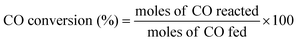Synthesis of methanol and dimethyl ether from syngas over Pd/ZnO/Al2O3 catalysts
Received
11th May 2012
, Accepted 16th June 2012
First published on 18th June 2012
Abstract
A Pd/ZnO/Al2O3 catalyst was developed for the synthesis of methanol and dimethyl ether (DME) from syngas with temperatures of operation ranging from 250 °C to 380 °C. High temperatures (e.g. 380 °C) are of interest when combining methanol and DME synthesis with a methanol to gasoline (MTG) process in a single reactor bed. A commercial Cu/ZnO/Al2O3 catalyst, utilized industrially for the synthesis of methanol at 220–280 °C, suffers from a rapid deactivation when the reaction is conducted at high temperature (> 320 °C). On the contrary, a Pd/ZnO/Al2O3 catalyst was found to be highly stable for methanol and DME synthesis at 375 °C. The Pd/ZnO/Al2O3 catalyst was thus further investigated for methanol and DME synthesis at P = 34–69 bar, T = 250–380 °C, GHSV = 5000–18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1, and molar feeds H2/CO = 1, 2, and 3. Selectivity to DME increased with decreasing operating temperature, and increasing operating pressure. Higher space velocity and H2/CO syngas feed ratios also enhanced DME selectivity. Undesirable CH4 formation was observed, however, it could be lessen through choice of process conditions and by catalyst design. By studying the effect of the Pd loading and the Pd:Zn molar ratio the formulation of the Pd/ZnO/Al2O3 catalyst was optimized. A catalyst with 5% Pd and a Pd:Zn molar ratio of 0.25:1 has been identified as the preferred catalyst. Results indicate that PdZn particles are more active than Pdo particles for the synthesis of methanol and less active for CH4 formation. A correlation between DME selectivity and concentration of acid sites has been established. Hence, two types of sites are required for the direct conversion of syngas to DME: (1) PdZn particles are active for the synthesis of methanol from syngas, and (2) acid sites which are active for the conversion of methanol to DME. Additionally, CO2 formation was problematic as PdZn was found to be active for the water-gas-shift (WGS) reaction, under all the conditions evaluated.
000 h−1, and molar feeds H2/CO = 1, 2, and 3. Selectivity to DME increased with decreasing operating temperature, and increasing operating pressure. Higher space velocity and H2/CO syngas feed ratios also enhanced DME selectivity. Undesirable CH4 formation was observed, however, it could be lessen through choice of process conditions and by catalyst design. By studying the effect of the Pd loading and the Pd:Zn molar ratio the formulation of the Pd/ZnO/Al2O3 catalyst was optimized. A catalyst with 5% Pd and a Pd:Zn molar ratio of 0.25:1 has been identified as the preferred catalyst. Results indicate that PdZn particles are more active than Pdo particles for the synthesis of methanol and less active for CH4 formation. A correlation between DME selectivity and concentration of acid sites has been established. Hence, two types of sites are required for the direct conversion of syngas to DME: (1) PdZn particles are active for the synthesis of methanol from syngas, and (2) acid sites which are active for the conversion of methanol to DME. Additionally, CO2 formation was problematic as PdZn was found to be active for the water-gas-shift (WGS) reaction, under all the conditions evaluated.
Introduction
Strained fossil fuel reserves and the increasing demand of emergent countries make the use of renewable energies attractive. Sun, wind and biomass are promising sources of renewable energies. Several technologies exist for converting biomass to energy such as heat, electricity or fuel. One approach involves biomass gasification to produce syngas. Syngas can be burned in gas turbines like natural gas, or it can be converted to high quality chemicals and fuels, such as methanol and dimethyl ether (DME). Methanol and DME are key intermediates in the methanol-to-gasoline (MTG) and methanol-to-olefins (MTO) processes.1,2
DME is often produced in a two-step process where methanol is first generated from syngas followed by methanol dehydration over a solid acid catalyst to produce DME. Alternatively, in a single-step conversion, DME is produced directly from syngas over a bifunctional or hybrid catalyst system employing both a methanol synthesis function and a methanol dehydration function.3,4 Producing DME directly from syngas has many economic and technical advantages, provided suitable catalyst(s) exist.5 In addition, thermodynamically, DME production from syngas is favored over methanol. Direct DME synthesis involves several competing reaction pathways. Methanol synthesis (eqn (1) and (2)) and the water-gas-shift (WGS, eqn (3)) are equilibrium reactions occurring over metal or mixed metal catalysts:
| | | CO + 2H2 ↔ CH3OH ΔH0 = −92.0 kJ mol−1 | (1) |
| | | CO2 + 3H2 ↔ CH3OH + H2O ΔH0 = −49.5 kJ mol−1 | (2) |
| | | CO + H2O ↔ CO2 + H2 (WGS) ΔH0 = −41.1 kJ mol−1 | (3) |
Dehydration of methanol to DME (
eqn (4)) is also equilibrium controlled and occurs over solid acid catalysts. By producing DME in the same step, methanol is continuously consumed in the reactor, pushing the conversion of syngas beyond that which would be achieved with methanol synthesis alone.
| | | 2CH3OH ↔ CH3OCH3 + H2O (dehydration) ΔH0 = −23.6 kJ mol−1 | (4) |
Methane forming reactions could occur from the hydrogenolysis of DME (
eqn (5)), hydrogenolysis of methanol (
eqn (6)) or from methanation reactions (
eqn (7) and (8)) over metal active sites. When operating at temperatures lower than approximately 280 °C, as is typically the case for direct DME synthesis, these reactions are not usually observed to any great extent.
5| | | CH3OCH3 + 2H2 → 2CH4 + H2O (hydrogenolysis) ΔH0 = −207.5 kJ mol−1 | (5) |
| | | CH3OH + H2 → CH4 + H2O (hydrogenolysis) ΔH0 = −115.4 kJ mol−1 | (6) |
| | | CO + 3H2 ↔ CH4 + H2O (CO methanation) ΔH0 = −206 kJ mol−1 | (7) |
| | | CO2 + 4H2 ↔ CH4 + 2H2O (CO2 methanation) ΔH0 = −165 kJ mol−1 | (8) |
Combined methanol/DME synthesis typically necessitates the use of a mixed catalyst system. Traditionally utilized are CuZnAl-type methanol synthesis catalysts and acidic zeolite or alumina to facilitate methanol dehydration.
6,7 The system operates at high pressure (5–10 MPa) and relatively low temperature (200–280 °C). The well known Topsoe Integrated Gasoline Synthesis (TIGAS) process has demonstrated successful integration of methanol and DME synthesis.
8 The TIGAS process consists of the methanol/DME production in one step and then in a second step methanol/DME is converted to gasoline using the zeolite-catalyzed MTG process.
8 Two separate synthesis reactors are used because the optimal temperatures are different for the two operations. Methanol synthesis favors temperatures around 250 °C, using conventional Cu methanol catalysts, whereas MTG proceeds at 350–400 °C.
