Acid hydrolysis of O-acetyl-galactoglucomannan
Bright T.
Kusema
,
Tea
Tönnov
,
Päivi
Mäki-Arvela
,
Tapio
Salmi
,
Stefan
Willför
,
Bjarne
Holmbom
and
Dmitry Yu.
Murzin
*
Process Chemistry Centre, Department of Chemical Engineering, Åbo Akademi University, FI-20500 Turku/Åbo, Finland. E-mail: dmurzin@abo.fi
First published on 20th July 2012
Abstract
Acid hydrolysis of O-acetyl-galactoglucomannan (GGM), the main non-cellulosic polysaccharide in coniferous wood, was studied at 90 °C in the pH range of 0.5–2.0 using different acids (hydrochloric acid, sulfuric acid, trifluoroacetic acid, oxalic acid, and formic acid) as homogeneous catalysts and cation exchange resins (Smopex 101 and Amberlyst 15) as heterogeneous catalysts. The hydrolysis rate was exclusively dependent on the pH of the solution, but independent of the concentration and type of acid. Analysis of the formation rates of oligomers with DP 2–5 revealed that GGM hydrolysis occurs rather randomly. The final products were mannose, galactose, and glucose. In the beginning of the reaction, the formation of galactose was slightly faster than those of other sugars, since galactose is a side substituent to the glucose–mannose main chain of GGM. However, with longer reaction times, the formation rates of sugar monomers were correlated with their initial abundances in the polysaccharide. Autocatalytic kinetics was observed in the presence of the heterogeneous catalysts: an induction time was observed, after which the reaction proceeded with the same rates as with homogeneous acids. The autocatalytic effect is probably explained by the increase of the hydrolysis rate constant, as the GGM chain length decreases and the molecule becomes more accessible for the active sites on the outer surface of the catalyst.
1. Introduction
The biorefinery concept involves transformation of biomass to energy, chemicals, and fuel components.1 Acid hydrolysis is one method2 among others (enzymatic hydrolysis, ultrasonic,3 hot4 and supercritical water treatments,5 mineral acid6,7 and alkaline treatment8 or the use of ionic9,10 and supercritical liquids as solvents) to obtain sugar monomers from lignocellulosic raw materials. The acid hydrolysis of cellulose is an old technology, but due to the semicrystalline structure of cellulose,11 more harsh conditions are needed for its depolymerisation compared to that of hemicelluloses. Hemicelluloses are heteropolysaccharides and the most important hemicelluloses in softwood (coniferous trees) are galactoglucomannans (20%), and arabinoglucuronoxylans (10%).12,13 Furthermore, softwoods contain minor amounts of other polysaccharides such as arabinogalactans, pectins and starch. Hemicelluloses are non-crystalline and thus lower temperatures and acid concentrations are needed to obtain sugars from them compared to the case when starting from cellulose as a raw material. If rather high temperatures are used during the hydrolysis of hemicelluloses under acidic conditions, sugars may react further to give 5-hydroxymethylfurfural and furfural, among other by-products, which may be undesirable.Acid hydrolysis of biomass to produce sugars can in principle be performed with either homogeneous or heterogeneous acids. When using inorganic mineral acids, such as hydrochloric acid (HCl) or sulfuric acid (H2SO4), the product mixture should be neutralized after the hydrolysis step to separate the sugar monomers.14 During the neutralization step, salts are precipitated. Thereafter, the sugar monomers can be separated by chromatography.14,15 If organic acids are applied as catalysts, they can be evaporated from the product mixture16 and thus no neutralization step is needed. The use of solid acids could be beneficial, especially for continuous production of sugars as a fixed bed operation. There exist, however, only demonstrations of batchwise hydrolysis of biomass.17,18 The drawback of using heterogeneous catalysts, such as ion exchange resins, is the presence of mass transfer limitations, which can, however, be diminished, when using a fibrous polymeric catalyst containing sulphonic acid groups (Smopex 101). This kind of catalyst geometry facilitates short diffusion pathways, and fibrous Smopex 101 is a promising catalyst for acid hydrolysis of hemicelluloses.17
