Non-enzymatic dynamic kinetic resolution of racemic α-arylalkanoic acids: an advanced asymmetric synthesis of chiral nonsteroidal anti-inflammatory drugs (NSAIDs)†
Isamu
Shiina
*,
Keisuke
Ono
and
Kenya
Nakata
Department of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: shiina@rs.kagu.tus.ac.jp; Fax: +81 3-3260-5609
First published on 4th July 2012
Abstract
An efficient protocol was developed to produce chiral 2-arylalkanoic esters in high yields (up to 99%) from racemic carboxylic acids utilizing the racemization of the mixed-anhydrides generated from acid components with pivalic anhydride in the presence of an acyl-transfer catalyst. The present DKR involves the enantio-discriminating esterification of the racemic 2-arylalkanoic acids and the rapid racemization of the chiral 2-arylalkanoic acids under suitable reaction conditions using pivalic anhydride, diisopropylethylamine, and benzotetramisole (BTM) in a polar solvent, and this method was successfully applied for the preparation of pharmacodynamically active (S)-enantiomers of nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen and naproxen.
The synthesis of optically active carboxylic acids and esters is a very important topic due to the significant role they play in the field of medicinal and pharmaceutical science.1 For instance, several 2-arylalkanoic acids, such as ibuprofen and naproxen, function as nonsteroidal anti-inflammatory drugs (NSAIDs),2,3 and the therapeutically relevant mechanism of action of these therapeutic agents has been found only for the (S)-enantiomers.4–6
We have recently discovered the kinetic resolution (KR) of racemic 2-arylalkanoic acids with achiral alcohols using carboxylic anhydride as the condensation reagent in the presence of a chiral acyl-transfer catalyst,7,8 such as (R)-benzotetramisole ((R)-BTM) or (S)-β-Np-BTM (Scheme 1) [eqn (1)].9,10 On the other hand, dynamic kinetic resolution (DKR) is a more attractive method than the KR because the theoretical yield of the target molecule becomes quantitative in the DKR through the rapid racemization of the reactants (Scheme 1) [eqn (2)].11 By using these successive reactions including racemization and selective esterification, a single enantiomer might be preferentially prepared in a high yield from a mixture of (R)- and (S)-2-arylalkanoic acids.
![Two methods for the preparation of the optically active 2-arylalkanoic acids. Our previous study (kinetic resolution (KR)) [eqn (1)] and this study (dynamic kinetic resolution (DKR)) [eqn (2)].](/image/article/2012/CY/c2cy20329d/c2cy20329d-s1.gif) | ||
| Scheme 1 Two methods for the preparation of the optically active 2-arylalkanoic acids. Our previous study (kinetic resolution (KR)) [eqn (1)] and this study (dynamic kinetic resolution (DKR)) [eqn (2)]. | ||
It is well known that the forced reaction conditions, such as the presence of strong bases at high temperature, are required for producing the interconversion between the enantiomers of 2-arylalkanoic acids.12 However, we anticipated that an effective protocol for the DKR might be developed if milder reaction conditions for the racemization of the reactants could be established. We now report a new method for the DKR of a variety of racemic 2-arylalkanoic acids by asymmetric esterification via the in situ formation of mixed-anhydrides using pivalic anhydride (Piv2O) as the coupling reagent.
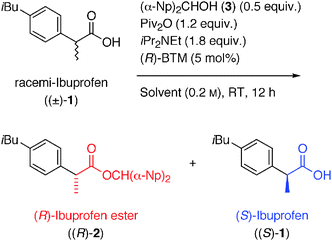
As a preliminary investigation, we examined the solvent effects in the original method for the KR of racemic ibuprofen ((±)-1) with 0.5 equiv. of (α-Np)2CHOH (3)13 using Piv2O and (R)-BTM at room temperature for 12 h, which was the optimized reaction conditions established in our previous studies (Table 1).10 As shown by entries 1–6, a variety of general nonpolar and less polar organic solvents (dipole moment = 0.37–2.69 D (debye)) were first used for the reaction, and good to high yields and enantioselectivities of the produced (R)-ibuprofen ester ((R)-2) (41–46% yield, 83–92% ee) and the recovered (S)-ibuprofen ((S)-1) (39–47% yield, 61–73% ee) were observed in all cases.
