Room temperature Baeyer–Villiger oxidation using molecular oxygen over mesoporous zirconium phosphate†
Apurba
Sinhamahapatra
a,
Ankita
Sinha
b,
Sandip Kumar
Pahari
a,
Narottam
Sutradhar
a,
Hari C.
Bajaj
a and
Asit Baran
Panda
*a
aDiscipline of Inorganic Materials and Catalysis, Central Salt and Marine Chemicals Research Institute (Council of Scientific and Industrial Research), G. B. Marg, Bhavnagar-364021, Gujarat, India. E-mail: abpanda@csmcri.org; Fax: +91 278-2567562; Tel: +91 278-2567760, Ext.: 704
bDepartment of Applied Chemistry, Indian School of Mines, Dhanbad-826004, India
First published on 24th July 2012
Abstract
Lactones have found wide applications as key molecules for the synthesis of important bioactive compounds, natural products and polymers; and represent a valuable family of synthons for various organic transformations. A series of lactones or esters are synthesized from their corresponding ketones employing Baeyer–Villiger (BV) oxidation at room temperature using mesoporous zirconium phosphate (m-ZrP) as a solid acid catalyst, molecular oxygen (O2)/benzaldehyde as an oxidizing agent, in a solvent free reaction medium. The oxidation reaction is studied in detail by varying the reaction parameters like molar ratio of the reactants, reaction temperature, time and catalyst loading. The m-ZrP showed high catalytic activity for the BV oxidation of cyclohexanone as well as other ketones in the presence of reduced amount of benzaldehyde with 100% selectivity for the corresponding lactones/esters. The protocol is suitable even for bulkier cyclic ketones like adamantanone. The m-ZrP catalyst showed excellent reusability.
1. Introduction
The oxidation of organic compounds with good selectivity is a challenging and important reaction for their vast applications in pharmaceutical, agricultural and natural product, resin, steroid and fine chemicals syntheses.1 Traditionally, these processes use stoichiometric reagents and end up with a large amount of toxic waste. Most of these processes are energy intensive and environmentally hazardous, as they require high temperature and pressure. Moreover, these stoichiometric homogeneous catalysts are not recoverable and reusable.Baeyer–Villiger (BV) oxidation, the transformation of ketones to their corresponding esters or lactones (for cyclic ketones), is one of the most important and widely used oxidation reactions.2 It is extensively used for the synthesis of antibiotics, steroids, pheromones, monomers for polymerization and various fine chemicals.3–5 One of the important uses of BV oxidation is the synthesis of ε-caprolactone, the monomer unit of an important biodegradable polymer, polycaprolactone. BV oxidation was first reported in 1899 for the oxidation of menthone and tetrahydrocarvone to the respective lactones using Caro's acid (monopersulfuric acid).2 Later on various peracids, such as monopersulfuric acid, perbenzoic acid, m-chloroperbenzoic acid (m-CPBA) and trifluoroperacetic acid, have been used for the BV oxidation reactions.4 These peracids are highly expensive, shock sensitive and environmentally malignant.4 Therefore, it was pertinent to replace peracids with greener alternatives for the BV oxidation. A suitable solid recyclable catalyst which operates in a homogeneous phase is essential3,6 for the BV oxidation. Among many reports, mainly two protocols have been used for investigating the replacement of organic peracid. One is the use of aqueous hydrogen peroxide as the stoichiometric oxidant3,4,7–10 with some shortcomings such as hydrogen peroxide is kinetically inert compared to peracids; and the presence of water in the reaction mixture leads to the hydrolysis of the product.11–13 In addition using high concentrations of hydrogen peroxide in organic solvents is not safe. Another important protocol is to conduct the BV oxidation using molecular oxygen through the in situ formation of peracid in combination with molecular oxygen and an aldehyde (as a sacrificial oxidant), known as the Mukaiyama method.14 Recently, some heterogeneous catalysts have been developed for effective BV oxidation with peracids and H2O2.7,8,15–17
However, reports of BV oxidation using molecular oxygen, benzaldehyde and a heterogeneous catalyst are scanty.18–26 Kaneda et al.18–20 have extensively studied the BV oxidation using molecular oxygen and various metal containing hydrotalcites in CCl4 or 1,2-dichloroethane as solvent. Later on Kawabata et al.21,22 reported the iron containing MCM-41 and magnesium–aluminium hydrotalcite as catalysts for the same purpose in acetonitrile at room temperature; however the duration of reaction was 15 h. Recently, Subramanian et al.25 and Chrobok26 have also reported the same reaction using iron containing MCM-48 as catalyst in ionic liquid in the presence of solvent or organic additives. Most of these developed procedures used a substrate to aldehyde ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and catalysts. The use of solvent leads to the generation of large amounts of organic hazardous waste. Moreover, the metal ion containing catalyst is associated with leaching and de-activation. So it is pertinent to develop an environment friendly protocol for the BV oxidation with a minimum amount of aldehydes, and high yield under solvent free conditions at room temperature (energy efficient) in a short reaction time.
:3 and catalysts. The use of solvent leads to the generation of large amounts of organic hazardous waste. Moreover, the metal ion containing catalyst is associated with leaching and de-activation. So it is pertinent to develop an environment friendly protocol for the BV oxidation with a minimum amount of aldehydes, and high yield under solvent free conditions at room temperature (energy efficient) in a short reaction time.