The acid hydrolysis of hemicelluloses has been studied using arabinogalactans,19O-acetyl-galactoglucomannans20 or glucurunoxylans21,22 as raw materials. GGM can be recovered from process waters of mechanical pulping processes23 and thus it is a potential feedstock for synthesis of valuable products. GGM can also be obtained by hot-water extraction of wood. The hydrolysis of water soluble O-acetyl-galactoglucomannan (GGM) has been studied in order to investigate its stability under acidic conditions for the production of dietary fibers, thus to preserve the polymeric form and not to produce monomers.20
Glucose, mannose, and galactose are the monosaccharides produced from GGM, which consists of a linear backbone of randomly distributed (1 → 4)-linked β-D-mannopyranosyl and (1 → 4)-linked β-D-glucopyranosyl units with α-D-galactopyranosyl units as single side units to mannosyl units as shown in Fig. 1. The hydroxyl groups at C-2 and C-3 in the mannose units are partially substituted by O-acetyl groups. The degree of acetylation of the isolated GGM was 20%, based on the molar ratio of acetyl groups per GGM sugar unit. The amount of acetic acid which may be produced from the acetyl groups during the hydrolysis of GGM is, however, minor to substantially influence the hydrolysis reaction and cannot be responsible for autocatalytic behaviour.
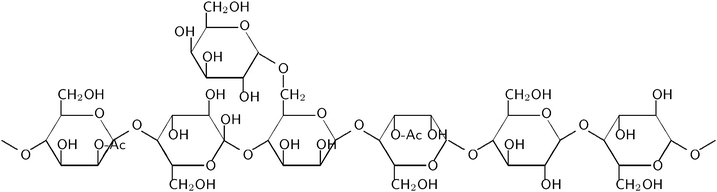 | ||
| Fig. 1 Structure of O-acetyl-galactoglucomannan (GGM). | ||
The average molar mass of GGM extracted from TMP process waters is around 20 kDa and the ratio of Gal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Glc:Man is equal to 0.5:1:4.20 However, the molar ratio is dependent on the means of isolation and purification.21
:Glc:Man is equal to 0.5:1:4.20 However, the molar ratio is dependent on the means of isolation and purification.21
Valuable sugars, especially D-mannose, which has applications as a drug, can be obtained from galactoglucomannan. D-Mannose is already being used as a drug to decrease the symptoms of lower urinary track diseases. This sugar is absorbed in the human body more slowly than glucose and thus it is transferred directly to the blood circulation. It can fix with bacteria, such as E. coli, thus removing bacteria from the human body.24–26 An advantage of D-mannose is that it can be used together with antibiotics. D-Mannose can also suppress the sugar and triglyceride levels of diabetic patients. Mannose is a growth accelerator for pigs27 and an indicator for the presence of a commonly used phosphomannose-isomerase (PMI) gene. Furthermore, concerning the utilization of GGM, glucose can be transformed via Lobry–de Bruyn–van Ekenstein transformation to mannose.28 Another valuable sugar in GGM is galactose, which has many applications in pharmaceutical and food related areas. It is, for example, used as a nutrient medium for maintaining viability of neural cells.29 Furthermore, galactose has been used in drugs exhibiting cytotoxic and anti-inflammatory properties.30
The aim of the current work was to determine the kinetics of acid hydrolysis of O-acetyl-galactoglucomannan to produce the corresponding monomeric sugar units in high yields. The monomers from GGM are rare sugars, i.e. valuable products which have a lot of applications in the food, pharmaceutical and cosmetic industries. The hydrolysis was conducted in the presence of different acid catalysts, both organic and inorganic acids, as well as heterogeneous catalysts. The main parameters for this investigation were the acid type and the acid concentration.