| Entry | Solvent | Dipole moment/D | Yield (2;1) [%] | ee(2;1)[%] |
|---|---|---|---|---|
 a The reaction was carried out using 1.2 equiv. of bis(α-naphthyl) methanol (3), 2.4 equiv. of Piv2O, 4.8 equiv. of iPr2NEt, and 5 mol% of (R)-BTM in DMF (0.2 M) at RT for 48 h.
a The reaction was carried out using 1.2 equiv. of bis(α-naphthyl) methanol (3), 2.4 equiv. of Piv2O, 4.8 equiv. of iPr2NEt, and 5 mol% of (R)-BTM in DMF (0.2 M) at RT for 48 h.
|
||||
| 1 | Toluene | 0.37 | 45;47 | 85;65 |
| 2 | Et2O | 1.12 | 45;45 | 83;67 |
| 3 | CH2Cl2 | 1.14 | 41;46 | 91;72 |
| 4 | THF | 1.70 | 42;42 | 92;61 |
| 5 | EtOAc | 1.88 | 46;40 | 86;73 |
| 6 | Acetone | 2.69 | 44;39 | 89;71 |
| 7 | MeCN | 3.44 | 46;46 | 85;47 |
| 8 | DMA | 3.72 | 38;47 | 94;13 |
| 9 | DMF | 3.86 | 39;42 | 92;10 |
| 10a | DMF | 3.86 | 93;4 | 91;12 |
| 11 | DMI | 4.07 | 19;48 | 93;7 |
| 12 | NMP | 4.09 | 31;49 | 92;12 |
| 13 | DMSO | 4.30 | 22;61 | 89;2 |
Fortunately, it was found that THF as an alternative medium was effective as the original reported CH2Cl2 (compare entries 3 and 4). When the reaction was carried out in MeCN, which is a comparable polar solvent (dipole moment = 3.44 D), the reaction smoothly proceeded and the desired (R)-ibuprofen ester ((R)-2) was also produced with a good ee. However, the enantiopurity of the recovered carboxylic acid (S)-1 had significantly decreased (entry 7, 47% ee). This tendency was more dramatic when using higher polar solvents, such as DMA, DMF, DMI, NMP, and DMSO (dipole moment = 3.72–4.30 D) (entries 8–13). Because the racemization of the remaining carboxylic acid was significantly enhanced in such polar solvents, it was postulated that these reaction conditions might be suitable for the DKR of the racemic NSAIDs in the presence of Piv2O and (R)-BTM. Actually, we successfully revealed that the racemization of the acid (S)-1 rapidly took place in the presence of sufficient amounts of reagents in the DMF solvent as shown by entry 10, and the drastic improvement in the yield of the desired ibuprofen ester (R)-2 was found (93%) without loss of its high enantiopurity (91% ee).
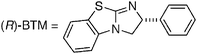
In order to explore the scope and limitation of this new method, we carried out a systematic examination of the electronic effects on the aromatic rings of the substrates (Scheme 2).14 For example, the DKR of (±)-1a–f with electron donating groups (EDGs), such as methyl and methoxy groups, on the aromatic rings at the o-, m-, and p-positions afforded good yields (85–96%) of the corresponding chiral ester (R)-2a–f with high enantiopurities (84–97% ee) except for the yields of (R)-2d and (R)-2f (76% and 50%, respectively). Because the DKR of (±)-1d and (±)-1f showed somewhat low conversions to form (R)-2d and (R)-2f under the standard reaction conditions, it was re-investigated by adding an excess amount of Piv2O to the mixture and prolonging the reaction time. Fortunately, we observed better yields of (R)-2d and (R)-2f (94% and 56%, respectively) from the racemic carboxylic acids (±)-1d and (±)-1f without any loss of enantioselectivities (87% ee and 97% ee, respectively).