In this regard, zirconium phosphate (ZrP) may be an important alternative solid catalyst for the BV oxidation, as it showed good catalytic activity for different organic transformations including oxidation reactions.27–30 Rocha et al.17 reported amorphous and layered zirconium phosphates catalyzed BV oxidation of d-valerolactone, p-methoxyphenol and 2,4,6-trimethylphenol using hydrogen peroxide and glacial acetic acid with good yields. Recently, we have reported the synthesis of mesoporous zirconium phosphate (m-ZrP) having high specific surface area and narrow pore size distributions with excellent catalytic activity towards different organic reactions under solvent free conditions.30–33 To the best of our knowledge, there are no reports on the use of mesoporous zirconium phosphate for the BV oxidation using molecular oxygen and benzaldehyde.
Herein, we report a detailed study of BV oxidation over m-ZrP catalyst using molecular oxygen and reduced amount of benzaldehyde in a solvent free reaction medium. The effect of molar ratio of reactants, amount of catalyst, reaction temperature and time, and oxygen was investigated. The activity of m-ZrP was also studied using other oxidizing agents. Reusability of the used m-ZrP was also investigated.
2. Experimental
2.1 Materials
Zirconium oxychlorideoctahydrate (ZrOCl2·8H2O, 96.0%), di-ammonium hydrogen ortho-phosphate [(NH4)2HPO4, 98.5%] and cetyltrimethylammonium bromide (CTAB, 98%) were purchased from s. d. Fine Chemicals, India, and used without further purification. Ammonium carbonate was purchased from Merck, India, and used as such. High purity organic reagents (98–99.5%) were purchased from Sigma-Aldrich, USA, and used as received.2.2 Synthesis of m-ZrP
The mesoporous zirconium phosphate was synthesized using a reported procedure.31 Synthesis was carried out using molar ratio of Zr4+/PO43−/CTAB/H2O = 1:2:0.25:1000.
In a typical synthetic procedure, the required amount of aqueous ammonium carbonate solution was slowly added to an aqueous solution of ZrOCl2·8H2O which resulted in a white precipitate of zirconium carbonate. After completion of precipitation, it was filtered, washed with copious amounts of de-ionized water till it was chloride free. The above precipitates of zirconium carbonate were re-dissolved in an aqueous solution of ammonium carbonate under controlled addition with constant stirring, which resulted in a clear solution of zirconium carbonate complex. An aqueous solution of di-ammonium hydrogen orthophosphate was added to this clear solution of zirconium carbonate complex followed by the addition of the combined reaction mixture to an aqueous solution of CTAB of desired concentration with constant stirring, which resulted in a white precipitate. After 12 h stirring at ambient conditions, the mixture was aged at 80 °C for 2 days and at 90 °C for 1 day, in a sealed glass bottle. Further, it was kept for one more day in an autoclave at 110 °C for hydrothermal treatment. After cooling, it was filtered, washed thoroughly with distilled water and dried. The dry sample was calcined at 550 °C for 6 h to remove CTAB; stored in a glass bottle under ambient conditions and used as a catalyst without any additional activation step.
2.3 Characterization of m-ZrP
The calcined m-ZrP was characterized by powder X-ray diffraction using a Rigaku MINIFLEX-II (FD 41521) powder diffractometer using Cu Kα (λ = 1.54178 Å) radiation and a Ni filter in the small (2θ = 1–7°) as well as wide (2θ = 10–80°) angle region. The BET surface area, total pore volume, and average pore diameter were measured using the N2 adsorption–desorption method using ASAP 2010 Micromeritics, USA, at −196 °C after degassing the sample under vacuum (10−2 Torr) at 300 °C for 6 h. The surface area was determined using the BET (Brunauer–Emmett–Teller) equation. Pore size distributions were determined using the BJH (Barrett–Joyner–Halenda) model of cylindrical pore approximation. Single point adsorption total pore volume was obtained at P/Po = 0.97. A scanning electron microscope (SEM) (Leo series 1430 VP) equipped with INCA was used to determine the morphology of the sample and experiments were performed with an acceleration voltage of 200 kV. The sample was supported on the aluminum stub and gold coated before microscopic observation. Transmission electron microscopy (TEM) images were collected on a JEOL JEM microscope with an acceleration voltage of 200 kV. The sample was prepared by mounting the ethanol dispersed sample on a carbon coated copper grid. The FTIR and DRIFT (diffuse reflectance FT-IR) spectra were obtained in the range of 400–4000 cm−1 and 1200–1800 cm−1, respectively, on a Perkin-Elmer GX spectrophotometer using KBr. For DRIFT study pyridine was absorbed on the sample prior to analysis. NH3-TPD (temperature-programmed desorption) measurements were carried out on Micromeritics 2020 (USA) to measure the acid strength of m-ZrP. For detailed characterization procedures please see ESI† (S1).2.4 General procedure of BV oxidation
Baeyer–Villiger oxidation (Scheme 1) was carried out in a two-necked round bottom flask (50 mL) fitted with a condenser and a magnetic stirrer. In a typical procedure, O2 was purged at a flow rate of 10 mL min−1 under constant stirring (500 rpm) to the m-ZrP (10 wt%, 98 mg) and benzaldehyde (17.5 mmol) mixture. After 20 min of stirring, cyclohexanone (10 mmol) was added. The reaction was carried out at a desired temperature using a preheated oil bath wherever required. After the completion of reaction, the reaction mixture was diluted with ethyl acetate and the catalyst was separated by centrifugation. The reaction mixture was neutralized by 0.5 M NaHCO3 solution and extracted using water and ethyl acetate. GCMS (GC-MS QP 2010, Shimadzu) was used for the product identification. GC (Varian-450 GC, fitted with a flame ionization detector, VS-5 factor 4 30 nm capillary column) was used for conversion and selectivity studies of the corresponding products (S2, ESI†). The term selectivity is expressed as selective formation of lactone or ester (product) with respect to conversion of ketone. n-Tetradecane was used as the internal standard to calculate the initial and final mole percentages of cyclohexanone. The blank reactions were performed in the absence of benzaldehyde, m-ZrP and molecular oxygen separately for comparison. The reaction was also carried out using other oxidants and m-ZrP as catalyst. | ||