2. Experimental section
GGM was isolated from the thermomechanical pulp (TMP) of Norway spruce (Picea abies) on laboratory scale according to the method reported in ref. 23. The GGM water extract was purified and concentrated using filtration and ultrafiltration techniques. Ethanol:water in a ratio of 4:1 was added to the concentrated GGM solution to precipitate GGM, followed by drying. The GGM was further purified by redissolving it in water and performing dialysis.
The experiments of acid hydrolysis of GGM were conducted isothermally in a glass-jacketed glass reactor. A 0.5 weight% GGM solution was prepared by dissolving 0.5 g of dried GGM in 100 mL distilled water. The solution was preheated and the pH was adjusted by adding 1 M of the mineral acid (HCl, H2SO4, CF3CO2H, H2C2O4) to the targeted pH values of 0.5, 0.8, 1, and 2. The solution was loaded into the reactor and the temperature set to the desired value. This moment was considered as the initial starting point of the experiment. Furthermore, Amberlyst 15 and Smopex 101 with the acid capacity of 4.7 and 3.6 meq g−1, respectively, were used as catalysts. Amberlyst 15 is an ion exchange resin with a particle diameter of 0.45–0.60 mm, whereas Smopex 101 is a fibrous resin catalyst, with the fiber diameter of 10 μm.17 The average pore size of Amberlyst 15 is 40–90 nm, whereas Smopex 101 is a non-porous catalyst made from polyethylene-grafted polystyrene containing sulfonic groups on its surface.31,32
Samples for analysis were periodically withdrawn from the reactor and neutralized with 1 M NaOH prior to the analysis. The samples were silylated and the monomers analyzed by gas chromatography (GC) using a flame ionization detector (FID), and a 25 m × 0.2 mm i.d. column coated with cross-linked methyl polysiloxane (HP-1). The column oven temperature was 100 °C, raised at 2 °C min−1 to 170 °C, and at 12 °C min−1 to 290 °C (4 min).33,34 The oligomers were also analyzed by GC-FID on a 7.5 m × 0.53 mm i.d. column coated with cross-linked methyl polysiloxane (HP-1) after direct silylation of the samples. The column oven temperature was 120 °C, raised at 6 °C min−1 to 300 °C (10 min). Low-molecular weight compounds which may appear due to further degradation of the monomers were analyzed by means of high precision liquid chromatography (HPLC), equipped with Bio-Rad Aminex HPX-87C and Aminex HPX-87H carbohydrate columns. The total amount of the products in the liquid phase was determined by a total organic carbon analyzer, TOC-V CSN.20,23
3. Kinetic results and discussion
3.1. General kinetic phenomena
The kinetics of sugar monosaccharides formation from galactoglucomannan at 90 °C and pH 1 (HCl) is shown in Fig. 2. Almost complete conversion of GGM was observed after 800 min. Under these conditions, complete hydrolysis of the GGM was achieved without any further degradation of the monomers. A gradual increase of the monomers was observed, with mannose having a faster initial formation rate than glucose and galactose.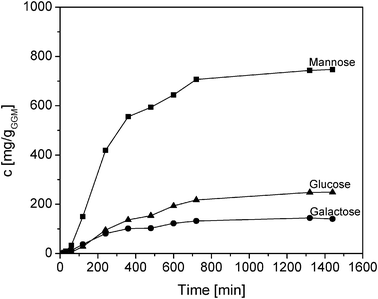 | ||
| Fig. 2 Kinetic curve showing released monomers during acid hydrolysis of GGM at 90 °C and pH 1 using HCl as catalyst. | ||
The rate of acid hydrolysis of hemicelluloses is influenced by the acid medium, the structure of the hemicellulose, and the anhydrosugar structure, i.e. whether it is in an α- or a β-anomer or if it is the furanose or pyranose form.35 The mechanism reported in the literature for the homogeneous acid-catalyzed hydrolysis of hemicelluloses as well as cellulose is based on the protonation of the glycosidic oxygen. The protonation is followed by breaking of the bond and simultaneous formation of a lower molar mass polymer chain, oligomer, or monosaccharide depending on the location of the catalytically hydrolyzed glycosidic bond, and a relatively unstable carbocation, which reacts with water leading to the regeneration of the proton and the formation of a monosaccharide. The acid hydrolysis of hemicelluloses is relatively easy and is faster than that of cellulose due to the crystalline structure of cellulose.