2
Being a capital intensive process, combining methanol/DME synthesis and gasoline production into a one synthesis reactor operation would be economically attractive. Several groups have attempted this integration using conventional methanol synthesis and zeolite materials.9,10 However, several technical hurdles exist. For one, there is a mismatch in pressure and temperature when combining conventional methanol synthesis and MTG. The high pressure (5–10 MPa) operating conditions required to synthesize methanol/DME will affect the final product distribution of the gasoline. The conversion of DME to gasoline requires relatively high temperature (350–400 °C),2 operating conditions at which methanol/DME synthesis by itself is thermodynamically unfavorable. Furthermore, the conventional Cu/ZnO/Al2O3 catalyst used for methanol synthesis suffers from instability at temperatures greater than approximately 270 °C.11 Hence, a catalyst both active and stable for the synthesis of methanol at high temperature is lacking for the direct conversion of syngas to gasoline.
In recent years a Pd/ZnO-type catalyst has been developed for the methanol steam reforming reaction (MSR). Pd/ZnO has been shown to be quite active and especially selective to direct production of H2 and CO2.12,13 Beyond methanol steam reforming, the PdZn-type catalyst was also found to be very active for both water gas shift14 and also for methanol synthesis,15 the latter having operating as high as 350 °C. Pd/ZnO catalysts have been shown to be significantly more stable for methanol steam reforming at higher temperatures (e.g. >280 °C) as compared to the conventional CuZnAl-type methanol catalyst.16 Hence, the more stable PdZn-type catalyst is a potential candidate for use in the direct synthesis of gasoline from syngas.
The present study investigated a Pd/Zn/Al2O3 bifunctional catalyst for the direct conversion of syngas to methanol and DME over a wide temperature range, 250–380 °C. The higher temperatures incorporate a regime that is not optimal for the methanol synthesis reaction since this reaction is thermodynamically limited. However, in a single step gasoline synthesis process equilibrium constraint to hydrocarbon product is alleviated, as described in more detail elsewhere (e.g. integrating Pd/ZnO/Al2O3 with zeolite).17,18 The, objectives here are to (1) demonstrate that the Pd/ZnO/Al2O3 catalyst is more stable than the Cu/ZnO/Al2O3 catalyst for methanol synthesis at high temperature (375 °C), and (2) to study the effect of the Pd loading and Pd:Zn molar ratio on catalyst performance in order to optimize catalyst formulation and understand what parameters affect the selectivity. Operating temperature, pressure, gas hour space velocity (GHSV), and syngas ratio (H2/CO) were process variables explored. Catalytic compositions were altered by varying Pd and Zn loadings on the alumina substrate, affecting catalytic activity and selectivity. The relationship of the bifunctional PdZn metal and acid sites and their impact on catalytic performance were investigated. A combination of synthesis experimentation and material characterization has led to insights regarding the reaction pathways.
Experimental section
1. Catalyst preparation
Two series of Pd/ZnO/Al2O3 catalysts were prepared by incipient wetness impregnation of an Al2O3 support (Engelhard, AL-3945E) with a Pd nitrate solution (21.21 wt% Pd in nitric acid) to which a Zn nitrate precursor (Sigma Aldrich) was added, as reported in a previous study.19 The catalysts were dried at 110 °C for 8 h and calcined at 350 °C for 3 h. For the first series, the Pd loading was equal to 8.8 wt% and the Pd:Zn molar ratio varied from 0.25:1 to 0.75:1. For the second series, the Pd loading varied from 2.5 to 20 wt% and the Pd:Zn molar ratio was kept constant and equal to 0.25:1. A Pd/Al2O3 catalyst with 8.8 wt% Pd was prepared according to the same method with no addition of Zn nitrate precursor to the Pd solution. The Pd/ZnO/Al2O3 catalysts were labeled xPd/ZnO/Al2O3 − y were x stands for the Pd loading and y stands for the Pd/Zn molar ratio. For example, 8.8Pd/ZnO/Al2O3−0.25 indicates a Pd loading of 8.8 wt% and a Pd:Zn content of 0.25:1 (molar). For comparison purpose a commercial Cu/ZnO/Al2O3 catalyst (Synetix, F51-8 PPT) was tested under the same conditions as the supported Pd catalysts. Note that the term “spent” refers to the catalyst after reaction.
2. BET surface area
Nitrogen adsorption was measured at 77 K with an automatic adsorptiometer (Micromeritics ASAP 2000). The samples were pretreated at 150 °C for 12 h under vacuum. The surface areas were determined from adsorption values for five relative pressures (P/P0) ranging from 0.05 to 0.2 using the BET method. The pore volumes were determined from the total amount of N2 adsorbed between P/P0 = 0.05 and P/P0 = 0.98. Prior to BET measurements the catalysts have been reduced under 10% H2/N2 at 400 °C for 2 h.
3. X-ray Diffraction (XRD)
XRD analysis of the spent catalysts (i.e. after methanol synthesis reaction conditions) was conducted using a Philips X'pert MPD (Model PW3040/00) diffractometer with a copper anode (Kα1 = 0.15405 nm) and a scanning rate of 0.01° per second between 2θ = 10°–70°. The diffraction patterns were analyzed using Jade 5 (Materials Data Inc., Livermore, CA) and the Powder Diffraction File database (International Center for Diffraction Data, Newtown Square, PA). Particle sizes of the samples were determined from the XRD patterns using the Debye-Sherrer relation (d = 0.89λ/ Bcosθ, where λ is the wavelength of Cu-Kα radiation, B is the calibrated half-width of the peak in radians, and θ is the diffraction angle of a crystal face). The metal dispersion was estimated from the particle size by assuming hemispherical geometry using the equation D = 1/d (D = dispersion and d = metal particle size).20
4. Scanning transmission electron microscopy (S/TEM)
Scanning Transmission Electron Microscopy (S/TEM) was performed with FEI Titan 80–300 operated at 300 kV. The FEI Titan is equipped with CEOS GmbH double-hexapole aberration corrector for the probe-forming lens, which allows imaging with ∼0.1 nm resolution in scanning transmission electron microscopy (STEM) mode. The STEM images were acquired on High Angle Annular Dark Field (HAADF) with inner collection angle of 52 mrad. In general, the TEM sample preparation involved mounting of powder samples on copper grids covered with lacey carbon support films and immediate loading them into the TEM airlock to minimize an exposure to atmospheric O2. Note that the samples were analyzed by S/TEM after ex situ reduction under 10% H2/N2 at 400 °C for 2 h.
5. Infrared spectroscopy
IR spectra were recorded with a Bruker spectrometer, equipped with a MCT detector (resolution: 4 cm−1, 256 scans). The samples pressed into a pellet were first pretreated under H2 for 2 h at 400 °C. During this pretreatment, the sample was alternatively exposed to H2 for 30 min and evacuated under vacuum for 15 min to simulate flow conditions. After that, the temperature was cooled down to room temperature and small doses of CO were progressively added until saturation of the catalyst surface occurred.