| Scheme 1 BV oxidation of cyclohexanone using molecular oxygen and benzaldehyde over m-ZrP as catalyst. | ||
2.5 Re-generation of used catalyst
The used m-ZrP was regenerated by treatment with H2O2. In a typical procedure, 1 g of used catalyst was refluxed in 40 ml 30% (w/v) H2O2 for 3 h at 80 °C under continuous stirring. Then the catalyst was filtered off, washed with distilled water, and dried overnight at 100 °C.3. Results and discussion
3.1 Characterization of m-ZrP
The BET specific surface area (SBET) was estimated using nitrogen adsorption–desorption measurement at liquid N2 temperature in a relative pressure range (P/P0) of 0.05 to 1.0. The obtained total surface area, pore volume and average pore diameter of the m-ZrP were 400 m2 g−1, 0.37 cm3 g−1 and 3.0 nm respectively. The nitrogen adsorption–desorption isotherm corresponds to type IV with clear formation of hysteresis indicating a typical mesoporous material (Fig. 1a) with narrow pore size distribution (inset Fig. 1a).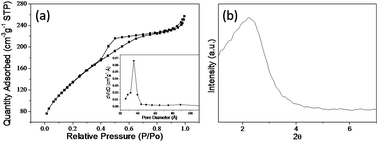 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherm and pore size distribution (inset) (a) and low angle XRD pattern (b) of the calcined m-ZrP. | ||
The XRD pattern of the calcined solid mass showed a single diffraction peak at 2θ = 2.2° in the low angle region (Fig. 1b) due to mesoporous structure of the zirconium phosphate. A single reflection does not allow structural assignment; although only one broad diffraction peak generally indicates that the present pores are in short range order. In the wide angle XRD, two broad peaks in the range of 10–40° and 40–70° depicted the amorphous nature of the synthesized m-ZrP catalyst (Fig. S1, ESI†).34
The SEM images of m-ZrP showed spherical particles with a smooth surface. The TEM images exhibited the mono-dispersed porous nature of the particles with worm-like pore structures with an average pore size of ∼3–4 nm, supporting pore size obtained from the surface area measurement. The SAED pattern of the calcined m-ZrP confirmed the amorphous nature of the synthesized materials, as observed from the wide angle XRD pattern (Fig. S2–S4, ESI†). The EDX analysis confirmed that the Zr/P ratio of the synthesized m-ZrP is 0.55.
The 31P MAS NMR spectroscopy (performed using H3PO4 as reference) was studied to identify the microenvironment of phosphorus in the calcined zirconium phosphate (Fig. 2a). The calcined zirconium phosphate gave one distinguishable resonance signal centered at ∼δ −21 ppm, attributed to the connectivity of phosphorus with 3 P–O–Zr bonds [(OH) P–(OZr)3]. The absence of signal at –24 ppm confirmed that the phosphorus with 4 P–O–Zr bonds was not formed on calcination at 550 °C.
 | ||
| Fig. 2 31P MAS NMR spectra (a) and the DRIFT spectra (b) of the calcined m-ZrP. | ||
The FT-IR spectra of m-ZrP depict a strong and sharp band centered at 1046 cm−1 corresponding to P–O stretching vibration. The broad peak at 2352 cm−1 corresponds to the (P)–O–H stretching vibration. The broad band around 3445 cm−1 may be attributed to OH stretching of a water molecule and the weak peak at 1634 cm−1 indicates the bending mode of the water molecule, however it is not proportionate with the total intensity of the corresponding stretching band (Fig. S5, ESI†).
The DRIFT analysis of pyridine-adsorbed m-ZrP and NH3-TPD was carried out to know the nature of acid sites and the total acid sites present in the synthesized sample, respectively.
In the DRIFT spectrum (Fig. 2b), the bands at 1634 and 1540 cm−1 indicated the presence of Brönsted acid sites, due to the interaction of the Brönsted acid site with pyridine. The Brönsted acidity mainly arises because of the P–OH groups present in the synthesized m-ZrP. The observed band at 1446 cm−1 is due to the interaction of Lewis acid sites of m-ZrP with pyridine. The band at 1489 cm−1 is the combined band for all the acid sites of different nature present. The intensity of the adsorption band due to the pyridine adsorbed on Lewis acid sites gradually decreased with the increase in temperature, however, the adsorption band due to Brönsted acid sites remained almost constant, indicating the presence of weak Lewis acid sites compared to the Brönsted acid sites. One distinct peak centered at 433 °C with a hump at ∼326 °C was observed in the NH3-TPD profile and desorption of NH3 continued even at 900 °C, confirming the presence of a significant amount of strong acidic sites. The total amount of desorbed NH3 was ∼1.8 mmol NH3 per gram of catalyst (Fig. S6, ESI†). For a detailed discussion on synthesis, characterization and the formation mechanism, one can refer to our previous report.31
3.2 BV oxidation
The primary aim of the present work was to explore the selective transformation of cyclohexanone to ε-caprolactone. The BV oxidation of cyclohexanone was carried out using molecular oxygen and benzaldehyde over m-ZrP in solvent free reaction media.In the first set of reaction, the BV oxidation of cyclohexanone was carried out by varying the amount of catalyst (5–15 wt% m-ZrP with respect to cyclohexanone) at room temperature (RT, ∼29 °C). 5 wt% m-ZrP resulted in low conversion of cyclohexanone, however, with an increase in catalyst loading the conversion gradually increased to 78% (with 10 wt% catalyst loading, Fig. 3a). Above 10 wt% the effect of catalyst on conversion was almost negligible. In all the performed reactions, the selectivity was 100%.
 | ||