Depending on the reaction conditions, the extent of the hydrolysis varies from partially hydrolyzed oligomers to a complete release of monomers. Degradation products of the sugars, which are 5-hydroxymethylfurfural from the hexoses, as well as further smaller degradation products may however be formed, if the reaction conditions become too severe (a combination of acidic medium and high temperatures). No side products were formed in any experiments performed in this study, since the reaction conditions were rather mild. The mass balance of the total organic carbon in the liquid phase products was complete.
Essentially random scission of GGM was observed during the acid hydrolysis as proposed by ref. 20 due to the fact that the initial rates of formation of oligomers with degree of polymerization (DP) 2–5 were about the same as those at the initial stages of the reaction (Fig. 3). The GGM oligomers were released from the hemicellulose and their concentrations increased during the first hour (DP 3–5) and for two hours (DP 2), passing through the maximum. Almost all the oligomers were hydrolyzed to the monomers after 720 min. It should be pointed out that higher oligomers, probably appearing at the initial stage of the reaction, were not analysed.
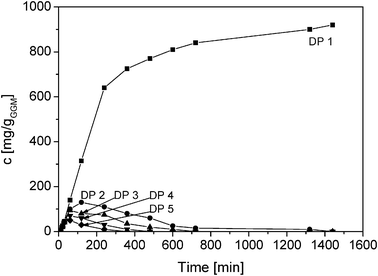 | ||
| Fig. 3 Released oligomers with DP 2–5 during acid hydrolysis of GGM at 90 °C and pH 1 using HCl as a catalyst. | ||
3.2. Effect of acid type – homogeneous and heterogeneous catalysts
Several inorganic and organic acids as well as heterogeneous acids were studied as catalysts for the title reaction, such as hydrochloric acid (HCl), sulfuric acid (H2SO4), trifluoroacetic acid (CF3CO2H), oxalic acid (H2C2O4), Amberlyst 15, and Smopex 101. There was no clear dependence of the GGM hydrolysis rate on the type of homogeneous acid used (Fig. 4, Table 1). The test plot for the hydrolysis of GGM is shown in Fig. 4, the slope of which gives the apparent rate constant. Only in this way we can get a reliable estimation of the overall rate constant. Analogous observations have been made for the hydrolysis of arabinogalactan, inulin and oligofructose.19,36 The GGM hydrolysis rates were influenced only by the proton concentration and not by the type of the homogeneous acid used, in agreement with the literature. Previous kinetic studies report that the rate constant of the hydrolysis of glycosidic bonds present in the hemicelluloses is independent of the acid, but dependent on the proton concentration only.35 However, differences in the performance of different acids have been observed on the dissolution power of glucan and decomposition of xylose. When using heterogeneous acid catalysts, such as the cation exchange resins Amberlyst 15 and Smopex 15, the hydrolysis experiments for these acids were performed at pH 1.5 and 2.0, respectively, since it is difficult to get very low pH levels for Smopex 15. The results showed that the first order rate constant for Amberlyst 15 (0.0001 min−1, calculated after 500 min reaction time) was lower even at pH 1.0 than that achieved for Smopex at pH 2.0 (0.0002 min−1), thus confirming analogous results as received in the acid hydrolysis of arabinogalactan showing the presence of mass transfer limitations for Amberlyst 15.17 It was concluded that the fibre catalyst Smopex-101 operated practically under diffusion-free conditions, whereas for Amberlyst 15 diffusional limitations appear as the effectiveness factor is less than 0.70. That is, however, the worst case scenario and such limitations might in reality be less prominent. Probably the reaction takes place at the outer surface of the catalyst only.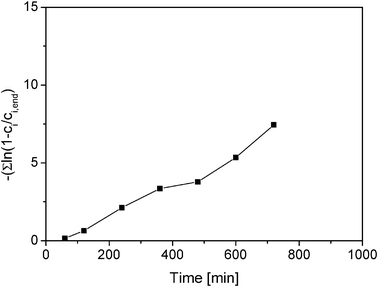 | ||