6. NH3 temperature programmed desorption (TPD)
NH3-TPD experiments were performed on an automated catalyst characterization unit (Micromeritics Autochem 2910) equipped with a TCD detector. The catalyst (0.1 g) was loaded in a U-type quartz tube. Then a 10% H2/Ar mixture was passed through the sample starting from 20 °C and heating up to 400 °C with a ramp of 5 °C min−1 and held at this temperature for 2 h under 10% H2/Ar mixture and one more hour under He. The temperature was cooled down to 100 °C under He flow and the adsorption of NH3 (16% NH3/He) was carried out at 100 °C for 2 h. After that, He flowed for 2 h at the same temperature, to remove the physisorbed NH3 from the surface of the catalyst. The catalyst was then heated to 650 °C (ramp 5 °C min−1) and held at this temperature for 1 h.
7. Catalytic activity
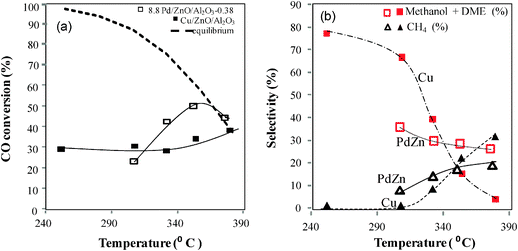
Catalytic activity tests were conducted in a 7.8 mm inner diameter fixed-bed reactor. The catalyst (0.6 g), diluted with SiC (3 g), was loaded between two layers of quartz wool inside the reactor. A dual K-type thermocouple was placed in the reactor for the measurement of inlet and catalyst bed temperatures. The catalyst was reduced at 400 °C for 2 h, using 10% H2/N2 gas mixture, prior to the test. A premixed gas containing H2, CO, CO2 and N2 was fed into the system using a Brooks Mass Flow Controller (5850E series). Four different premixed gas compositions with syngas ratios H2/CO = 1, 2 or 3 (mol) were used and are listed in Table 1. The catalysts were tested at temperatures between 250–380 °C over a range of gas hour space velocity (5000–20000 h−1) and pressures (34.5–69 bar). The gas products were separated using MS-5A and PPU columns and analyzed on-line by means of an Agilent Micro GC equipped with a TCD detector. The catalysts activities were compared using the following definitions:
where Pi is a certain product and υ is the number of carbon atoms/molecule in Pi. e.g. if P = CO2, υCO2 = 1, while for P = CH3OCH3, υCH3OCH3 = 2. Chemcad (version 5.6) was used to estimate the equilibrium CO conversion.
Table 1 Premix gas compositions
|
|
H2/CO |
H2 (%) |
CO (%) |
CO2(%) |
N2 (%) |
| Premix 1 |
2 |
59.73 |
32.18 |
4.15 |
3.94 |
| Premix 2 |
1 |
41.50 |
41.44 |
12.97 |
4.09 |
| Premix 3 |
2 |
58.03 |
28.98 |
8.89 |
4.10 |
| Premix 4 |
3 |
66.11 |
21.94 |
7.75 |
4.20 |
Results and discussion
1. Catalyst compositions and textural properties
Compositional information and the surface area and pore volume of the catalysts are shown in Table 2. For a given Pd loading, the surface area and pore volume increase with an increase of Pd:Zn ratio (i.e. decrease in the ZnO content). This could be due to a blocking of the pores of the Al2O3 support by ZnO and by the PdZn particles. The PdZn particle size decreases with the Pd:Zn ratio (see Table 3). Similarly, the decrease of the surface area and pore volume with the increase of the Pd loading from 2.5 to 20% is likely due to the increase of the PdZn particle size and ZnO content.
Table 2 Catalysts composition and textural properties
| Catalyst |
Pd loading (wt%) |
Pd:Zn (molar) |
Surface area (m2 g−1) |
Pore volume (cm3 g−1) |
| 8.8Pd/ZnO/Al2O3−0.25 |
8.8 |
0.25:1 |
171.8 |
0.37 |
| 8.8Pd/ZnO/Al2O3−0.38 |
8.8 |
0.38:1 |
183.4 |
0.43 |
| 8.8Pd/ZnO/Al2O3−0.75 |
8.8 |
0.75:1 |
197.2 |
0.46 |
| 8.8Pd/Al2O3 |
8.8 |
1:0 |
229.2 |
0.55 |
| 2.5Pd/ZnO/Al2O3−0.25 |
2.5 |
0.25:1 |
213.6 |
0.59 |
| 5Pd/ZnO/Al2O3−0.25 |
5.0 |
0.25:1 |
192.8 |
0.5 |
| 20Pd/ZnO/Al2O3−0.25 |
20 |
0.25:1 |
81.05 |
0.12 |
Table 3 Particles size of the Pd/ZnO/Al2O3 catalysts determined by XRD and dispersion measurements
| Catalyst |
PdZn Particle size (nm) |
Dispersiona (%) |
|
dispersion calculated from PdZn particle size using the equation D = 1/d with D = dispersion and d = PdZn particle size.
Peaks characteristic of bimetallic PdZn particles were not detected by XRD indicating that the particle size is below the XRD detection limit (i.e. < 4 nm).
|
| 8.8Pd/ZnO/Al2O3−0.25 |
13.8 |
7.2 |
| 8.8Pd/ZnO/Al2O3−0.38 |
8.7 |
11.5 |
| 8.8Pd/ZnO/Al2O3−0.75 |
7.3 |
13.7 |
| 2.5Pd/ZnO/Al2O3−0.25 |
<4b |
NA |
| 5Pd/ZnO/Al2O3−0.25 |
8.6 |
11.6 |
| 20Pd/ZnO/Al2O3−0.25 |
14.8 |
6.8 |
2. X-ray diffraction
For the 8.8Pd/ZnO/Al2O3−0.38 catalyst exposed to methanol synthesis reaction conditions, the results have shown that the PdZn particles size increases during the first 12 h on stream (from 4.0 nm to 7.5 nm) but does not significantly increase for TOS > 12 h. We have thus examined the spent catalysts by XRD to determine the PdZn particle size. Fig. 1(a) shows the XRD patterns for the spent Pd/ZnO/Al2O3 catalysts with different Pd loading and same Pd:Zn molar ratio, measured at 2θ = 40° to 48°. For the samples with a Pd loading > 2.5%, peaks characteristic of bimetallic PdZn at 41.2° and 44.1° are observed. When the Pd loading increases these peaks become more intense and their bandwidths decrease indicating an increase of the PdZn particle size (see Table 3). For the 2.5Pd/ZnO/Al2O3−0.25 catalyst, one broad peak is detected between 2θ = 40.5° and 42.5°. It is likely that this peak is characteristic of Al2O3. However, the presence of small PdZn particles that could contribute to this broad peak is not ruled out. None of the XRD patterns shows peaks characteristic of Pdo (expected at 40.2°), suggesting that the samples present only bimetallic PdZn particles. Fig. 1(b) displays the XRD patterns for the Pd/ZnO/Al2O3 catalysts with 8.8% Pd and different Pd:Zn molar ratios. The XRD pattern obtained for the 8.8Pd/Al2O3 catalyst is presented in Fig. 1(b) as well. The XRD patterns for the Pd/ZnO/Al2O3 catalysts show only peaks characteristic of PdZn, again suggesting the absence of metallic Pdo particles. Note that the bandwidth of the PdZn peak at 41.2° increases with the Pd:Zn ratio, indicating a decrease in the bimetallic PdZn particle size. The PdZn particle size and dispersion calculated from these XRD measurements are presented in Table 3. The dispersion increases with the increase in Pd:Zn molar ratio and decreases with an increase in the Pd loading.