| Fig. 3 Effect of catalyst loading at room temperature (a) and reaction temperature with 10 wt% catalyst loading (b) on the BV oxidation using molecular oxygen and benzaldehyde over m-ZrP. Reaction conditions: molar ratio of cyclohexanone and benzaldehyde, 1:3; cyclohexanone addition time, 20 min; and time, 3 h. | ||
Although our prime objective was to conduct the reaction at RT, the effect of temperature on the BV oxidation of cyclohexanone was also examined. The reaction was performed at the temperature range from RT to 60 °C (Fig. 3b). With the increase in the reaction temperature from RT to 60 °C, the conversion of cyclohexanone increased gradually (83%). However, the selectivity of ε-caprolactone decreased from 100 to 91% at 60 °C due to the formation of side products, 2-benzylidene cyclohexanone and 2,6-di-benzylidene cyclohexanone. These products are formed via Claisen–Schmidt (CS) condensation of cyclohexanone and benzaldehyde in the presence of m-ZrP as the acid catalyst (S3, ESI†). Hence, further reactions were performed at RT.
The prior articles18–26 on the BV oxidation using molecular oxygen and benzaldehyde over m-ZrP were not focused on the amount of benzaldehyde. In all cases, 3 mol of benzaldehyde was used for 1 mol of ketone. As benzaldehyde leads to benzoic acid at the end of reaction, one should be interested in the minimization of benzaldehyde requirement for the maximum yield. To investigate the optimum requirement of benzaldehyde for the BV oxidation of cyclohexanone, the molar ratio of cyclohexanone and benzaldehyde was varied from 1:4 to 1:1 keeping the other parameter constant (Fig. 4). The selectivity of ε-caprolactone was 100% under the studied conditions. However the conversion was optimum (78%) at the ratio of 1:4 to 1:1.75 and with further decrease in ratio the conversion decreased. 35% conversion was achieved when the ratio was 1:1. Although theoretically 1:1 ratio of cyclohexanone to benzaldehyde is required for the reaction, due to self-oxidation of benzaldehyde to benzoic acid in the presence of molecular oxygen, higher amount of benzaldehyde is needed. Here, it should be mentioned that under the employed reaction conditions using m-ZrP as catalyst, the rate of self-oxidation is not as much and maximum conversion with 100% selectivity resulted with lower amounts of benzaldehyde compared to the prior articles.18–26
 | ||
| Fig. 4 Effect of amount of benzaldehyde on the BV oxidation using molecular oxygen and benzaldehyde over m-ZrP. Reaction conditions: addition time, 20 min; catalyst loading, 10 wt%; RT and time, 3 h. | ||
The progress of the BV oxidation of cyclohexanone in the presence of O2/benzaldehyde over m-ZrP was studied by analyzing the reaction product at different time intervals (Fig. 5) under optimized parameters. During the progress of reaction, initially the conversion of cyclohexanone increased with time and the reaction was almost complete in 1.5 h. After 1.5 h, the conversion was constant (78%) with 100% selectivity of ε-caprolactone.
 | ||
| Fig. 5 Reaction profile for the BV oxidation using molecular oxygen and benzaldehyde over m-ZrP. Reaction conditions: molar ratio of cyclohexanone and benzaldehyde, 1:1.75; addition time, 20 min; catalyst loading, 10 wt%; and RT. | ||
Thus the optimized reaction conditions for BV oxidation of cyclohexanone to ε-caprolactone in the presence of molecular O2/benzaldehyde as an oxidant over m-ZrP at RT are as follows: molar ratio of cyclohexanone to benzaldehyde 1:1.75, reaction time 1.5 h and catalyst loading 10 wt%.
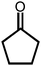
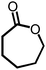
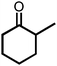
Due to the encouraging catalytic activity of m-ZrP for the BV oxidation of cyclohexanone using O2/benzaldehyde at RT, the catalyst was screened for the BV oxidation of a series of ketones (cyclic as well as open chain) under optimized reaction conditions (Table 1). All the ketones showed good conversion with 100% selectivity, except 2-methylcyclohexanone (entry 3, Table 1). The cyclic ketone, cyclopentanone gave maximum yield (92%) for the most stable 6-membered lactone. The bulkier ketone such as 4-methyl cyclohexanone (86%) and adamantanone (96%) (entries 4 and 5, respectively, Table 1) also gave good yield. There are reports where some catalysts showed inferior results for bulkier ketones although good activity was observed for other ketones, like cyclohexanone.20,21 The high surface area and large pore size of m-ZrP allowed the reaction of these bulky molecules with the peracid, which resulted in selective formation of corresponding lactones with high yield. In the case of 2-methyl cyclohexanone mainly 6-methyl caprolactone (92%) was formed as its transition state is more stable than that of 2-methyl caprolactone (7%). The group which is able to stabilize the positive charge developed on the carbonyl carbon is more prone to migration.25 This methodology is also applicable for the acyclic ketones as acetophenone and 4-methoxy acetophenone (entries 6 and 7, respectively, Table 1) yielded 31% and 44% of corresponding esters, respectively. The above results clearly indicated that the present methodology is highly effective for selective transformation of ketones to their corresponding lactones or esters. Efficiency of m-ZrP was also studied with other oxidizing agents such as O2, m-CPBA, H2O2, and TBHP for BV oxidation of cyclohexanone (Table 2). O2 (alone), H2O2 and TBHP were found to be inactive at RT, whereas m-CPBA showed good oxidizing ability for BV oxidation of cyclohexanone in the presence of m-ZrP, almost equivalent to that of the O2/benzaldehyde system.
| Entry | Ketone | Product | Conversion (%), selectivity (%) | |
|---|---|---|---|---|
| m-ZrP | Blankb | |||
|
a Reaction conditions: ketone:benzaldehyde, 1:1.75; addition time, 20 min; catalyst loading, 10 wt% with respect to ketone; RT and time, 1.5 h.
b Blank reaction performed without catalyst, keeping the other parameters same.
c The other product is 2-methyl caprolactone.
|
||||
| 1 |
|