| Fig. 4 Apparent first-order rate constants plotted as a function of time for hydrolysis of GGM with HCl pH 1 at 90 °C. | ||
| pH | HCl | CF3CO2H | ||
|---|---|---|---|---|
| k/min−1 | k/10−pH | k/min−1 | k/10−pH | |
| n.d. – not determined. | ||||
| 0.5 | 0.013 | 0.041 | 0.009 | 0.028 |
| 0.8 | 0.0035 | 0.022 | 0.0034 | 0.021 |
| 1.0 | 0.0035 | 0.022 | n.d. | n.d. |
| 1.5 | 0.0003 | 0.0095 | 0.0003 | 0.0095 |
The formation kinetics of mannose and galactose using HCl, H2SO4, and CF3CO2H as catalysts are shown in Fig. 5a and b. When plotting the concentrations of glucose and galactose as a function of galactose concentration, it can be seen that galactose was released faster initially compared to glucose, since the ratio between the concentrations of glucose to galactose changed from the initial value of 0.7 to 1.6 after 240 min (Fig. 5c). A similar trend was visible for the ratio mannose-to-galactose varying from 4.6 to 6 (Fig. 5d). Selective acid hydrolysis of the side chain has also been reported for acid hydrolysis of gum arabic37 and arabinogalactans,19 for which selective hydrolysis of the side chain sugar units was achieved.
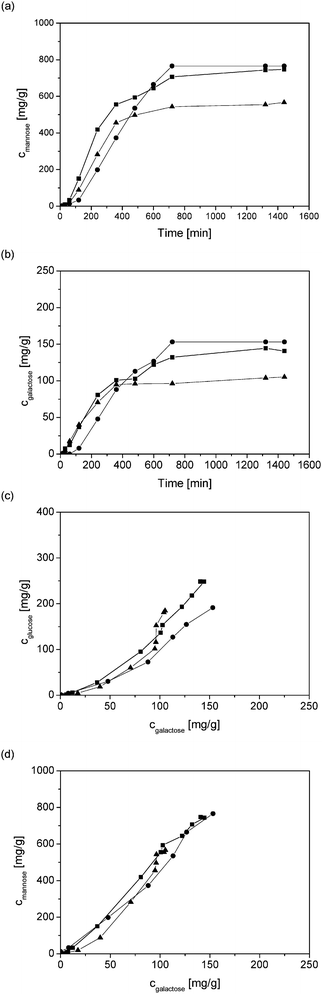 | ||
| Fig. 5 Formation of (a) mannose and (b) galactose as a function of time, formation of (c) glucose and (d) mannose as a function of formation of galactose in acid hydrolysis of GGM using different acids at pH = 1. Symbols: (■) HCl, (●) H2SO4 and (▲) CF3CO2H. | ||
Two heterogeneous solid acid catalysts were applied in this study, the non-porous fibrous catalyst Smopex 101 containing grafted sulfonic acids and Amberlyst 15, which is a conventional porous cation exchange resin (Fig. 6). It was shown that in the case of Amberlyst 15, the presence of internal mass transfer limitations could not be avoided. An autocatalytic behaviour was observed: the solid acid catalysts seemed to need an introductory time during which the hydrolysis rate of GGM was very slow. After 500 min, the reaction started to proceed with the expected rate, for which the first-order rate constants were calculated. The conversion level of GGM for Amberlyst 15 at pH 1 was around 50% after 6000 min, whereas a complete conversion was achieved with HCl at pH 1 in 800 min. There is no direct comparison of the conversion for Smopex 101, since this experiment was performed at pH 2. The final conversion under these conditions was about 60% after 4300 min. However, Smopex 101 is a much more active catalyst than Amberlyst 15 as revealed by Fig. 6a. The longer reaction time needed for Smopex 101 compared to that needed for homogeneous catalysts can be compensated with the benefit of easy catalyst separation and possibility to conveniently perform the reaction in a continuous mode.