 |
| | Fig. 1 XRD patterns for the spent Pd/ZnO/Al2O3 catalysts with different Pd loading and same Pd:Zn molar ratio (a) and for the catalysts with 8.8% Pd and different Pd:Zn molar ratio (b). | |
3. Scanning transmission electron microscopy (STEM)
The Pd/ZnO/Al2O3 catalysts were analyzed using STEM. Fig. 2a shows bimetallic PdZn particles supported by ZnO/γ-Al2O3 for the 2.5Pd/ZnO/Al2O3−0.25 catalyst. Detailed high-resolution imaging in Fig. 2b confirms that the particles are PdZn bimetallic and have an ordered tetragonal structure with L10 type ordering. Nevertheless, some of the PdZn particles, such as the one shown in Fig. 2b, exhibit a contrast variation at the nanoscale indicating some compositional or structural inhomogeneities. The observations of bimetallic PdZn particles with tetragonal L10 type ordering are common for all of the analyzed Pd/ZnO/Al2O3 catalysts.
 |
| | Fig. 2 (a) General view of the supported PdZn particles for the spent 2.5% Pd/ZnO/Al2O3 0.25:1 catalyst. (b) High resolution HAADF image revealing the crystallographic nature of the PdZn intermetallic particles. | |
4. Infrared spectroscopy
Fig. 3(a) shows the infrared spectra recorded between 1800–2125 cm−1, after saturation of the catalyst surface with CO at room temperature, for the Pd/ZnO/Al2O3−0.25 catalysts with different Pd loadings. For all the catalysts, the IR spectra present one main band between 2069–2077 cm−1 ascribed to the vibration of CO linearly adsorbed on the PdZn alloy particles.21,22 The shift observed between the spectra for the different catalysts for the band at 2069–2077 cm−1 is not understood yet. In addition, a closer look at the spectrum obtained for the 2.5Pd/ZnO/Al2O3−0.25 catalyst shows one broad band between 1800–2000 cm−1 due to multi-bonded CO species21 and characteristic of Pd° particles.22 Note that the band at 1800–2000 cm−1 is hardly detectable for the 2.5Pd/ZnO/Al2O3−0.25 catalyst, suggesting that the amount of Pd° is low compared to the amount of bimetallic PdZn particles. Note that the presence of a band due to linearly CO species adsorbed on Pd° for the catalysts with Pd > 2.5% can be ruled out since for Pd°/ZnO/Al2O3 catalyst band due to linearly adsorbed CO between 2100–2000 cm−1 is accompanied by a more intense band between 2000–1800 cm−1.22
 |
| | Fig. 3 Infrared spectra recorded after CO adsorption at room temperature, after saturation of the surface by CO for the Pd/ZnO/Al2O3 catalysts with different Pd loading and Pd:Zn molar ratio. | |
Fig. 3(b) shows the infrared spectra recorded between 1800–2125 cm−1, for the 8.8Pd/ZnO/Al2O3 catalysts with different Pd:Zn molar ratios. All the spectra present one band at 2069–2077 cm−1 characteristic of PdZn alloy. The spectrum recorded for the 8.8Pd/ZnO/Al2O3−0.75 catalyst also shows one more band between 1800–2000 cm−1 which is attributed to Pd°. These results suggest that the amount of Pd° increases with the Pd:Zn molar ratio. Note that contrary to the IR measurements, the XRD patterns did not indicate the presence of Pd° for the 8.8Pd/ZnO/Al2O3−0.75. It can be due to the fact that the Pd° particles are too small to be detected by XRD or to the fact that IR spectroscopy is sensitive to the surface composition of the catalyst, whereas XRD technique informs on the structure/bulk composition of the catalyst.
5. Catalytic activity
5.1 Thermodynamics of methanol synthesis and dehydration reactions.
Fig. 4(a) presents the equilibrium CO conversion for the synthesis of methanol at T = 225–400 °C and P = 34.5–69 bar and H2/CO/CO2 of 2/1/0.13 (premix 1 in Table 1). For these calculations two cases are considered: (1) methanol was the only product, and (2) both methanol and DME as products. For the first case, equilibrium CO conversion decreases with increasing temperature from 75% at 225 °C, to 0% at 400 °C. For the second case, with both methanol and DME as products, the same trend of decreasing conversion with increasing temperature is seen, but overall conversions are higher. This demonstrates the benefit in thermodynamic driving force when both methanol synthesis and methanol dehydration are employed in tandem. Fig. 4(a) also shows how equilibrium CO conversion increases with pressure. For example, at 375 °C, CO conversion increases from 10% at P = 34.5 bar, to 38% at P = 69 bar.
 |
| | Fig. 4 (a) Evolution of the equilibrium CO conversion as a function of the temperature for methanol synthesis considering the formation of CH3OH and DME as products for P = 69 bar, P = 51.8 bar, P = 34.5 bar, and considering only the formation of CH3OH for P = 69 bar, H2/CO/CO2 = 2/1/0.13 (premix 1, Table 1). (b) Evolution of the CO conversion, CO2 selectivity, DME selectivity and CH3OH selectivity at equilibrium as a function of the temperature for P = 69 bar and H2/CO = 2, considering the formation of CH3OH and DME as products. | |
Fig. 4(b) shows the equilibrium CO conversion and selectivities to methanol, DME, and CO2 at T = 225–400 °C and P = 69 bar, when considering both methanol and DME as products. Equilibrium selectivity to DME decreases from 64% at 225 °C, to 40% at 400 °C. On the other hand, equilibrium selectivity to CO2 increases from 31% at 225 °C, to 54% at 400 °C. Equilibrium methanol selectivity remains quite level across the entire temperature range at approximately 5%.
5.2 Comparison of catalytic activity for commercial Cu/ZnO/Al2O3 and 8.8Pd/ZnO/Al2O3 catalysts for methanol and DME synthesis from syngas.