 | ||
| Scheme 2 Examination of the electronic effects on the aromatic rings of the substrates using racemic 2-arylalkanoic acids (±)-1a–i. | ||
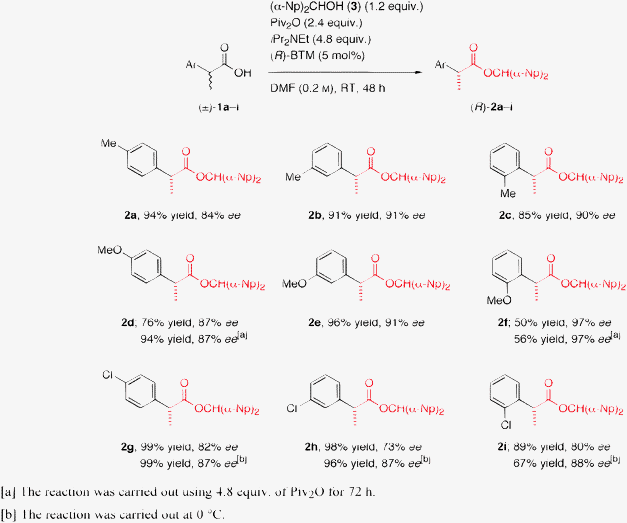
We next performed the DKR of a variety of 2-arylpropanoic acids ((±)-1j–r) including several nonsteroidal anti-inflammatory drugs (NSAIDs) under the optimized reaction conditions to assess the general applicability of this novel method (Scheme 3).14 All the reactions provided very good results, and the desired carboxylic esters (R)-2j–r were obtained in excellent yields (88–96%) with high enantiopurities (81–93% ee) irrespective of the substitution pattern and the electronic effects on the aromatic rings of the substrates. It is noteworthy that the DKR of several NSAIDs, such as flurbiprofen (1o), ketoprofen (1p), fenoprofen (1q), and naproxen (1r), was also easily achieved to produce the corresponding esters with good enantiopurities.
 | ||
| Scheme 3 DKR of a variety of racemic 2-arylalkanoic acids (±)-1j–r. | ||
We further examined the reaction pathway for the DKR of racemic 2-arylalkanoic acids based on the kinetic studies as shown in Fig. 1 and 2. In the presence of 5 mol% of (R)-BTM, the racemic deuterated 2-phenylpropanoic acid-d3 was treated with 1.2 equiv. of 3, 2.4 equiv. of Piv2O, and 4.8 equiv. of iPr2NEt in DMF-d7 at room temperature, and the relative amounts of four species: (i) the remaining deuterated 2-phenylpropanoic acid-d3; (ii) initially formed mixed anhydride (MA); (iii) symmetrical anhydride (SA), having two moieties of the deuterated 2-phenylpropanoic acid-d3; and (iv) the formed ester, were monitored by 1H NMR. The quantity of the deuterated 2-phenylpropanoic acid-d3 gradually decreased in proportion to the reaction time and almost all of the acid components, such as the free acid, MA, and SA, finally disappeared after 50 h (3000 min). On the other hand, the quantity of the desired ester time-dependently increased and reached 97% after 50 h, and the favorable enantioselectivity of this DKR in DMF-d7 was found to provide the corresponding (R)-ester in 92% ee. The magnified graph from 0 to 200 min (Fig. 2) showed that the MA was first generated by the reaction of the deuterated 2-phenylpropanoic acid-d3 with Piv2O, and the quantity of MA increased with the maximum peak at 40 min after the beginning of the reaction. From 40 to 3000 min, the formed MA was gradually consumed and then transformed into the desired (R)-ester in accordance with the decrease in the quantity of the starting deuterated 2-phenylpropanoic acid-d3 (Fig. 1). The formation of a small amount of SA, which was generated from MA via disproportionation, was also detected in the 1H NMR analysis, and almost all of the SA disappeared within 1 h as shown in Fig. 2.