|
92, 100 | 36, 100 |
| 2 |

|
|
78, 100 | 15, 100 |
| 3 |
|
|
75, 92c | 26, 55c |
| 4 |

|

|
86, 100 | 31, 100 |
| 5 |

|

|
96, 100 | Trace |
| 6 |

|

|
31, 100 | Trace |
| 7 |

|

|
44, 100 | Trace |
| Entry | Oxidant | Conversion (%) | Selectivity (%) |
|---|---|---|---|
|
a Reaction conditions: cyclohexanone:benzaldehyde, 1:1.75; catalyst loading, 10 wt%; RT and time, 1.5 h.
b Reaction conditions: catalyst loading, 10 wt%; RT and time, 1.5 h.
c Reaction conditions: cyclohexanone:m-CPBA, 1:1.75; catalyst loading, 10 wt%; RT and time, 1.5 h.
d Reaction conditions: cyclohexanone:H2O2, 1:44; catalyst loading, 10 wt%; temperature, 40 °C and time, 1.5 h.
e Reaction conditions: cyclohexanone:H2O2:benzonitrile, 1:44:3; catalyst loading, 10 wt%; temperature, 40 °C and time, 1.5 h.
f Reaction conditions: cyclohexanone:tertiary butyl hydrogen peroxide, 1:3; catalyst loading, 10 wt%; temperature, 40 °C and time, 1.5 h.
|
|||
| 1 | Molecular O2/benzaldehydea | 78 | 100 |
| 2 | Molecular O2b | No reaction | — |
| 3 | m-Chloro perbenzoic acidc | 76 | 100 |
| 4 | H2O2d | Trace | — |
| 5 | H2O2/benzonitrilee | 37 | 72 |
| 6 | Tertiary butyl hydrogen peroxidef | Trace | — |
We have also extended the present work to explore the catalytic activity of m-ZrP using m-CPBA as the oxidant at RT for other ketones (Table 3). A control reaction was also carried out without m-ZrP using m-CPBA as an oxidant. The results clearly indicated that the presence of m-ZrP was vital for better conversion and selectivity. All the substrates end up giving better yields using m-CPBA as an oxidizing agent and m-ZrP as catalyst at RT. Slight conversion was observed at RT when only H2O2 was used as an oxidant. However, in the presence of benzonitrile, H2O2 showed 37% conversion of cyclohexanone with 73% selectivity for ε-caprolactone; and 27% selectivity for 6-hydroxyhexanoic acid (Scheme S2, ESI†). As an inspiration of the present result and literature reports13,35,36 the activity of m-ZrP was investigated for the BV oxidation of cyclohexanone using H2O2 in the presence of benzonitrile (S4, ESI†). The study reveals that with the increase in temperature the conversion of cyclohexanone increased up to 90% but its selectivity to formation of ε-caprolactone is very less. Rather selective synthesis of 6-hydroxy-hexanoic acid (79%) can be achieved by tuning the reaction conditions.
| Entry | Ketone | Product | Conversion (%), Selectivity (%) | |
|---|---|---|---|---|
| m-ZrP | Blankb | |||
|
a Reaction conditions: ketone:m-CPBA, 1:1.75; addition time, 20 min; catalyst loading, 10 wt% with respect to ketone; RT and time, 1.5 h.
b Blank reaction performed without catalyst keeping the other parameters same.
c The other product is 2-methyl caprolactone.
|
||||
| 1 |
|