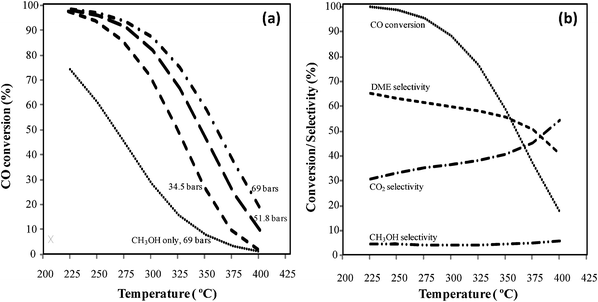
Industrially, the Cu/ZnO/Al2O3 catalyst is used for the synthesis of methanol from syngas.11 We have compared a commercial Cu/ZnO/Al2O3 catalyst to the 8.8Pd/ZnO/Al2O3−0.38 catalyst for the synthesis of methanol, focusing on catalyst stability at relatively high pressure (i.e. 69 bar) and temperature (i.e. 375 °C). These reaction conditions are suitable for the direct conversion of syngas17 to gasoline and the temperature is significantly higher than that employed in conventional methanol synthesis. Fig. 5 presents CO conversion versus time for a period of 125 h on stream for the two catalysts. Note that the stability test was conducted at higher GHSV (i.e. 8340 h−1) for the 8.8Pd/ZnO/Al2O3−0.38 catalyst to ensure the CO conversion to be below the equilibrium CO conversion. It is clear that the Cu/ZnO/Al2O3 suffers from rapid deactivation under these conditions. This deactivation is not surprising and is due to sintering of the Cu particles.23 This is in stark contrast with the trend observed for the 8.8Pd/ZnO/Al2O3−0.38 catalyst. CO conversion is quite stable with time-on-stream for the supported PdZn catalyst. Catalytic activity as a function of temperature is shown in Fig. 6(a) for both catalysts. Note that for each catalyst, the temperature was increased progressively from 250 °C to 380 °C. The activity was measured after a 12 h plateau at 250 °C and a 3–4 h plateau at 310 °C, 330 °C, 355 °C and 380 °C. For the 8.8Pd/ZnO/Al2O3−0.38 catalyst, CO conversion increases with temperature until 360 °C, beyond which it decreases due to equilibrium constraints. At temperature ≥330 °C the CO conversion is greater for the 8.8Pd/ZnO/Al2O3−0.38 catalyst than for Cu/ZnO/Al2O3. For both catalysts, CO2, C2H6, CH4, methanol and DME were produced during reaction. For Cu/ZnO/Al2O3, the CO conversion is relatively flat, as compared to the 8.8Pd/ZnO/Al2O3−0.38 catalyst. This is due to a progressive deactivation occurring during data collection. The activity was measured over a 24 h period during which the catalyst deactivates as evidenced from Fig. 5.
 |
| | Fig. 5 Evolution of the CO conversion with time on stream for the Cu/ZnO/Al2O3 and the 8.8P/ZnO/Al2O3−0.38 catalysts. Reaction T = 375 °C, P = 69 bar, H2/CO = 2 (premix 3), GHSV = 3500 h−1 for Cu/ZnO/Al2O3 and GHSV = 8340 h−1 for 8.8Pd/ZnO/Al2O3−0.38. | |
 |
| | Fig. 6 Evolution of (a) the CO conversion and (b) the methanol, DME and CH4 selectivity as a function of the temperature for the Cu/ZnO/Al2O3 (Cu) and the 8.8Pd/ZnO/Al2O3−0.38 catalysts. opened symbols: 8.8Pd/ZnO/Al2O3−0.38 (PdZn), filled symbols: Cu/ZnO/Al2O3. P = 69 bar, GHSV = 10000 h−1 and H2/CO = 2 (premix 1). | |
Fig. 6(b) shows the evolution of the methanol, DME, and CH4 selectivities as a function of temperature. The remaining selectivity (not shown) is for CO2. Note that for simplification and because both are the desired products, methanol and DME are lumped together and shown in the graph as “methanol + DME”. The compositional breakdown between methanol and DME is described below and is also shown in Table 4. As shown in Fig. 6(b) selectivity to both methanol and DME decreases dramatically with increasing temperature. For the Cu/ZnO/Al2O3 catalyst, methanol and DME selectivity decreases from 75% at 240 °C, to 4.3% at 380 °C. For the 8.8Pd/ZnO/Al2O3−0.38 catalyst, the selectivity to methanol and DME decreases from 36% at 240 °C, to 26% at 380 °C, but is significantly higher than for the Cu/ZnO/Al2O3 catalyst at 380 °C. At 380 °C, the selectivity to methanol is 1.8% and 4.5% for the Cu/ZnO/Al2O3 and 8.8Pd/ZnO/Al2O3−0.38 catalysts, respectively, whereas the DME selectivity is 2.5 and 21.5%, respectively. This represents a seven-fold selectivity advantage for the 8.8Pd/ZnO/Al2O3−0.38 catalyst at 380 °C. For both catalysts, selectivity to undesirable CH4 increases with temperature and reaches 32% and 21.4% at 380 °C for the Cu/ZnO/Al2O3 and 8.8Pd/ZnO/Al2O3−0.38 catalysts, respectively. CH4 production is an undesired byproduct and could also lead to the formation of coke. At temperatures above 350 °C, the Cu/ZnO/Al2O3 catalyst produces a substantially greater amount of CH4 and lesser amount of methanol and DME compared to the 8.8Pd/ZnO/Al2O3−0.38 catalyst.
Table 4 Effect of the temperature, pressure and GHSV, on the conversion and selectivity for the 8.8Pd/ZnO/Al2O3−0.38 catalyst
| Temperature (°) |
Pressure (bar) |
GHSV (h−1) |
CO conversion (%) |
Selectivity (%) |
| CO2 |
CH4 |
C2H6 |
Methanol |
DME |
| 307 |
69 |
10000 |
22.9 |
54.2 |
7.6 |
2.3 |
8.8 |
27.1 |
| 332 |
69 |
10000 |
41.9 |
53.5 |
12.9 |
3.9 |
8.4 |
21.1 |
| 352 |
69 |
10000 |
49.7 |
51.6 |
15.8 |
4.4 |
6.0 |
22.0 |
| 380 |
69 |
10000 |
44.2 |
52 |
17.5 |
4.5 |
4.5 |
21.4 |
| 380 |
34.5 |
10000 |
20.6 |
66.1 |
16.4 |
2.2 |
3.3 |
12 |
| 380 |
51.8 |
10000 |
39 |
57.4 |
19.6 |
4.2 |
4.1 |
14.7 |
| 380 |
69 |
5000 |
63.7 |
57.7 |
28.4 |
8.0 |
2.1 |
3.8 |
| 380 |
69 |
18000 |
40.3 |
52.8 |
15.1 |
3.2 |
5.2 |
23.7 |
These results clearly show the 8.8Pd/ZnO/Al2O3−0.38 catalyst to be preferred over the Cu/ZnO/Al2O3 catalyst for methanol and DME synthesis from syngas at temperatures above 350 °C. In contrast to the Cu/ZnO/Al2O3 catalyst, the 8.8Pd/ZnO/Al2O3−0.38 catalyst does not suffer from deactivation at these relatively high temperatures. In addition, higher selectivity to desirable DME and methanol, and lower selectivity to undesired CH4 is observed for the 8.8Pd/ZnO/Al2O3−0.38 catalyst. Optimizing the formulation of the Pd/ZnO/Al2O3 catalyst in order to potentially suppress the production of CH4 while facilitating methanol and DME formation appears thus very relevant.
