Preparation of ethane-bridged organosilica group and keggin type heteropoly acid co-functionalized ZrO2 hybrid catalyst for biodiesel synthesis from eruca sativa gars oil
Fang
Su
,
Ling
Ma
,
Yihang
Guo
* and
We
Li
*
School of Chemistry, Northeast Normal University, Changchun, 130024, P. R. China. E-mail: guoyh@nenu.edu.cn; Fax: +86 431 85098705; Tel: +86 431 85098705
First published on 6th July 2012
Abstract
A series of mesostructured ZrO2-based hybrid catalysts, functionalized by both an ethane-bridged organosilica group and the Keggin type heteropoly acid, H3PW12O40–ZrO2–Si(Et)Si, were prepared by one-step sol-gel-hydrothermal method in the presence of a triblock copolymer surfactant (F127). The materials were well characterized by spectroscopy methods such as FT-IR and MAS NMR, transmission electron microscopy, and nitrogen gas physisorption measurements to confirm the structural integrity of the Keggin units and ethane-bridged organosilica groups in the hybrid materials, and to test the mesostructure, morphology, and porosity of the materials. The materials were subsequently utilized as environmentally-friendly solid acid catalysts in the transesterification of low-cost Eruca Sativa Gars oil with methanol to produce fatty acid methyl esters under atmosphere refluxing. Compared with alkyl-free H3PW12O40–ZrO2, the prepared H3PW12O40–ZrO2–Si(Et)Si exhibited higher catalytic activity toward the target reaction. This enhanced acid-catalytic activity after the introduction of both acidic and hydrophobic functionalities within the ZrO2 matrix was explained in terms of strong Brϕnsted acidity, excellent porosity, and enhanced hydrophobicity of the hybrid materials.
1. Introduction
Biodiesel, a mixture of C12–C22 fatty acid monoalkyl esters (FAMEs), obtained from base- or acid-catalyzed transesterification of triglycerides (TGs) with short-chain alcohols, is a sustainable, non-toxic, and biodegradable petrol fuel replacement. A wide range of feedstocks including edible oils, used vegetable oils, animal fats, and non-edible plant oils can potentially be employed for biodiesel production.1 From the viewpoint of biodiesel production in a large scale, low-cost feedstock-like non-edible plant oils are paid particular attention. These low-cost feedstocks generally contain high content free fatty acids (FFAs), and therefore pre-esterification of the FFAs also produces FAMEs.Base-catalyzed transesterification is much faster and requires a lower temperature compared with the acid-catalyzed reaction. However, the method suffers from some severe drawbacks, including sensitivity to water, as well as FFAs that widely exist in the low-cost feedstocks. For homogeneous base-catalyzed transesterification from low-cost feedstocks, saponification is inevitable, leading to not only consumption of the catalyst, but also causing the product separation problem. As for heterogeneous base-catalyzed transesterification from low-cost feedstocks, considerable decreased catalytic activity was found because the basic sites were poisoned by strong adsorption of FFAs and water on the surface sites. Therefore, refined vegetable oils with less than 0.5 wt% of FFA content are required for the base-catalyzed biodiesel production, which leads to an impractical and uneconomical process due to high feedstocks cost and priority as food resources.
For feedstocks with high FFA content, acid catalysis is preferable to base catalysis because it allows simultaneous esterification of FFAs and transesterification of triglycerides under the appropriate reaction conditions, without the formation of soap. As a consequence, biodiesel manufacturing process would be further simplified. However, the inherent drawbacks for the use of homogeneous mineral acid catalysts (e.g., sulfuric acid, hydrofluoric acid, and p-toluenesulfonic acid) such as slow reaction rate, requirement of high temperature, high molar ratio of alcohol to oil, separation of the catalysts, and serious environmental and corrosion related problems also limit their practical applications for biodiesel production.2 Accordingly, the development of recyclable and readily separable heterogeneous acid catalysts to convert low-cost feedstocks to biodiesel has attracted much attention. Up till now, the heterogeneous acid catalysts used for biodiesel production mainly include zeolites,3 mixed metal oxides like sulphated zirconia,4 carbonhydrate-derived solid acid catalysts,5 ion-exchange resin catalysts,6 and cesium partly substituted Keggin type heteropoly acids (CsxH3−xPW12O40, x = 2.0–2.3).7 Most of the above solid acid-catalyzed biodiesel production reactions are performed under high temperature and high pressure (e.g. 200 °C, 600 psi) due to the normally quite low catalytic activity. Moreover, high viscosity and poor miscibility of feedstocks with light alcohols severely hamper the reaction rate of biodiesel production. Therefore, the development of environmentally benign solid acid catalysts that can work efficiently towards simultaneous esterification and transesterification under mild conditions is still a challenge.