|
90, 100 | 66, 100 |
| 2 |

|

|
76, 100 | 65, 100 |
| 3 |

|

|
69, 92c | 46, 59c |
| 4 |
|

|
89, 100 | 61, 100 |
| 5 |

|

|
91, 100 | Trace |
| 6 |

|

|
23, 100 | Trace |
| 7 |

|

|
32, 100 | Trace |
The comparative analysis of catalytic activity of m-ZrP with other reported solid catalysts for BV oxidation of cyclohexanone to ε-caprolactone using O2/benzaldehyde as an oxidant (Table 4) clearly exhibited the better catalytic activity of m-ZrP as a solid acid catalyst over the reported catalytic systems: Fe–MCM-41, Fe–MCM-48, MgAl–Cu–CO3 and MgAl–CO3 with respect to the yield of ε-caprolactone. However, MgAl–Fe–CO3 and Fe/Mg3Al showed better catalytic activity than m-ZrP. Still, they have some disadvantages with respect to m-ZrP such as, MgAl–Fe–CO3 showed better catalytic activity for cyclohexanone, but for bulkier ketones its activity was inferior; for example adamantanone gave 54% yield. Moreover, a long reaction time (15 h) was required for completion of reaction when Fe/Mg3Al was used as catalyst. The activity of the catalyst towards cyclopentanone was poor. All the mentioned protocols used different organic solvents, higher amounts of benzaldehyde (cyclohexanone:benzaldehyde is 1:3) for better conversion; and some used higher reaction temperature rather than RT. Whereas, in the present methodology, using m-ZrP as catalyst, the cyclohexanone to benzaldehyde ratio is only 1:1.75 and the reaction was performed at RT without using any additional solvent.
| Entry | Catalyst | Solvent | Temperature (°C) | Time (h) | Cyclohexanone:benzaldehyde |
Conversion (%) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a DCE: 1,2-dichloroethane. b DMF: dimethylformamide. | ||||||||
| 1 | m-ZrP | None | RT | 2 | 1:1.75 |
78 | 78 | — |
| 2 | Fe–MCM-41 | MeCN | 25 | 15 | 1:3 |
85 | 77 | 29 |
| 3 | Fe–MCM-48 | DCEa | 50 ± 5 | 2 | 1:3 |
92 | 62 | 24 |
| 4 | MgAl–Fe–CO3 | DCE | 40 | 5 | 1:3 |
100 | 100 | 19 |
| 5 | MgAl–Cu–CO3 | DCE | 40 | 5 | 1:3 |
72 | 72 | 19 |
| 6 | MgAl–CO3 | DCE | 40 | 5 | 1:3 |
66 | 65 | 19 |
| 7 | Fe/Mg3Al | DMFb | 15 | 1:3 |
>99 | 21 | ||
The m-ZrP catalyst was reused after a simple activation step by refluxing it with 30% (w/v) H2O2 for 3 h at 80 °C. The results are presented in Fig. 6. Up to the fourth cycle, there was no significant decrease in conversion (74%) and selectivity (99%).
 | ||
| Fig. 6 Reuse of m-ZrP for the BV oxidation using molecular oxygen and benzaldehyde. Reaction conditions: molar ratio of cyclohexanone and benzaldehyde, 1:1.75; catalyst loading, 10 wt%; RT and time, 1.5 h. | ||
3.3 Reaction mechanism of the BV oxidation
The mechanistic pathway of BV oxidation has been investigated extensively. Criegee proposed a two step basic mechanism of the reaction in 1948.37 In the first step the peracid attacks the carbonyl carbon, leading to the Criegee intermediate, and in the second step, one of the adjacent carbon–carbon bonds migrates to the perester oxygen, rearranging the carbonyl with loss of a proton and O–O bond cleavage. The BV oxidation reaction is highly influenced by the acidic conditions.38 It is established that the BV oxidation in the presence of solvent proceeds through an ionic mechanism.39 An alternative neutral concerted mechanistic pathway is also proposed without any ionic species formation.40 For a solid acid catalyst, Nunez et al.41 performed the reaction using potassium peroxomonosulfate supported on an acidic silica gel in dichloromethane; and proposed a reaction pathway considering the involvement of the solid surface and the interaction of cyclohexanone with the Brönsted acid site of the catalyst. Kaneda et al.18 had reported that in the presence of molecular oxygen, the benzaldehyde transformed to perbenzoic acid and initiated the BV oxidation. Kawabata et al.21 and very recently Subramanian et al.25 performed the BV oxidation using molecular oxygen and benzaldehyde in 1,2-dichloroethane using Fe containing MCM-41 and MCM-48, respectively, and postulated that the reaction proceeded via the coordination of carbonyl of ketone to Fe3+ as Lewis acid sites. However, there are no reports for the BV oxidation using molecular oxygen and benzaldehyde under solvent free conditions over a mesoporous acid catalyst. We believe that in the present case, BV oxidation using m-ZrP as a solid acid catalyst under solvent free conditions, the reaction proceeds through a similar pathway (Scheme 2) as reported by Nunez et al.41 The ketone is first adsorbed on the Brönsted acid site of m-ZrP by acid–base interaction. Then the activated carbonyl group reacts with the peracid leading to the Criegee type intermediate. Finally, in the presence of catalyst the R-group migrated to the peroxy oxygen which resulted in the formation of a lactone or an ester. The formation of a Criegee type intermediate can also be interpreted by the formation of 6-methyl caprolactone as a major product in BV oxidation of 3-methyl cyclohexanone.25 Generally, in a regio-selective rearrangement, a group is more prone to migration that can stabilize the positive carbonyl carbon. The intermediate leading to 6-methyl caprolactone is more stable in comparison to the 2-methyl caprolactone. | ||