5.3 Comparison of catalytic activity for the commercial Cu/ZnO/Al2O3 catalysts, 8.8Pd/ZnO/Al2O3 catalyst and Al2O3 support for methanol dehydration to DME.
As discussed above for both the Cu/ZnO/Al2O3 and 8.8Pd/ZnO/Al2O3−0.38 catalysts, formation of DME was observed. It is well known that dehydration of methanol proceeds over acidic sites offered by solid acid catalysts such as alumina and zeolites.24 Thus, the acid sites of the Al2O3 support likely promote DME formation via methanol dehydration once methanol is formed from the syngas. To investigate this further, we compared catalytic activity for the methanol-to-DME dehydration reaction (eqn (4)) at 250–425 °C and at 1 bar. Activities of Cu/ZnO/Al2O3, 8.8Pd/ZnO/Al2O3−0.38, and Al2O3 alone (the support used for the PdZn catalysts) were compared. Fig. 7 presents methanol conversion versus temperature for the three catalysts. Conversion increases dramatically with increasing temperature for both Cu/ZnO/Al2O3 and 8.8Pd/ZnO/Al2O3−0.38 and complete methanol conversion was achieved at approximately 350 °C for both. For the Al2O3 support, conversion increases just slightly with temperature, from 29% to 35%. The significant increase of the conversion with the temperature for the Cu/ZnO/Al2O3 and 8.8Pd/ZnO/Al2O3−0.38 catalysts, compared to the Al2O3 alone, is due to a considerable increase of their activity for methanol decomposition. Fig. 8(a) and (b) present selectivities for the different products (DME, CH4, CO and CO2) as a function of the temperature. For the Al2O3 support, as expected, DME is the only product observed up to 400 °C. At 425 °C, CH4 is produced in addition to DME. Also at this temperature a small amount of H2 and CO2 was detected. It is thus possible that CH4 was produced, at least in part, from DME hydrogenolysis or CO2 methanation. Note that H2 and CO2 were likely produced from methanol steam reforming rather than methanol decomposition since no CO was detected. This is in contrast with the results obtained for the Cu/ZnO/Al2O3 catalyst where CH4 is produced along with CO and CO2, with little DME formation. It appears that methanol is primarily decomposed to CO and H2 (the reverse of eqn (1)). The small amount of CO2 formed is likely due to the water-gas-shift reaction (eqn (3)). The CH4 is produced from CO (and/or CO2) methanation, DME decomposition or DME hydrogenolysis. Interestingly, for the Pd/ZnO/Al2O3−0.38 catalyst, formation of CH4 is not observed over the entire range investigated. However, DME formation is observed, with an optimum temperature at approximately 310 °C (20% selectivity) and decreases with increasing temperature. Like the Cu/ZnO/Al2O3 catalyst, the majority product formed over the entire temperature range is CO, as a result of methanol decomposition.
 |
| | Fig. 7 Methanol-to-DME reaction. Evolution of the CH3OH conversion with the reaction temperature for P = 1 bar, GHSV = 5000 h−1 and. CH3OH = 36.1% in N2. | |
 |
| | Fig. 8 Methanol-to-DME reaction. Evolution of the DME selectivity (a), CH4 selectivity (a), CO selectivity (b) and CO2 selectivity (b) as a function of the temperature for the dehydration of methanol. P = 1 bar, GHSV = 5000 h−1, CH3OH = 36.1% in N2. Filled symbols represent DME (a) and CO (b) selectivities. Opened symbols represent CH4 (a) and CO2 (b) selectivities. | |
These results indicate that the Pd/ZnO/Al2O3−0.38 catalyst is more active for the dehydration of methanol to DME as compared to commercial Cu/ZnO/Al2O3. This explains why when syngas feed is used (see Fig. 6b), the selectivity to DME is higher for the Pd/ZnO/Al2O3−0.38 catalyst than the Cu/ZnO/Al2O3 catalyst. As seen in Fig. 8, the selectivity toward DME is lower than the selectivity toward CO and CO2 whatever the temperature (between 250–410 °C), for the Pd/ZnO/Al2O3−0.38 catalyst. This is due to the fact that the methanol dehydration reaction experiments were conducted at atmospheric pressure. At high pressure (i.e. 69 bar), equilibrium selectivity to methanol from syngas is favored via methanol synthesis (eqn (2)). Thus, with increased methanol synthesis, as opposed to methanol reforming, increased DME production will result.
5.4 Effect of the temperature, pressure and gas hour space velocity (GHSV) on the syngas conversion and selectivity for the 8.8Pd/ZnO/Al2O3−0.38 catalyst.
The effects of the temperature, pressure, and GHSV on the reactivity for syngas conversion to methanol and DME were examined for the 8.8Pd/ZnO/Al2O3−0.38 catalyst and the results are presented in Table 4. The effect of temperature on conversion and selectivity has already been discussed above. As expected, CO conversion increases with pressure. Keeping the operating temperature constant at 380 °C, CO conversion increases from 20.6% to 44.2% when increasing the pressure from 34.5 bar to 69.0 bar. Selectivity to methanol and DME does increase somewhat with pressure. Methanol selectivity increases from 3.3% to 4.5% and DME increases from 12.0% to 21.4%. An increase in methanol production is indeed predicted as the forward methanol synthesis reaction rates (eqn (1) and (2)) are favored with increasing pressure. CH4 and C2H6 production did not increase substantially with pressure. When the pressure increased from 34.5 bar to 69 bar the CH4 and C2H6 selectivities increased only slightly from 16.4% to 17.5% and from 2.2% to 4.5%, respectively.
GHSV was varied to determine the time dependence of conversion and selectivity, with the results shown in Table 4. As expected, CO conversion decreases with increasing GHSV. GHSV's of 5000, 10000, and 18000 h−1 resulted in CO conversions of 63.7%, 44.2%, and 40.3%, respectively. Selectivities to CO2, CH4 and C2H6 increase with decreasing GHSV, whereas selectivity toward DME and methanol decrease. For example, at GHSVs of 5000 and 18000 h−1 selectivity to DME was 3.8% and 23.7%, respectively. Hence, shorter residence time favor the formation of DME relative to side products. Methane formation reactions such as DME hydrogenolysis (eqn (5)) become increasingly dominant at longer residence time. The fact that increased throughputs enhance selectivity to methanol and DME is an important finding from a practical standpoint.
