Xie, Fuel, 2010, 89, 1348 CrossRef CAS.
- J. Agrell, H. Birgersson, M. Boutonnet, I. Melián-Cabrera, R. M. Navarro and J. L. G. Fierro, J. Catal., 2003, 219, 389 CrossRef CAS.
- J. Agrell, M. Boutonnet, I. Melián-Cabrera and J. L. G. Fierro, Appl. Catal., A, 2003, 253, 201 CrossRef CAS.
- G. C. Chinchen, K. C. Waugh and D. A. Whan, Appl. Catal., 1986, 25, 101 CrossRef CAS.
- G. C. Chinchen, P. J. Denny, D. G. Parker, M. S. Spencer and D. A. Whan, Appl. Catal., 1987, 30, 333 CrossRef CAS.
- W. X. Pan, R. Cao, D. L. Roberts and G. L. Griffin, J. Catal., 1988, 114, 440 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20423a |
| This journal is © The Royal Society of Chemistry 2012 |