| Scheme 2 Proposed pathway for the BV oxidation of cyclohexanone using molecular oxygen and benzaldehyde over m-ZrP as catalyst. | ||
Now, it is also important to put some light on the reaction of benzaldehyde with oxygen to form peracid and the role of catalyst in this reaction. The reaction follows a radical pathway, as the reaction was inhibited by addition of 2,2,6,6 tetramethylpiperidinyloxy (TEMPO, a radical scavenger). A similar pathway has also been reported by different researchers.42 To study the role of catalyst, two different sets of reactions were performed for the oxidation of benzaldehyde by molecular oxygen in the presence or absence of catalyst. The reaction was monitored by GC. Results are summarized in Fig. 7. It was observed that both perbenzoic acid and benzoic acid were formed in the presence or absence of catalyst, but the conversion of benzaldehyde was more in the presence of catalyst in the same reaction time. However, more perbenzoic acid was formed with respect to benzoic acid in the presence of catalyst, while in the absence of catalyst benzoic acid was formed more in the same reaction time. The results clearly indicate that the m-ZrP facilitates the peracid formation. This may be due to the uptake of one electron from benzaldehyde by the Lewis acid sites of m-ZrP facilitating free radical reaction. In the same time, the m-ZrP prevents the transformation of perbenzoic acid to benzoic acid by the trace amount of water present in the system through sequestering the water molecules in its porous system.
 | ||
| Fig. 7 Oxidation of benzaldehyde using molecular oxygen in the presence (a) and absence (b) of m-ZrP. Reaction conditions: benzaldehyde, 20 mmol; catalyst loading, 10 wt%; RT and time, 1.5 h. | ||
To support the above mentioned results, a few separate BV oxidation reactions of cyclohexanone were performed by varying the reaction conditions. A set of reactions were performed by varying the addition time of the ketone. It was observed that the conversion varies with reaction time (S5, ESI†). Initially conversion increased with increase in addition time and decreased after 20 min. Another reaction was performed where O2 was bubbled in benzaldehyde in the absence of catalyst and after 20 min catalyst and ketone were added. As expected, lower yield (60%) of lactone was obtained within the same reaction time. All the results support that m-ZrP has influence on the peracid formation; and there is some induction time for maximum peracid formation (20 min) to give better yield.
4. Conclusions
Mesoporous zirconium phosphate showed excellent catalytic activity for the synthesis of lactones as well as esters by Baeyer–Villiger (BV) oxidation of corresponding ketones using molecular oxygen/benzaldehyde as an oxidizing agent with reduced amount of benzaldehyde in a solvent free protocol at room temperature. All the used ketones showed 100% selectivity with reasonably high conversion with a cyclohexanone to benzaldehyde ratio of 1:1.75. The m-ZrP and O2/benzaldehyde protocol is also effective even for bulkier cyclic ketones like adamantanone (96% yield of corresponding lactone). The catalyst m-ZrP was also effective for the BV oxidation using other oxidants e.g. m-CPBA and H2O2 in the presence of benzonitrile. The m-ZrP is easily recoverable from the reaction system and can be reused without considerable change in catalytic activity, at least up to four cycles. Finally, the performed synthetic method was advantageous over other methods, as it provides good conversion with 100% selectivity with molecular O2 and with reduced amount of benzaldehyde. m-ZrP as a solid acid catalyst is reusable in solvent free media at RT, which makes the procedure economical for industrial purposes.
Acknowledgements