One can see from Table 4 that a considerable amount of CO2 is produced during reaction. Indeed, whatever the reaction temperature (300–380 °C), pressure (34–69 bar) and GHSV (5000–18000 h−1), the selectivity to CO2 is always approximately 50%. Under the present reaction conditions, the water-gas-shift activity is significant, limiting methanol and DME formation.
5.5 Effect of the Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :Zn molar ratio on the reactivity of the Al2O3 supported catalysts with 8.8% Pd.
Changes in Pd/Zn composition and the resulting effect on catalytic activity were also examined. Fig. 9(a) presents the evolution of the conversion as a function of the Pd:Zn molar ratio at 380 °C and 69 bar. CO conversion increases from 36% to 44% when the Pd:Zn molar ratio increases from 0.25:1 to 0.38:1. CO conversion decreases for higher Pd:Zn molar ratios and is equal to 26% for the 8.8Pd/Al2O3 catalyst with a Pd:Zn = 1:0. As evident from Fig. 9(a) there exists an optimum in Pd:Zn ratio for CO conversion. The higher CO conversion observed for the 8.8Pd/ZnO/Al2O3−0.38 catalyst, compared to the 8.8Pd/ZnO/Al2O3−0.25 catalyst, is probably due to a higher PdZn dispersion (see Table 3). From Fig. 9(a), it can be seen that for Pd:Zn > 0.25:1, the CO conversion decreases with an increase in Pd:Zn ratio. The IR measurements have shown an increase of the amount of Pdo with the increase of the Pd:Zn ratio. Consequently, these results strongly suggest Pdo particles to be less active than PdZn particles for the synthesis of methanol.
:Zn molar ratio on the reactivity of the Al2O3 supported catalysts with 8.8% Pd.
Changes in Pd/Zn composition and the resulting effect on catalytic activity were also examined. Fig. 9(a) presents the evolution of the conversion as a function of the Pd:Zn molar ratio at 380 °C and 69 bar. CO conversion increases from 36% to 44% when the Pd:Zn molar ratio increases from 0.25:1 to 0.38:1. CO conversion decreases for higher Pd:Zn molar ratios and is equal to 26% for the 8.8Pd/Al2O3 catalyst with a Pd:Zn = 1:0. As evident from Fig. 9(a) there exists an optimum in Pd:Zn ratio for CO conversion. The higher CO conversion observed for the 8.8Pd/ZnO/Al2O3−0.38 catalyst, compared to the 8.8Pd/ZnO/Al2O3−0.25 catalyst, is probably due to a higher PdZn dispersion (see Table 3). From Fig. 9(a), it can be seen that for Pd:Zn > 0.25:1, the CO conversion decreases with an increase in Pd:Zn ratio. The IR measurements have shown an increase of the amount of Pdo with the increase of the Pd:Zn ratio. Consequently, these results strongly suggest Pdo particles to be less active than PdZn particles for the synthesis of methanol.
 |
| | Fig. 9 Evolution of (a) the conversion and (b) the selectivity with the Pd:Zn molar ratio for the supported 8.8% Pd catalysts. Reaction T = 380 °C, P = 69 bar, H2/CO = 2 (premix 1) and GHSV = 10000 h−1. | |
The effect of Pd:Zn molar ratio on product selectivity is displayed in Fig. 9(b). The CO2 selectivity is similar for all Pd:Zn molar ratios investigated and is equal to ∼54%. One can also see that the CH4 and DME selectivities follow opposite trends. Indeed, the DME selectivity decreases from 24.5% to 10.5% with increasing Pd:Zn molar ratio. Consistent with this, a significant increase of the CH4 selectivity, from 13 to 33%, is observed with increasing Pd:Zn molar ratio. As the Pd:Zn ratio increases from Pd:Zn = 0.38 to 1.0 more Pdo sites are present on the surface of the catalyst, as shown by the IR results described above. Hence, these results show Pdo to facilitate CH4 formation.
Methanol dehydration to DME is catalyzed by acid catalysts. Al2O3 alone is active for the formation of DME from methanol.25 However, Pd/ZnO is inactive for the dehydration of methanol to DME.26,27 It is thus reasonable to assume that for the Pd/ZnO/Al2O3 catalysts, the alumina support is the source of acidity. NH3-TPD experiments were conducted to determine the concentration of the acid sites for the 8.8Pd/ZnO/Al2O3 catalysts and the 8.8Pd/Al2O3 catalyst. The NH3-TPD profiles (not shown) have indicated the presence of one single peak located at 180 °C for all the Pd/ZnO/Al2O3 catalysts. Fig. 11(a) shows the evolution of the amount of NH3 desorbed as a function of the Pd:Zn ratio (for the catalyst with 8.8% Pd loading). The amount of NH3 desorbed decreases with increasing Pd:Zn ratio and is the lowest for the 8.8Pd/Al2O3 sample (with Pd:Zn = 1:0). This signifies that the concentration of acid sites decreases with increasing Pd:Zn ratio. Since the dispersion increases with the Pd:Zn ratio (see Table 3), the coverage of the Al2O3 support increases and the number of accessible acid sites decreases. Note that there is a correlation between the DME selectivity and the amount of acid sites. It indicates that for the 8.8Pd/ZnO/Al2O3 catalysts, the acid sites of the Al2O3 support are active for the dehydration of methanol to DME.
5.7 Effect of the feed syngas ratio on the catalytic activity of the 5Pd/ZnO/Al2O3−0.25 catalyst.
The effect of feed syngas ratio on the catalytic performance of this composition is examined on 5Pd/ZnO/Al2O3−0.25, identified above as the most selective catalyst under these conditions. H2/CO molar ratios of 1.0, 2.0, and 3.0 were evaluated and the results are shown in Fig. 12. CO conversion increases whereas the CO2 selectivity decreases with the increase of the H2/CO ratio [Fig. 12(a)]. DME selectivity increases from approximately 11% to 32% when increasing the H2/CO ratio from 1.0 to 3.0. The methanol selectivity is approximately 4% at H2/CO = 1 then approaches zero at higher H2/CO ratios. Interestingly, CH4 production also decreases with feed syngas ratio, from approximately 6% to 2% for H2/CO = 1.0 and H2/CO = 3.0, respectively. Methanol synthesis and subsequent methanol dehydration is thus favored over CO methanation or DME hydrogenolysis under these higher hydrogen-containing feeds.