In the search for efficient solid acid catalysts for biodiesel synthesis, herein, we focus on the preparation of a series of novel hybrid solid acid catalysts, i.e., mesostructured ZrO2 functionalized by both ethane-bridged organosilica groups and Keggin type heteropoly acid (HPA), H3PW12O40–ZrO2–Si(Et)Si. HPAs are known as one of the strong Brϕnsted acids, and they exhibit excellent catalytic activity in a wide variety of acid-catalyzed reactions including the alkylation and acylation of hydrocarbons, the hydration of alkenes, and the polymerization of tetrahydrofuran.3b The disadvantages of HPAs such as small specific surface area (1–10 m2 g−1) and high solubility in polar media can be overcome by dispersing them throughout inorganic porous materials. For heterogenizing HPAs to the hybrid solid acids, ZrO2 attracts our interest since it is an acidic support. After the formation of HPA–ZrO2 hybrid materials, the Brϕnsted acidity is expected to be enhanced significantly due to the strong interaction between HPA cluster and ZrO2 support, which can promote the release of the protons.8 This enhanced Brϕnsted acidity will play the paramount role to obtain high FAMEs yields under mild conditions. Additionally, the performance of the heterogeneous catalysts is also determined to a great extent by their textural properties. The fabrication of solid acid catalysts with perfect porous structure, including large surface area as well as uniform pore-size distribution, is expected to improve the catalytic activity by increasing active site numbers and the accessibility of active sites to the substrates. For the designed H3PW12O40–ZrO2–Si(Et)Si hybrid catalysts, their porosity can be adjusted by the introduction of ethane-bridged organosilica groups. Finally, the introduction of ethane-bridged organosilica groups can also improve the surface hydrophobicity of H3PW12O40–ZrO2, which is another key goal in the development of solid acid catalysts for both esterification and transesterification. The hydrophilic nature of the polar H3PW12O40–ZrO2 surface means it is not ideal for reactions of apolar organic molecules (e.g. TGs or FFAs). For the esterification reaction, hydrophobic ethane-bridged organosilica groups can create a hydrophobic environment within pores and thereby ensure that the yielded water is excluded from the active sites effectively; as for the transesterification reaction, more efficient adsorption of hydrophobic TGs and easy desorption of hydrophilic glycerol (by product) can be realized.9
In the current work, H3PW12O40–ZrO2–Si(Et)Si with various percentages of ethane-bridged organosilica groups were prepared by a one-step sol-gel co-condensation-hydrothermal treatment with the aid of a triblock copolymer surfactant, F127. By using the one-step preparation route, drawbacks that existed in the post-synthesis method, such as poor control over H3PW12O40 loading, H3PW12O40 leaching, and the loss of homogeneity due to minor changes in the structure, can be overcome. The heterogeneous acid-catalytic activity of H3PW12O40–ZrO2–Si(Et)Si was evaluated by direct transesterification of Eruca Sativa Gars (ESG) oil with methanol under atmosphere refluxing and 65 °C for biodiesel production. ESG oil is a kind of non-edible oil, which is competitive for the commercial use of biodiesel production because of its low price and high yield in the drought regions and half drought regions of the world.7b
2. Experimental
2.1. Materials
H3PW12O40·xH2O, Pluronic F127 (EO106PO60EO106, where EO = –CH2CH2O–, PO = –CH2(CH3)CHO–, Mr = 12 600), 1,2-bis-(trimethoxysilyl)ethane (BTMSE, 97%) and zirconyl chloride (ZrOCl2·8H2O, 30% in hydrochloric acid) were purchased from Sigma–Aldrich and used without further purification. ESG oil is commercially available. All other chemicals were analytical reagents and obtained from Beihua Fine Chemical, Beijing, China.2.2. Preparation of catalyst
A series of H3PW12O40–ZrO2–Si(Et)Si hybrid materials with different percentages of ethane-bridged silica groups (–Si–CH2–CH2–Si–) were synthesized as follows: F127 (1.19 g) was dissolved in ethanol (19.3 mL) under vigorous stirring at room temperature. Subsequently, BTMSE (0.3 mL–1.8 mL), ZrOCl2·8H2O (1.6 mL), and aqueous H3PW12O40 (0.151 g–0.347 g in 2 mL ethanol) were added dropwise to the above solution at hourly intervals, successively. The resulting mixture was stirred at room temperature for 1 h and then it was aged at 40 °C for 24 h. The hydrogel thus obtained was subjected to hydrothermal treatment at 80 °C for another 48 h at a heating rate of 2 °C min−1. The resulting white gel was air-dried at 100 °C overnight, and then it was extracted by boiling ethanol for 12 h to remove F127. The product is denoted as H3PW12O40–ZrO2–Si(Et)Si-x, where x represents the mol percentage (mol%) of ethane-bridged silica groups in the products. The loading of H3PW12O40 in all products is ca. 10 wt%, determined by a Leeman Prodigy Spec ICP-AES.2.3. Characterization of catalyst