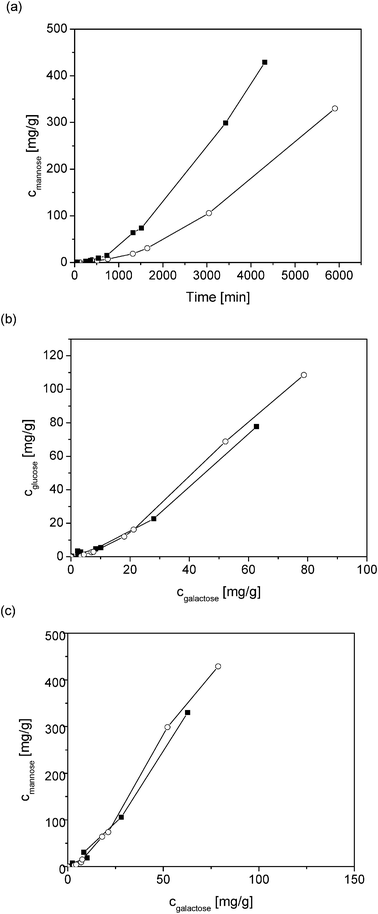 | ||
| Fig. 6 Formation of mannose (a), concentration of glucose (b) and mannose (c) as a function of concentration of galactose on Smopex-101 at pH 2.0 (■) and Amberlyst 15 at pH 1.0 (○) and at 90 °C. | ||
The formation rates of different sugars can be compared by plotting the sugar concentrations against each other (Fig. 6b and c). The ratio between the concentrations of glucose to galactose increased from the initial one being 0.8 until 1300 min for Smopex 101 to 1.3, whereas for the ratio between concentration of mannose to concentration of galactose the corresponding values increased from 3.5 to 4.6. These results showed, analogously to the use of homogeneous catalysts, that initially galactose release was favoured, but with increasing reaction times, their ratio corresponded to the original concentration ratio of different monomers. Smopex 101 and Amberlyst 15 had a very similar behaviour concerning the product distribution, which shows that the ratios of the hydrolysis rate constants of mannose, glucose and galactose are independent of the cation exchange catalyst (Fig. 6b and c).
3.3. Effect of pH
Generally, it is accepted that the acid hydrolysis rate increases with an increasing acid concentration. The double logarithmic plot of the reaction rate constant as a function of [H+] concentration during the hydrolysis of GGM at 90 °C gives a straight line with a slope ≈ 1 (Fig. 4, Table 1). This indicates a first order reaction with respect to [H+] at 90 °C over the studied pH range.The formation of mannose as a function of pH using HCl as a catalyst is shown in Fig. 7a. The rate of the release of the monomeric sugar units increased as the pH value of the acid decreased from 1.5 to a more acidic 0.5. The maximum mannose yield was achieved within 600 min at pH 0.5 without further degradation of the monomers. The product distribution of the sugar monomers showed that galactose, which is located in the side chain of glucomannan, is released with comparative rates as glucose and galactose, as well as mannose and galactose (Figs. 7b and c). The relative product distributions were independent of the pH, which is expected, since the mechanism of the formation of each monomer is the same.