 |
| | Fig. 11 Evolution of the DME selectivity and the amount of NH3 desorbed from the catalysts surface as a function of (a) the Pd:Zn molar ratio and (b) the Pd loading. Reaction T = 380 °C, P = 69 bar, GHSV = 10000 h−1 and H2/CO = 2 (premix 1). | |
 |
| | Fig. 12 Effect of the feed gas composition on (a) the conversion and (b) on the selectivity for the 5Pd/ZnO/Al2O3−0.25 catalyst. For H2/CO = 1 (premix 2), for H2/CO = 2 (premix 3) and for H2/CO = 3 (premix 4). Reaction T = 380 °C, P = 69 bar and GHSV = 10000 h−1. | |
Conclusion
In this work we have demonstrated the use of a Pd/ZnO/Al2O3 catalyst for the high-temperature production of methanol and DME from syngas and contrasted its activity with that of a commercial Cu/ZnO/Al2O3 catalyst. The PdZn-formulation outperforms the Cu-based formulation at these conditions which are directly relevant to the potential single-step syngas-to-gasoline concept, which combines methanol/DME synthesis with MTG in a single reactor. By studying the influence of the Pd loading and Pd:Zn molar ratio, a catalyst with 5% Pd and a Pd:Zn molar ratio of 0.25:1 has been identified as the best performing catalyst. Since Pdo promotes the formation of methane over methanol it is critical that catalyst synthesis avoid generation of Pdo. A direct relationship between DME selectivity and concentration of acid sites was shown. Hence, two types of sites are required for the direct conversion of syngas to DME: (1) PdZn particles which are active sites for the synthesis of methanol from syngas, and (2) acid sites which are active for the conversion of methanol to DME. The results of this study have shown that a non negligible amount of undesired CH4 is produced during reaction at high temperature of operation. Also, under the conditions tested, the PdZn particles were quite active for the water-gas-shift reaction, leading to the formation of a large amount of CO2 at the expense of DME formation. To consider Pd/ZnO/Al2O3 as a catalyst for direct conversion of syngas to gasoline it will be necessary to further investigate the parameters that could favor the methanol synthesis reaction and further lower methane formation.
Acknowledgements
The authors gratefully acknowledge financial support for this work provided by the Energy Conversion Initiative, funded internally by the Pacific Northwest National Laboratory (PNNL). This work was also supported by the National Advanced Biofuels Consortium (NABC) which is funded by the Department of Energy's Office of Biomass Program with recovery act funds. PNNL funding was provided under contract DE-AC05-76RL01830. Finally, the authors would like to acknowledge that a portion of this work was done in the Environmental Molecular Sciences Laboratory (EMSL), a DOE sponsored user facility located at PNNL in Richland, WA.
References
- F. Keil, Microporous Mesoporous Mater., 1999, 29, 49–66 CrossRef CAS.
- T. Mokrani and M. Scurell, Catal. Rev. Sci. Eng., 2009, 51, 1–145 CAS.
- J. Hu, K. Brooks, J. Holladay, D. Howe and T. Simon, Catal. Today, 2007, 125, 103–110 CrossRef CAS.
- S. H. Ahn, S. H. Kim, K. B. Jung and H. S. Hahm, Korean J. Chem. Eng., 2008, 25, 466–470 CrossRef CAS.
- C. Arcoumanis, C. Bae, R. Crookes and E. Kinoshita, Fuel, 2008, 87, 1014–1030 CrossRef CAS.
- S. Jiang, J. S. Hwang, T. Jin, C. Tianxi, W. Cho, Y. S. Baek and S. E. Parkd, Bull. Korean Chem. Soc., 2004, 25, 185–189 CrossRef CAS.
- M. Mollavali, F. Yaripou, H. Atashi and S. Sahebdelfar, Ind. Eng. Chem. Res., 2008, 47, 3265–3273 CrossRef CAS.
-
J. Topp-Jorgensen, in Methane Conversion, ed. D. M. Bibby, C. D. Chang, R. F. Howe and S. Yurchak, Elsevier Science Publishers, 1988, p. 293 Search PubMed.
- K. Fujimoto, H. Saima and H. Tominaga, J. Catal., 1985, 94, 16–23 CrossRef CAS.
- J. Erena, J. M. Arandes, J. Bilbao, M. Olazar and H. I. de Lasa, J. Chem. Technol. Biotechnol., 1998, 72, 190–196 CrossRef CAS.
-
C. Satterfield, in Heterogeneous Catalysis in Industrial Practice, ed. C. Satterfield, Krieger Publishing Company, Malabar, Florida, 2nd edn, 1996, p. 446 Search PubMed.
- Y.-H. Chin, R. Dagle, J. Hu, A. C. Dohnalkova and Y. Wang, Catal. Today, 2002, 77, 79–88 CrossRef CAS.
- N. Iwasa, S. Masuda, N. Ogawa and N. Takezawa, Appl. Catal., A, 1995, 125, 145 CrossRef CAS.
- R. Dagle, A. Platon, D. Palo, A. Datye, J. Vohs and Y. Wang, Appl. Catal., A, 2008, 342, 63–68 CrossRef CAS.
-
J. Hu, R. Dagle, J. Holladay, C. CaO, Y. Wang, J. White, E. Douglas and D. Stevens, U.S. Patent, 7 858 667, 2010 Search PubMed.
- D. Palo, R. Dagle and J. Holladay, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS.
- R. A. Dagle, J. A. Lizarazo, L. V. M., M. J. Gray, J. F. White, D. L. King and D. R. Palo, Submitted to Applied Catalysis A: General, 2012 Search PubMed.
- Y. Zhu, S. B. Jones, M. J. Biddy, R. A. Dagle and D. R. Palo, Bioresour. Technol., 2012, 117, 341–351 CrossRef CAS.
- G. Xia, J. D. Holladay, R. A. Dagle, E. O. Jones and Y. Wang, Chem. Eng. Technol., 2005, 28, 515 CrossRef CAS.
-
M. Boudart and G. Djega-Mariadassou, The Kinetics of Heterogeneous Catalytic Reactions, Princeton University Press, Princeton, NJ, 1984 Search PubMed.
-
N. Sheppard and T. T. Nguyen, in Advances in Infrared and Raman spectroscopy, ed. R. E. Hester and R. J. H. Clarke, Heyden and Son, London, 1978, vol. 5, p. 67 Search PubMed.
- V. Lebarbier, R. A. Dagle, T. Conant, J. M. Vohs, A. Datye and Y. Wang, Catal. Lett., 2008, 122, 223–227 CrossRef CAS.
- J. T. Sun, I. S. Metcalfe and M. Sahibzada, Ind. Eng. Chem. Res., 1999, 38(10), 3868–3872 CrossRef CAS.
- F. S. Ramos, A. M. D. de Farias, L. E. P. Borges, J. L. Monteiro, M. A. Fraga, E. F. Sousa-Aguiar and L. G. Appel, Catal. Today, 2005, 101, 39–44 CrossRef CAS.
- F. Yaripour, F. Baghaei, I. Schmidt and J. Perregaard, Catal. Commun., 2005, 6, 147–152 CrossRef CAS.
- N. Iwasa, O. Yamamoto, T. Akazawa, S. Ohyama and N. Takezawa, J. Chem. Soc., Chem. Commun., 1991, 18, 1322–1323 RSC.
- Y. A. Ryndin, R. F. Hicks, A. T. Bell and Y. I. Yermakov, J. Catal., 1981, 70, 287–297 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.