Low angle XRD (LXRD) patterns were obtained on a D/max-2200 VPC diffractometer using Cu Kα radiation. FTIR spectra were recorded on a Nicolet Magna 560 IR spectrophotometer. TEM was performed on a JEM-2100F high resolution transmission electron microscope at an accelerating voltage of 200 kV. Nitrogen porosimetry measurements were performed on a Micromeritics ASAP 2020M surface area and porosity analyzer after the samples were outgassed under vacuum at 363 K for 1 h and 373 K for 12 h. The surface areas were calculated using the Brunauer–Emmett–Teller (BET) equation, while pore size distribution curves were calculated using Barrett–Joyner–Halenda (BJH) desorption branch of the isotherms, and the pore volume was accumulated up to P/P0 = 0.99. 31P MAS NMR, 13C CP-MAS NMR, and 29Si MAS NMR spectra were recorded on a Bruker AVANCE III 400 WB spectrometer equipped with a 4 mm standard bore CP MAS probehead. The dried and finely powdered samples were packed in the ZrO2 rotor closed with Ke–F cap which were spun at 12KHz rate. Chemical shifts for all 31P MAS NMR, 13C CP-MAS NMR, and 29Si MAS NMR spectra were referenced to the signal of monoammonium phosphate (NH4H2PO4) standard (δ = 0.00), adamantane (C10H16) standard (δCH2 =38.5), and 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt standard (δ = 0.0), respectively.2.4. Catalytic tests
The catalysts were dried for 2 h at 120 °C in vacuum before the catalytic tests.All reactions were carried out in a three-necked round bottomed glass flask fitted with a water cooled condenser. For each reaction, 100 mg (5 wt%) of air-exposed catalyst was added. Transesterification of ESG oil was performed by stirring the mixture of ESG oil (494 mg) and methanol (2 mL) at 65 °C for 24 h. At periodical intervals, 0.1 mL of the reaction mixture was withdrawn and then diluted with acetone to 5 mL. The diluted suspension was centrifuged, and the clear solution was analyzed by a Shimadzu 2014C gas chromatograph (GC) to obtain the concentrations of the produced FAMEs. The GC was equipped with a HP-INNOWAX capillary column (film thickness, 0.5 μm; i.d., 0.32 mm; length, 30 m) and flame ionization detector. The operation temperature was 250 °C, flow rate of nitrogen gas was 1.0 mL min−1, and ethyl laurate was applied as an internal standard.
3. Results and discussion
3.1. Characterizations of catalyst
TEM observations are consistent with the above LXRD testing result, revealing that the H3PW12O40–ZrO2–Si(Et)Si hybrid materials tailored by F127 possess 3D wormhole-like pore morphology with uniform pore size but interconnecting pore channels (Fig. 1a). Ethane-free H3PW12O40–ZrO2 also exhibits 3D wormhole-like pore morphology, however, its porosity is much worse compared with H3PW12O40–ZrO2–Si(Et)Si (Fig. 1b).
 | ||
| Fig. 1 TEM images of H3PW12O40–ZrO2–Si(Et)Si-60 (a), and H3PW12O40–ZrO2 (b). | ||
The nitrogen sorption isotherms of ethane-bridged H3PW12O40–ZrO2–Si(Et)Si are Type IV with H2-type hysteresis loops, implying they possess regular even mesopores with interconnecting channels (Fig. 2a). Additionally, the adsorbed nitrogen gas amount increased with the percentage of ethane-bridged organosilica groups. In the case of ethane-free H3PW12O40–ZrO2, the hysteresis loop is hardly observed, together with low nitrogen gas adsorbed amount. The pore size distribution curves displayed in Fig. 2b indicate ethane-bridged H3PW12O40–ZrO2–Si(Et)Si exhibit uniform pore sizes with a diameter of ca. 3.5 nm; and pore volume increased with the percentage of the incorporated ethane-bridged organosilica groups. Nevertheless, the porosity of H3PW12O40–ZrO2 is poor. Through analyzing the textural parameters summarized in Table 1 it is found that the BET surface area and pore volume of the H3PW12O40–ZrO2–Si(Et)Si hybrid materials continuously increased with the percentage of ethane-bridged organosilica groups from 40 mol% to 80 mol%. For the ethane-free H3PW12O40–ZrO2, its BET surface area is extremely small (3 m2 g−1); meanwhile, its pore volume is not detected. The above results are in good agreement with those of TEM and LXRD, implying that the porosity of H3PW12O40–ZrO2 was improved obviously due to the introduction of ethane-bridged organosilica groups within its framework.