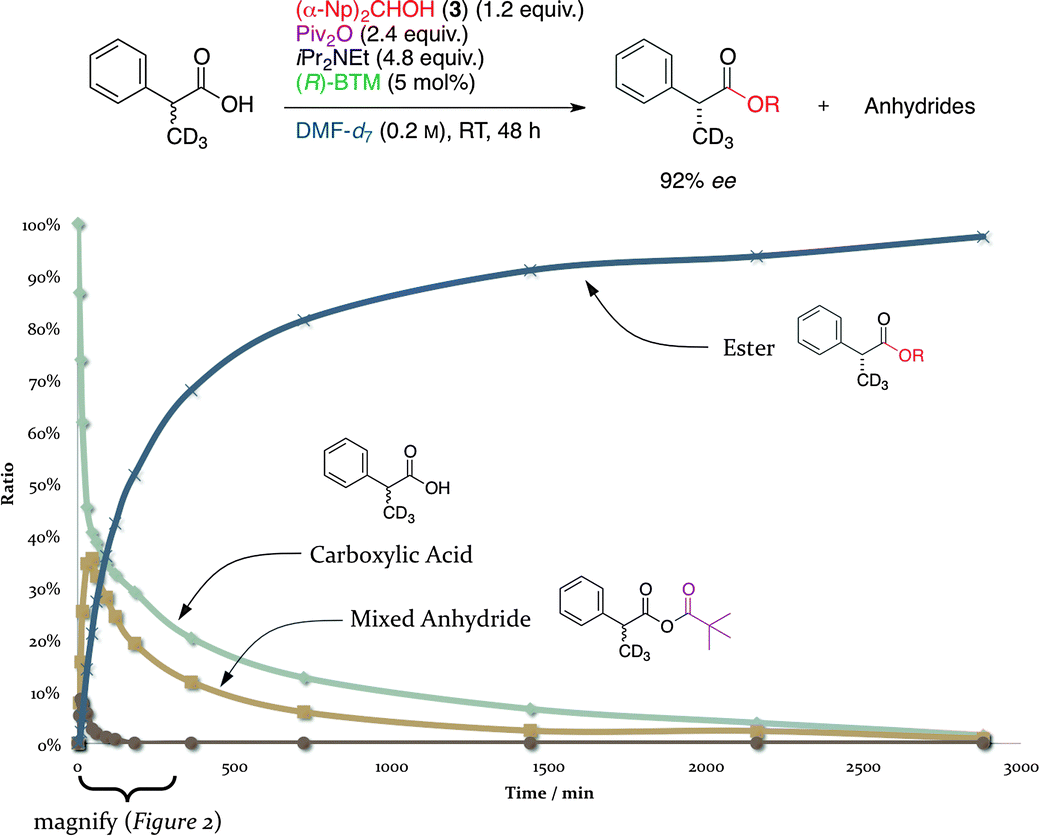 | ||
| Fig. 1 Time course of the DKR of deuterated 2-phenylpropanoic acid-d3 (0–3000 min). | ||
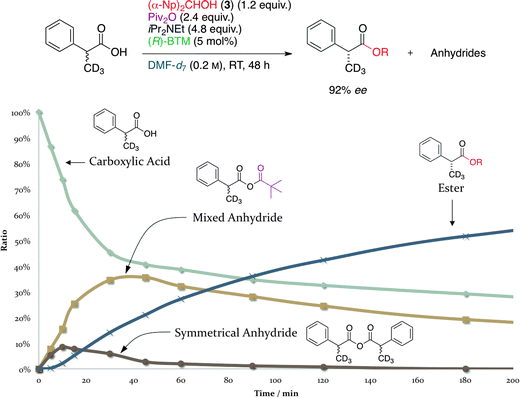 | ||
| Fig. 2 Time course of the DKR of deuterated 2-phenylpropanoic acid-d3 (0–200 min). | ||
Next, the time course of the racemization process was investigated without adding bis(α-naphthyl)methanol (3), a nucleophile, to the reaction mixture. The ee's of the racemized 2-phenylpropanoic acid derived from the “chiral” (S)-2-phenylpropanoic acid (99% ee) were determined after conversion into 2-phenyl-1-propanol, and the ee's of the alcohol were plotted on the graph (Fig. 3). There was no difference between the ee's of the racemized products starting from (S)-2-phenylpropanoic acid regardless of the sense of the chirality of the acyl-transfer catalysts, (R)- and (S)-BTM, during this racemization test. A similar racemization occurred using only Piv2O with iPr2NEt, but at a lower rate than the reaction with (R)- or (S)-BTM. We also found that no racemization proceeded without using Piv2O even though iPr2NEt and BTM were present in the reaction mixture. These results showed that the occurrence of the racemization requires the existence of Piv2O with iPr2NEt and/or BTM, and it was revealed that almost all of the racemization process could be completed within 4 h (240 min) when Piv2O was used with both iPr2NEt and BTM. Fig. 4 shows the time course of the formation of MA and SA derived from 2-phenylpropanoic acid-d3 without adding 3 to the reaction mixture. In situ generation of ca. 50 mol% of MA and ca. 10 mol% of SA was observed after 1 h, and it was found that there is an equilibrium among these three species via the facile transacylation process.