The authors are thankful to Department of Science and Technology, India (DST, SR/S1/IC-33/2011 and SR/S1/PC-1/2010), for financial support. Authors also acknowledge the analytical discipline and centre instrumentation facilities of CSMCRI for materials characterization. S. K. P. and N. S. acknowledge CSIR, India, for a research fellowship. We are thankful to the referees for suggesting to find out the role of catalyst in the in situ formation of peracid.Notes and references
- R. A. Sheldon and J. K. Kochi, Metal Catalyzed Oxidation of Organic Compounds, Academic Press, New York, 1981, p. 318 Search PubMed.
- A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1899, 32, 3625 CrossRef.
- G. Strukul, Angew. Chem., Int. Ed., 1998, 37, 1198 CrossRef.
- G. J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105 CrossRef.
- R. A. Michelin, P. Sagarbossa, A. Scarso and G. Strukul, Coord. Chem. Rev., 2010, 254, 646 CrossRef CAS.
- M. D. Mihovilovic, B. Muller and P. Stanetty, Eur. J. Org. Chem., 2002, 3711 CrossRef CAS.
- M. Ricci and E. Battistel, Chim. Ind. (Milan, Italy), 1997, 79, 879 CAS.
- S. Kirumakki, S. Samarajeewa, R. Harwell, A. Mukherjee, R. H. Herber and A. Clearfield, Chem. Commun., 2008, 5556 RSC.
- A. Cavarzan, A. Scarso, P. Sgarbossa, R. A. Michelin and G. Strukul, ChemCatChem, 2010, 2, 1296 CrossRef CAS.
- C. Chen, J. Peng, B. Li and L. Wang, Catal. Lett., 2009, 131, 618 CrossRef CAS.
- C. W. Jones, in Applications of Hydrogen Peroxide and its Derivatives, ed. J. H. Clark, RSC Clean Technology Monographs, RSC, Cambridge, 1999, p. 21 Search PubMed.
- G. Strukul, Catalytic Oxidations with Hydrogen Peroxide as Oxidant, Kluwer, Dordrecht, 1992 Search PubMed.
- C. J. Sanchidrian and J. R. Ruiz, Tetrahedron, 2008, 64, 2011 CrossRef.
- T. Yamada, T. Takai, O. Rhode and T. Mukaiyama, Chem. Lett., 1991, 1 CrossRef CAS.
- R. Mello, A. Olmos, J. P. Carbonell, M. E. Gonzalez-Nunez and G. Asensio, Green Chem., 2009, 11, 994 RSC.
- R. Llamas, C. J. Sanchidrian and J. R. Ruiz, Appl. Catal., B, 2007, 72, 18 CrossRef CAS.
- G. M. S. R. O. Rocha, T. M. Santos and C. S. S. Bispo, Catal. Lett., 2011, 141, 100 CrossRef CAS.
- K. Kaneda, S. Ueno, T. Imanaka, E. Shimotsuma, Y. Nishiyama and Y. Ishii, J. Org. Chem., 1994, 59, 2915 CrossRef CAS.
- K. Kaneda, S. Ueno and T. Imanaka, Chem. Commun., 1994, 797 RSC.
- K. Kaneda, S. Ueno and T. Imanaka, J. Mol. Catal. A: Chem., 1995, 102, 135 CrossRef CAS.
- T. Kawabata, Y. Ohishi, S. Itsuki, N. Fujisaki, T. Shishido, K. Takaki, Q. Zhang, Y. Wang and K. Takehira, J. Mol. Catal. A: Chem., 2005, 236, 99 CrossRef CAS.
- T. Kawabata, N. Fujisaki, Y. T. Sishido, K. Namura, T. Sano and K. Takehira, J. Mol. Catal. A: Chem., 2006, 253, 279 CrossRef CAS.
- S. I. Murahashi, Y. Oda and T. Naota, Tetrahedron Lett., 1992, 33, 7757 Search PubMed.
- R. Raja, J. M. Thomas and G. Sankar, Chem. Commun., 1999, 525 RSC.
- H. Subramanian, E. G. Nettleton, S. Budhi and R. T. Koodali, J. Mol. Catal. A: Chem., 2010, 330, 66 CrossRef CAS.
- A. Chrobok, Tetrahedron, 2010, 66, 2940 CrossRef CAS.
- K. Segawa, Y. Kurusu, Y. Nakajima and M. Kinishita, J. Catal., 1985, 94, 491 CrossRef CAS.
- A. Clearfield and D. S. Thakur, Appl. Catal., 1986, 26, 1 CrossRef CAS.
- J. Xiao, J. Xu and Z. Gao, Catal. Lett., 1999, 57, 37 CrossRef CAS.
- A. Sinhamahapatra, N. Sutradhar, S. K. Pahari, P. Pal, H. C. Bajaj, M. Jayachandran and A. B. Panda, ChemCatChem, 2011, 3, 1447 CrossRef CAS.
- A. Sinhamahapatra, N. Sutradhar, B. Roy, A. Tarafdar, H. C. Bajaj and A. B. Panda, Appl. Catal., A, 2010, 385, 22 CrossRef CAS.
- A. Sinhamahapatra, N. Sutradhar, S. Pahari, H. C. Bajaj and A. B. Panda, Appl. Catal., A, 2011, 394, 93 CrossRef CAS.
- A. Sinhamahapatra, N. Sutradhar, B. Roy, H. C. Bajaj and A. B. Panda, Appl. Catal., B, 2011, 103, 378 CrossRef CAS.
- T. Z. Ren, Z. Y. Yuan and B. L. Su, Chem. Commun., 2004, 2730–2731 RSC.
- R. Lalamas, C. J. Sanchidrian and J. R. Ruiz, Tetrahedron, 2007, 63, 143 Search PubMed.
- R. Lalamas, C. J. Sanchidrian and J. R. Ruiz, Appl. Catal., B, 2007, 72, 18 CrossRef.
- R. Criegee and J. Liebigs, Ann. Chem., 1948, 560, 128 Search PubMed.
- F. Grein, A. C. Chen, D. Edwards and C. M. Crudden, J. Org. Chem., 2006, 71, 861 CrossRef CAS.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, Hoboken, NJ, 6th edn, 2007, p. 1617 Search PubMed.
- N. M. Diez, S. Keller and R. A. Idaboy, Org. Biomol. Chem., 2009, 7, 3682 Search PubMed.
- M. E. G. Nunez, R. Mello, A. Olmos and G. Asensio, J. Org. Chem., 2005, 70, 10879 CrossRef.
- C. J. Sanchidrián and J. R. Ruiz, Tetrahedron, 2008, 64, 2011 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of m-ZrP, calculation of conversion and selectivity, C–S condensation over m-ZrP, BV oxidation using H2O2 over m-ZrP. See DOI: 10.1039/c2cy20404e |
| This journal is © The Royal Society of Chemistry 2012 |