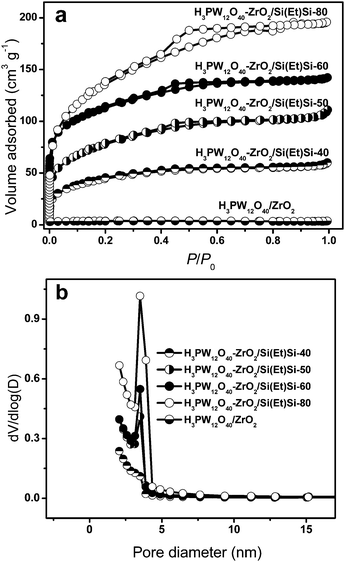 | ||
| Fig. 2 Nitrogen gas adsorption-desorption isotherms (a), and pore size distribution profiles of various H3PW12O40–ZrO2–Si(Et)Si materials (b). | ||
| Catalysts | S BET/ | D p a/ | V p b/ |
|---|---|---|---|
| m2 g−1 | nm | cm3 g−1 | |
| a Pore diameter (Dp) was estimated from BJH desorption determination. b Pore volume (Vp) was estimated from the pore volume determined using the adsorption branch of the N2 isotherm curve at P/P0 = 0.99 single point. | |||
| H3PW12O40–ZrO2 | 3 | — | — |
| H3PW12O40–ZrO2–Si(Et)Si-40 | 161 | 3.0 | 0.09 |
| H3PW12O40–ZrO2–Si(Et)Si-50 | 248 | 3.5 | 0.15 |
| H3PW12O40–ZrO2–Si(Et)Si-60 | 265 | 3.5 | 0.16 |
| H3PW12O40–ZrO2–Si(Et)Si-80 | 492 | 3.5 | 0.30 |
FT-IR spectra of H3PW12O40–ZrO2–Si(Et)Si can provide valuable structural information about the Keggin units and organic bridging groups (Fig. 3). Four vibrational peaks situated at 1100 cm−1 to 800 cm−1 originate from the stretching vibrations of the tetrahedral P–O bonds (1080 cm−1), terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (982 cm−1), and two types of bridging W–O–W bonds (815 cm−1 and 895 cm−1) of the cluster.10 The above four characteristic vibrational peaks can also been found in the H3PW12O40–ZrO2–Si(Et)Si hybrid materials, however, the peak intensities decrease significantly. Generally, the stretching vibrations of Si–O bonds also appear at ca. 1080 cm−1, therefore, the IR peak at 1080 cm−1 may also come from the contribution of the Si–O bonds of the ethane-bridged organosilica groups.11 Additionally, compared with the vibrational spectrum of ethane-free H3PW12O40–ZrO2, some new peaks related to the ethane-bridged organosilica groups are found in the ethane-bridged H3PW12O40–ZrO2–Si(Et)Si hybrid materials. That is, the peak at 1150 cm−1 is assigned to the stretching vibrations of Si–C bonds, and the peak at 1452 cm−1 is assigned to the deformation vibrations of C–H bonds of ethane groups connected to Si atoms.11,12 The peak at 1266 cm−1 is attributed to other types of deformation vibrations of these C–H bonds.12 As for the two weak peaks at 2976 cm−1 and 2873 cm−1, they originate from the stretching vibrations of C–H bonds in the –CH2CH2– and –CH2CH3 moieties of the ethane-bridged organosilica groups.
O bonds (982 cm−1), and two types of bridging W–O–W bonds (815 cm−1 and 895 cm−1) of the cluster.10 The above four characteristic vibrational peaks can also been found in the H3PW12O40–ZrO2–Si(Et)Si hybrid materials, however, the peak intensities decrease significantly. Generally, the stretching vibrations of Si–O bonds also appear at ca. 1080 cm−1, therefore, the IR peak at 1080 cm−1 may also come from the contribution of the Si–O bonds of the ethane-bridged organosilica groups.11 Additionally, compared with the vibrational spectrum of ethane-free H3PW12O40–ZrO2, some new peaks related to the ethane-bridged organosilica groups are found in the ethane-bridged H3PW12O40–ZrO2–Si(Et)Si hybrid materials. That is, the peak at 1150 cm−1 is assigned to the stretching vibrations of Si–C bonds, and the peak at 1452 cm−1 is assigned to the deformation vibrations of C–H bonds of ethane groups connected to Si atoms.11,12 The peak at 1266 cm−1 is attributed to other types of deformation vibrations of these C–H bonds.12 As for the two weak peaks at 2976 cm−1 and 2873 cm−1, they originate from the stretching vibrations of C–H bonds in the –CH2CH2– and –CH2CH3 moieties of the ethane-bridged organosilica groups.
 | ||
| Fig. 3 FT-IR spectra of the starting H3PW12O40, ZrO2 matrix, and H3PW12O40–ZrO2–Si(Et)Si materials. | ||
31P MAS NMR spectra further confirm the structure integrity of the Keggin units after formation of the hybrid materials (Fig. 4). The stronger peak at 16.2 ppm or 16.3 ppm originates from the resonance of PO4 units within the bulk H3PW12O40 environment.7a The shoulder peak at 11.4 ppm (H3PW12O40–ZrO2), 14.0 ppm (H3PW12O40–ZrO2–Si(Et)Si-50), or 9.8 ppm (H3PW12O40–ZrO2–Si(Et)Si-60) is attributed to the PO4 unit of H3PW12O40 located near the ZrO2 surface. Owing to the strong interaction between the Keggin units and ZrO2, distortion of the Keggin cage at the interface of the two components occurred, creating a new chemical environmental that is slightly different from the PO4 unit of bulk H3PW12O40.