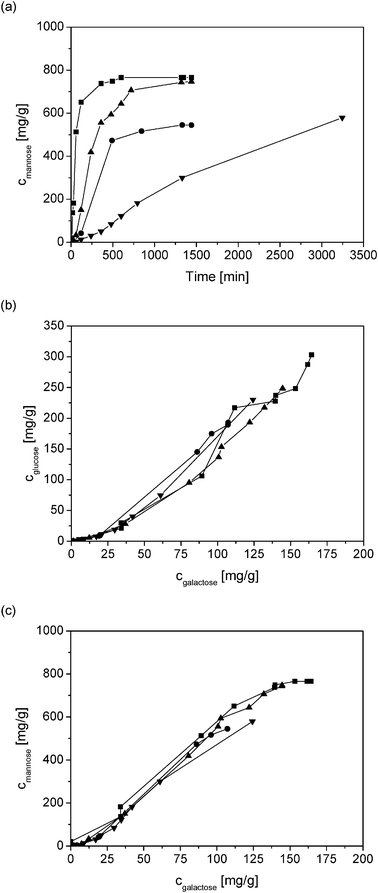 | ||
| Fig. 7 The formation rate of mannose (a), concentration of glucose as a function of galactose (b) and mannose as a function of galactose (c) in acid hydrolysis of GGM using HCl as catalyst with pH 0.5 (■), 0.8 (●), 1.0 (▲) and 1.5 (▼). | ||
4. Conclusions
The acid hydrolysis of O-acetyl-galactoglucomannan was investigated at 90 °C in the pH range of 0.5–2.0. Several homogeneous and heterogeneous catalysts, of both organic and inorganic acid types, were applied. The only reaction products were mannose, glucose and galactose and no side products were observed during the acid hydrolysis of GGM at 90 °C in the pH range of 0.5–2.0. The initial formation rates of oligomers were about the same, indicating that the scission of GGM occurs randomly in the beginning of the hydrolysis process.The hydrolysis rate was first order with respect to the acid concentration, but independent of the acid type. For the heterogeneous catalysts (Amberlyst 15 and Smopex 101), an autocatalytic behaviour was observed. Smopex 101 was a much more active heterogeneous catalyst than Amberlyst 15, because Smopex 101 has a fibrous structure, which diminishes the internal mass transfer resistance. The relative product distributions of mannose, glucose and galactose were independent of pH. In the beginning of the reaction, the formation of galactose was faster than that of glucose and galactose, which can be explained by the fact that galactose is a side substituent to the main chain of GGM.
Acknowledgements
This work is part of the activities at the Åbo Akademi University Process Chemistry Centre within the Finnish Centre of Excellence programmes (2000–2011) appointed by the Academy of Finland. The financial support from Forestcluster Ltd., the Future Biorefinery programme is also acknowledged.References
- F. Cherubini, Energy Convers. Manage., 2010, 51, 1412 CrossRef CAS.
- P. Mäki-Arvela, T. Salmi, B. Holmbom, S. Willför and D. Yu. Murzin, Chem. Rev., 2011, 111, 5638 CrossRef.
- A. Tayal and S. Khan, Macromolecules, 2000, 33, 9488 CrossRef CAS.
- D. Severian, Polysaccharides, 2005, 2, 893 Search PubMed.
- M. Sasaki, Z. Fang, Y. Fukushima, T. Adschiri and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 2883 CrossRef CAS.
- L. T. Fan, M. M. Gharpuray and Y.-H. Lee, Cellulose hydrolysis, Springer-Verlag, Berlin, 1987 Search PubMed.
- O. Akpinar, K. Erdogan and S. Bostanci, Carbohydr. Res., 2009, 344, 660 CrossRef CAS.
- M. Ishida, K. Otsuka, I. Takenaka and I. Yamanaka, J. Chem. Technol. Biotechnol., 2005, 80, 2814 CrossRef.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 134, 4974 CrossRef.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen and J.-P Mikkola, Ind. Crops Prod., 2010, 32, 175 CrossRef.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 108, 6712 CrossRef.
- E. Sjöström, Puukemia, Otakustantmo, Espoo, Finland, 1978 Search PubMed.
- C. Laine, Structures of hemicelluloses and pectins in wood and pulp, PhD Thesis, Helsinki University of Technology, 2005 Search PubMed.