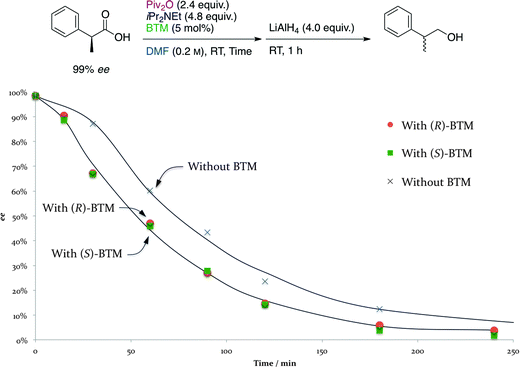 | ||
| Fig. 3 Time course of the racemization of (S)-2-phenylpropanoic acid (99% ee). | ||
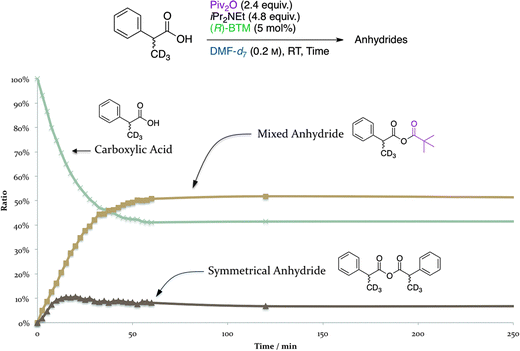 | ||
| Fig. 4 Time course of the formation of MA and SA derived from 2-phenylpropanoic acid-d3. | ||
Based on the above results, we concluded that the racemization of 2-phenylpropanoic acid proceeded faster than the chiral acyl-transfer reaction of MA with 3 involving the stereo-discrimination step. Initially, a small amount of (R)-2-phenylpropanoic acid in the racemic compound could have been consumed by the (R)-BTM-mediated kinetic resolution to afford the corresponding (R)-ester, and then the remaining (S)-2-phenylpropanoic acid could immediately racemize to supply the extra (R)-2-phenylpropanoic acid for further kinetic resolution in DMF. It is noteworthy that the present successful development of the DKR of a variety of the racemic 2-arylalkanoic acids was realized by the preferable assistance of the rapid racemization of 2-arylalkanoic acids under suitable conditions to form MA using Piv2O with iPr2NEt and BTM in a polar solvent.
We finally tried to develop an effective protocol for the synthesis of the enantiomerically pure (S)-ibuprofen ((S)-1) from racemic ibuprofen ((±)-1) using the DKR and KR combined systems as depicted in Scheme 4. The enantiomerically pure (S)-1 has been commercialized in Australia and Switzerland as dexibuprofen in 1994.15–17 The optical purity of the chiral product (S)-1 (92% ee), which was prepared in 95% yield (2 steps) from 2.06 g of (±)-1 using DKR in DMF followed by hydrogenation of the resulting ester, was further increased using not DKR but KR in toluene as a less polar solvent, and the optically pure (S)-2 (99% ee) was obtained in 90% yield from (S)-1 (92% ee). Iterative hydrogenation of the thus formed (S)-2 eventually provided the optically pure (S)-1 in 90% yield without any loss of chirality (99% ee). Through the present DKR–KR combined protocol, 1.58 g of the desired chiral (S)-ibuprofen ((S)-1) was successfully produced in a high yield (77%) with excellent purity (99% ee) from 2.06 g of racemic ibuprofen ((±)-1).