 | ||
| Fig. 4 31P MAS MNR spectra of H3PW12O40–ZrO2 (a), H3PW12O40–ZrO2–Si(Et)Si-50 (b), and H3PW12O40–ZrO2–Si(Et)Si-60 (c). | ||
From the 29Si MAS NMR spectrum shown in Fig. 5a it is observed that there are chemical shifts at δ −63.0 ppm and −59.3 ppm, which are due to the silicon species of SiC(OSi)3 (3T) and SiC(OSi)2 (2T) within the ethane-bridged organosilica groups, respectively.13 Virtually no signals due to Q-species on the scale of −90 ppm to −120 ppm were observed in the 29Si MAS NMR spectrum, indicating there is essentially no evidence for Si–C bonds cleavage by using the current one-step preparation route.14 From the representative 13C CP-MAS NMR spectrum shown in Fig. 5b, one strong signal (δ 4.9 ppm) and three weak signals (δ 70.7 ppm, 57.6 ppm, and 17.1 ppm) are seen, which are attributed to the carbon atoms within the ethane-bridged organosilica groups, carbon species in residual F127, and ethoxy groups formed during the F127 extraction process, respectively.15
 | ||
| Fig. 5 29Si MAS MNR (a) and 13C CP-MAS NMR (b) spectra of H3PW12O40–ZrO2–Si(Et)Si-60. | ||
From the combination of the above spectroscopy data it is inferred that: (1) both inorganic and organic functional groups are introduced into the ZrO2 framework by using the current preparation route; (2) considering the well-matched electronegativity and ionic radius of the Zr4+ ion (1.33, 0.072 nm) and the W6+ ion (1.70, 0.060 nm), the Keggin units interact with the ZrO2 framework through Zr–O–W covalent bonds at the interface of the two components. This strong interaction of surface ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ZrOH groups with WO bonds of the Keggin units led to the formation of (ZrOH2)n+[H3−nPW12O40]n− species; and (3) the ethane-bridged organosilica groups are, as a bridging component, directly introduced into the 3D interconnected ZrO2 framework through –Zr–O–Si–C–C–Si–O– linkages. Based on the above discussion, a proposed schematic representation of the H3PW12O40–ZrO2–Si(Et)Si hybrid material is shown in Scheme 1.
ZrOH groups with WO bonds of the Keggin units led to the formation of (ZrOH2)n+[H3−nPW12O40]n− species; and (3) the ethane-bridged organosilica groups are, as a bridging component, directly introduced into the 3D interconnected ZrO2 framework through –Zr–O–Si–C–C–Si–O– linkages. Based on the above discussion, a proposed schematic representation of the H3PW12O40–ZrO2–Si(Et)Si hybrid material is shown in Scheme 1.
 | ||
| Scheme 1 The wall composition of mesoporous H3PW12O40–ZrO2–Si(Et)Si material and the process of the H3PW12O40–ZrO2–Si(Et)Si-catalyzed biodiesel synthesis from Eruca Sativa Gars oil. | ||
3.2. Catalytic tests
The prepared H3PW12O40–ZrO2–Si(Et)Si hybrid materials, which combine strong Brϕnsted acidity, excellent porosity, and increased hydrophobicity compared with H3PW12O40–ZrO2, are expected to exhibit high catalytic activity towards biodiesel synthesis from ESG oil. Herein, the H3PW12O40–ZrO2–Si(Et)Si-catalyzed transesterification of ESG oil with methanol was performed under the conditions of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 ESG oil to MeOH molar ratio, 5 wt% catalyst, 24 h, 65 °C, and atmosphere refluxing. For comparison, ethane-free H3PW12O40–ZrO2 was also tested under the same conditions. Through GC-MS analysis, four FAMEs including methyl palmitate (MP), methyl stearate (MS), methyl oleate (MO), and methyl lioleate (ML) were the main products in the current system.1h Among them, MP came from the esterification of free palmic acid existing in ESG oil {142.6 μmol g−1, analyzed by an Agilent 1200 series liquid chromatograph system coupled with Bruker micrOTOF II mass spectrometry (LC-MS) (source type, ESI; ion polarity, positive; set nebulizer, 1.5 Bar; set dry heater, 180 °C; set dry gas, 80L min−1)}, and the other three FAMEs were produced from the transesterification of TGs in the ESG oil. The activity of the catalysts was evaluated quantitatively by the yield of each of the above FAMEs (% Y), Y = (MD/MT) × 100, where MD and MT are the number of mols of each FAME produced and expected, respectively.
:90 ESG oil to MeOH molar ratio, 5 wt% catalyst, 24 h, 65 °C, and atmosphere refluxing. For comparison, ethane-free H3PW12O40–ZrO2 was also tested under the same conditions. Through GC-MS analysis, four FAMEs including methyl palmitate (MP), methyl stearate (MS), methyl oleate (MO), and methyl lioleate (ML) were the main products in the current system.1h Among them, MP came from the esterification of free palmic acid existing in ESG oil {142.6 μmol g−1, analyzed by an Agilent 1200 series liquid chromatograph system coupled with Bruker micrOTOF II mass spectrometry (LC-MS) (source type, ESI; ion polarity, positive; set nebulizer, 1.5 Bar; set dry heater, 180 °C; set dry gas, 80L min−1)}, and the other three FAMEs were produced from the transesterification of TGs in the ESG oil. The activity of the catalysts was evaluated quantitatively by the yield of each of the above FAMEs (% Y), Y = (MD/MT) × 100, where MD and MT are the number of mols of each FAME produced and expected, respectively.
The catalytic activity of the H3PW12O40–ZrO2–Si(Et)Si hybrid materials with different percentages of ethane-bridged organosilica groups (0, 40 mol%, 50 mol%, 60 mol%, and 80 mol%) was compared firstly. From the results shown in Fig. 6a it was found that the yields of the four FAMEs continuously increased with the percentage of ethane-bridged organosilica groups from 0, to 40 mol%, to 50 mol%, to 60 mol%, however, when further increasing the percentage to 80 mol%, the activity began to decrease. However, all the ethane-bridged H3PW12O40–ZrO2–Si(Et)Si hybrid materials possess higher catalytic activity than that of ethane-free H3PW12O40–ZrO2. For example, after the reaction proceeded for 24 h, the yields of MP, MS, MO, and ML reached to 99.9%, 67.8%, 73.5%, and 75.3%, respectively, for the most active H3PW12O40–ZrO2–Si(Et)Si-60. For the ethane-free H3PW12O40–ZrO2, the yields of MP, MS, MO, and ML reached 35.2%, 18.4%, 18.9%, and 17.5%, respectively, under the same conditions.
 | ||
| Fig. 6 Catalytic activity of H3PW12O40–ZrO2 and various H3PW12O40–ZrO2–Si(Et)Si materials towards transesterification of ESG oil with methanol (a), kinetics study on H3PW12O40–ZrO2–Si(Et)Si-60 catalyzed transesterification of ESG oil with methanol (b) ESG oil:0.55 mmol, molar ratio of oil:MeOH = 1:90, 5 wt% catalyst, 24 h, 65 °C, and atmosphere refluxing. | ||
Kinetic study of H3PW12O40–ZrO2–Si(Et)Si-60-catalyzed transesterification of ESG oil further confirms the excellent acid catalytic activity of the as-prepared hybrid solid acids (Fig. 6b).
The above catalytic tests suggest that introduction of ethane-bridged organosilica groups can improve the acid catalytic activity of the H3PW12O40–ZrO2, allowing the acid-catalyzed transesterification reaction from low-cost feedstocks to be performed under mild conditions. This excellent performance makes the prepared H3PW12O40–ZrO2–Si(Et)Si material an ideal catalyst for the biodiesel production from low-cost biomass. The acid catalytic activity of the H3PW12O40–ZrO2–Si(Et)Si is mainly due to the Brϕnsted acidity of the H3PW12O40–ZrO2, which can ensure the transesterification reaction proceeds at an acceptable rate. ZrO2 itself shows Brϕnsted acidity, and its Brϕnsted acidity was enhanced by the interaction with Keggin units. The formed (ZrOH2)n+[H3−nPW12O40]n− species at the interface between ZrO2 and H3PW12O40 led to electron transfer from the terminal oxygen atoms within WO groups to ZrO2. Accordingly, the release of the protons in the hybrid solid acids became more easy compared with the starting Keggin units and ZrO2.
Additionally, the excellent porosity of the H3PW12O40–ZrO2–Si(Et)Si hybrid materials, including 3D interconnected mesostructure, large BET surface area, uniform pore size distribution, and high pore volume, also contributes to the enhanced catalytic activity of the H3PW12O40–ZrO2–Si(Et)Si compared with ethane-free H3PW12O40–ZrO2. A larger BET surface area and high pore volume can provide better dispersion of H3PW12O40 throughout the hybrid material, resulting in an increase of the population of the active sites and accessibility of the active sites to the reactants, while uniform pore size distributions can minimize diffusion limitation of reactants or products. Therefore, the 3D interconnected mesostructure together with the homogeneous dispersion of Keggin units within the pore channels and organic groups in the pore walls can allow more efficient transport of guest species via much more direct and easier diffusion to the network sites. From the results shown in Table 1 it can be seen that at a similar H3PW12O40 loading the BET surface area of the H3PW12O40–ZrO2–Si(Et)Si hybrid materials increased with the increasing percentage of the ethane-bridged organosilica groups. Accordingly, the acid-catalytic activity of the H3PW12O40–ZrO2–Si(Et)Si hybrid materials towards the transesterification of ESG oil increased with the percentage of the bridging organosilica groups. However, although the H3PW12O40–ZrO2–Si(Et)Si-80 material possesses the best surface textural properties, the yields of FAMEs are the lowest among all tested H3PW12O40–ZrO2–Si(Et)Si hybrid materials. This is due to the fact that a higher percentage of bridging ethane groups decreases the number of acid sites. The poor catalytic activity of H3PW12O40–ZrO2 is mainly due to its porosity being worse.