 | ||
| Scheme 4 A new synthetic method for the production of biologically active (S)-ibuprofen ((S)-1) (dexibuprofen). | ||
In conclusion, we have developed the first facile and novel dynamic kinetic resolution (DKR) of racemic 2-arylalkanoic acids with achiral alcohols using Piv2O and (R)-BTM. The choice of the solvent is very important for this reaction, and it was found that the racemization is drastically enhanced in a polar solvent with a high dipole moment (≥ ca. 3.7 D). The kinetics of the present method for the DKR of racemic 2-phenylpropanoic acid were systematically examined, and it was shown that the DKR consists of two efficient reactions: that is, (i) enantio-discriminating esterification of the racemic 2-phenylpropanoic acid with bis(α-naphthyl)methanol (3) in the presence of Piv2O and BTM, and (ii) rapid racemization of the chiral 2-phenylpropanoic acid via the formation of the mixed anhydride (MA) with Piv2O in the presence of iPr2NEt and BTM. This protocol was then successfully utilized for the effective DKR of racemic ibuprofen, flurbiprofen, ketoprofen, fenoprofen, and naproxen to yield a variety of optically active nonsteroidal anti-inflammatory drugs (NSAIDs), and their derivatives.
Acknowledgements
This study was partially supported by a Research Grant from Toray Science Foundation.Notes and references
- K. M. J. Brands and A. J. Davies, Chem. Rev., 2006, 106, 2711 CrossRef CAS and references therein.
- J.-P. Rieu, A. Boucherle, H. Cousse and G. Mouzin, Tetrahedron, 1986, 42, 4095 CrossRef CAS and references therein.
- H. R. Sonawane, N. S. Bellur, J. R. Ahuja and D. G. Kulkarni, Tetrahedron: Asymmetry, 1992, 3, 163 CrossRef CAS and references therein.
- A. J. Hutt and J. Caldwell, J. Pharm. Pharmacol., 1983, 35, 693 CrossRef CAS.
- J. Caldwell, A. J. Hutt and S. Fournel-Gigleux, Biochem. Pharmacol., 1988, 37, 105 CrossRef CAS.
- H. Hao, G. Wang, J. Sun, Z. Ding, X. Wu and M. Roberts, Biol. Pharm. Bull., 2005, 28, 682 CAS.
- K. Kacprzak and J. Gawronski, Synthesis, 2001, 961 CrossRef CAS and references therein.
- S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985 CrossRef CAS and references therein.
- I. Shiina, K. Nakata and Y. Onda, Eur. J. Org. Chem., 2008, 5887 CrossRef CAS.
- I. Shiina, K. Nakata, K. Ono, Y. Onda and M. Itagaki, J. Am. Chem. Soc., 2010, 132, 11629 CrossRef CAS.
- H. Pellissier, Tetrahedron, 2008, 64, 1563 CrossRef CAS and references therein.
- E. J. Ebbers, G. J. A. Ariaans, J. P. M. Houbiers, A. Bruggink and B. Zwanenburg, Tetrahedron, 1997, 53, 9417 CrossRef CAS and references therein.
- Only 3 functions as an effective nucleophile in this reaction because the stability of the transition state in the enantio-discriminating step is significantly affected by the structure of alcohol. See ref. 10.
- Experimental details are available as ESI†.
- D. Loew, O. Schuster and J. Lukas, Ibuprofen-containing medicament, European Patent 0 267 321 1988 Search PubMed; US Patent, 5,541,227, 1996 Search PubMed.
- A. Sunshine and E. M. Laska, A pharmaceutical composition containing S(+) ibuprofen substantially free of its R(–) antipode, PCT Patent Application International Publication Number WO 89/00421, 1989; US Patent, 4,851,444 1989 Search PubMed.
- G. Birrenbach and R.-D. Juch, Drugs containing S(+)-ibuprofen, European Patent, 0 607 467 1994; US Patent, 5,631,296 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20329d |
| This journal is © The Royal Society of Chemistry 2012 |