Finally, the introduction of ethane-bridged organosilica groups can improve the surface hydrophobicity of H3PW12O40–ZrO2, which has a positive influence on the acid-catalyzed transesterification reaction. Compared with H3PW12O40–ZrO2, the enhanced surface hydrophobicity of H3PW12O40–ZrO2–Si(Et)Si selectively creates an unsuitable environment for hydrophilic products (i.e. water or glycerol), leading to relatively weak adsorption of water or glycerol molecules and strong adsorption of TGs on the hybrid catalyst surface. Accordingly, the accessibility of active sites to the hydrophobic reactants (i.e. TGs or FFAs) is increased, which facilitates the transesterification reaction proceeding at a fast rate. At the same time, acid sites deactivation due to the strong adsorption of hydrophilic materials on the catalyst surface is inhibited. As a consequence, higher activity was obtained compared to ethane-free H3PW12O40–ZrO2. The above discussion is supported by the following experimental observation. When the H3PW12O40–ZrO2 was used as the transesterification catalyst, phase separation was observed in the reaction system with a transparent methanol layer and an opalescent oil layer containing the catalyst. However, a uniform blend of methanol layer, oil layer, and catalyst was obtained after replacement of H3PW12O40–ZrO2 with H3PW12O40–ZrO2–Si(Et)Si, leading to good miscibility of methanol with the TGs, which play an important role in the transesterification.
3.3. Regeneration and reusability
From the viewpoint of practical applications of the catalyst, H3PW12O40–ZrO2–Si(Et)Si-60 was selected to evaluate the reusability and to study the regeneration method of as-prepared hybrid materials. After the first catalytic run, the used catalyst was separated from the reaction mixture by centrifugation, and then they were washed with tetrahydrofuran (THF) three times. After being dried at 60 °C, the recovered catalyst was used for the second and third catalytic run, respectively, under the same experimental conditions and regeneration method. As shown in Fig. 7, the hybrid material showed a good catalytic stability maintaining a similar level of reactivity after three catalytic cycles. Furthermore, leaching of H3PW12O40 during each catalytic cycle was monitored by ICP-AES method. As expected, P and W were not detected in the catalyst-free reaction solutions. The above results prove that the hybrid material works effectively as a recyclable water-tolerant solid acid catalyst in the transesterification of ESG oil to produce FAMEs. They also show that the decrease of the reactivity after the first and the second run is due to the loss of the catalyst powder during the separation and recovery processes. This catalytic stability is due to the strong chemical, rather than physical, interaction existing between the Keggin unit and the ZrO2 support. Moreover, deactivation of the catalyst due to the strong adsorption of alkyl-free ZrO2-based catalyst to glycerol and water was inhibited since the hydrophobicity of the catalyst was enhanced due to the introduction of ethane-bridged organosilica moieties. | ||
| Fig. 7 Recyclability of H3PW12O40–ZrO2–Si(Et)Si-60 catalyst for the transesterification of ESG oil with methanol. ESG oil:0.55 mmol, molar ratio of ESG oil:MeOH = 1:90; 5 wt% catalyst, 24 h, 65 °C, and atmosphere refluxing. | ||
4. Conclusions
By using the current one-step sol-gel-hydrothermal method, ZrO2-based hybrid catalysts co-functionalized by ethane-bridged organosilica groups and the Keggin type heteropoly acid were successfully prepared. Compared with ethane-free H3PW12O40–ZrO2, the as-prepared H3PW12O40–ZrO2–Si(Et)Si hybrid catalysts exhibited higher heterogeneous acid catalytic activity towards biodiesel synthesis from low-cost Eruca Sativa Gars oil, regardless of the incorporating percentages of the ethane-bridged organosilica groups. This excellent catalytic activity of the hybrid acid catalysts is due to the combination of strong Brϕnsted acidity, 3D interconnected mesostructure, and enhanced hydrophobicity; and among these three factors, the inherent acidic properties of H3PW12O40–ZrO2 play a dominant role in the catalytic activity. The as-prepared multifunctionalized hybrid materials can be reused three times at least without significant loss of the catalytic activity, and they are considered as promising environmentally-friendly solid acid catalysts for application in acid-catalyzed biodiesel production from low-cost feedstocks under mild conditions.Acknowledgements
This work was supported by the Natural Science Fund Council of China (21173036) and the Fundamental Research Funds for the Central Universities (09QNTD004).Notes and references
- (a) L. L. Xu, Y. H. Wang, X. Yang, X. D. Yu, Y. H. Guo and J. H. Clark, Green Chem., 2008, 10, 746–755 RSC; (b) H. Q. Gu, Y. J. Jiang, L. Y. Zhou and J. Guo, Energy Environ. Sci., 2011, 4, 1337–1344 RSC; (c) H. J. Berchmans and S. Hirata, Bioresour. Technol., 2008, 99, 1716–1721 CrossRef CAS; (d) E. E. Kwon, J. Seo and H. Yi, Green Chem., 2012, 14, 1799–1804 RSC; (e) G. Knothe, Green Chem., 2011, 13, 3048–3065 RSC; (f) Y. C. Sharma, B. Singh and J. Korstad, Green Chem., 2011, 13, 2993–3006 RSC; (g) P. D. Patil, H. Reddy, T. Muppaneni, A. Mannarswamy, T. Sohuab, F. O. Holguin, P. Lammers, N. Nirmalakhandan, P. Cooke and S. G. Deng, Green Chem., 2012, 14, 809–818 RSC; (h) W. Li, F. Y. Ma, F. Su, L. Ma, S. Q. Zhang and Y. H. Guo, ChemSusChem, 2011, 4, 744–756 CrossRef CAS.
- (a) E. Lotero, Y. J. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Jr., Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS; (b) J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 11, 1285–1308 RSC.
- (a) A. A. Kiss, A. C. Dimian and G. Rothenberg, Adv. Synth. Catal., 2006, 348, 75–81 CrossRef CAS; (b) T. Okuhara, Chem. Rev., 2002, 102, 3641–3666 CrossRef CAS.
- (a) K. Suwannakarn, E. Lotero, J. Goodwin and C. Q. Lu, J. Catal., 2008, 255, 279–286 CrossRef CAS; (b) G. D. Yadav and A. D. Murkute, J. Catal., 2004, 224, 218–223 CrossRef CAS; (c) M. L. Grecea, A. C. Dimian, S. Tanase, V. Subbiah and G. Rothenberg, Catal. Sci. Technol., 2012, 2, 1500–1506 RSC.
- (a) M. Hara, ChemSusChem, 2009, 2, 129–135 CrossRef CAS; (b) M. Hara, Energy Environ. Sci., 2010, 3, 601–607 RSC; (c) W. Y. Lou, M. H. Zong and Z. Q. Duan, Bioresour. Technol., 2008, 99, 8752–8758 CrossRef CAS.
- (a) N. Shibasaki-Kitakawa, H. Honda, H. Kuribayashi, T. Toda, T. Fukumura and T. Yonemoto, Bioresour. Technol., 2007, 98, 416–421 CrossRef CAS; (b) Y. H. Feng, B. Q. He, Y. H. Cao, J. X. Li, M. Liu, F. Yan and X. P. Liang, Bioresour. Technol., 2010, 101, 1518–1521 CrossRef CAS.
- (a) K. Narasimharao, D. R. Brown, A. F. Lee, A. D. Newman, P. F. Siril, S. J. Tavener and K. Wilson, J. Catal., 2007, 248, 226–234 CrossRef CAS; (b) F. Chai, F. H. Cao, F. Y. Zhai, Y. Chen, X. H. Wang and Z. M. Su, Adv. Synth. Catal., 2007, 349, 1057–1065 CrossRef CAS.
- (a) K. Jacobson, R. Gopinath, L. C. Meher and A. K. Dalai, Appl. Catal., B, 2008, 85, 86–91 CrossRef CAS; (b) C. F. Oliveira, L. M. Dezaneti, F. A. C. Garcia, J. L. De Macedo and J. A. Dias, Appl. Catal., A, 2010, 372, 153–161 CrossRef CAS.
- (a) L. L. Xu, W. Li, J. L. Hu, X. Yang and Y. H. Guo, Appl. Catal., B, 2009, 90, 587–594 CrossRef CAS; (b) B. Karimi, H. M. Mirzaei and A. Mobaraki, Catal. Sci. Technol., 2012, 2, 828–834 RSC.
- A. D. Newman, A. F. Lee, K. Wilson and N. A. Young, Catal. Lett., 2005, 102, 45–50 CrossRef CAS.
- C. W. Young, P. C. Servais, C. C. Currie and M. J. Hunter, J. Am. Chem. Soc., 1948, 70, 3758–3764 CrossRef CAS.
- Y. S. Li, Y. Wang and S. Ceesay, Spectrochim. Acta, Part A, 2009, 71, 1819–1824 CrossRef.
- L. Zhao, G. S. Zhu, D. L. Zhang, Y. Di, Y. Chen, O. Terasaki and S. L. Qiu, J. Phys. Chem. B, 2005, 109, 764–768 CrossRef CAS.
- A. P. Kapoor, Q. H. Yang and S. Inagaki, Chem. Mater., 2004, 16, 1209–1213 CrossRef.
- K. J. Shea, G. Cerrato, S. Ardizzone, C. L. Bianchi, M. Signoretto and F. Pinna, Phys. Chem. Chem. Phys., 2002, 4, 3136–4507 RSC.
| This journal is © The Royal Society of Chemistry 2012 |