- J. Antila, V. Ravanko and P. Walliander, International Patent, WO 99/10542, 2003 Search PubMed.
- J. Kuusisto, I. Lindqvist and J. Antila, International Patent, WO 99/10384, 2009 Search PubMed.
- G. F. Fanta, T. P. Abbott, A. I. Herman, R. C. Burr and W. M. Doane, Biotechnol. Bioeng., 1984, 16, 1122 CrossRef.
- B. T. Kusema, G. Hilmann, P. Mäki-Arvela, S. Willför, B. Holmbom, T. Salmi and D. Yu. Murzin, Catal. Lett., 2011, 141, 408 CrossRef CAS.
- A. E. Abasaaed and Y. Y. Lee, Chem. Eng. Technol., 1995, 18, 440 CrossRef.
- B. T. Kusema, C. Xu, P. Mäki-Arvela, S. Willför, B. Holmbom, T. Salmi and D. Yu. Murzin, Int. J. Chem. React. Eng., 2010, 8, 1 Search PubMed.
- C. Xu, A. Pranovich, L. Vähäsalo, J. Hemming, B. Holmbom, H. A. Schols and S. Willför, J. Agric. Food Chem., 2008, 56, 2429 CrossRef CAS.
- V. T. Rovenskii, E. I. Sokol, G. I. Druzhinina, L. P. Radul and S. N. Kolesov, Vopr. Khim. Khim. Tekhnol., 1987, 83, 93 Search PubMed.
- J. Beinart and V. V. Kirilova, Koksnes Kim., 1976, 5, 36 Search PubMed.
- S. Willför, P. Rehn, A. Sundberg, K. Sundberg and B. Holmbom, Tappi J., 2003, 2, 27 Search PubMed.
- M. Aronson, O. Medalia, L. Schori, D. Mirelman, N. Sharon and I. Ofek, J. Infect. Dis., 1979, 139, 329 CrossRef CAS.
- E. K. Michaels, J. S. Chmiel, B. J. Plotkin and A. J. Schaeffer, Urol. Res., 1983, 11, 97 CrossRef CAS.
- P. O'Hanley, D. Lark, S. Falkow and G. Schoolnik, J. Clin. Invest., 1985, 75, 347 CrossRef CAS.
- M. E. Davis, C. V. Maxwell, G. F. Erf, D. C. Brown and T. J. Wistuba, J. Anim. Sci., 2004, 82, 1882 CAS.
- M. Miljković, Carbohydrates: Synthesis, Mechanisms, and Stereoelectronic Effects, Springer Verlag, 1st edn, 2009, ch. 4. Isomerization of Sugars, 94 Search PubMed.
- G. J. Brewer, US2003077564, 2003 Search PubMed.
- J. Raveth, K. Yoshikatus and N. Falk, WO2007/117505, 2007 Search PubMed.
- J. Lilja, D. Yu. Murzin, T. Salmi, J. Aumo, P. Mäki-Arvela and M. Sundell, J. Mol. Catal. A: Chem., 2002, 181–182, 555 CrossRef.
- J. Lilja, J. Aumo, T. Salmi, D. Yu. Murzin, P. Mäki-Arvela, M. Sundell, K. Ekman, R. Peltonen and H. Vainio, Appl. Catal., A, 2002, 228, 253 CrossRef CAS.
- S. Willför, A. Pranovich, T. Taminen, J. Puls, C. Laine, A. Suurnäkki, B. Saake, K. Uotila, H. Simolin, J. Hemming and B. Holmbom, Ind. Crops Prod., 2009, 29, 571 CrossRef.
- F. Bertaud, A. Sundberg and B. Holmbom, Carbohydr. Polym., 2002, 48, 319 CrossRef CAS.
- Y.-Z. Lai, Chemical degradation, in Wood and Cellulosic Chemistry, ed. D. N.-S. Hon and N. Shiraishi, Dekker: Basel, Switzerland, 2nd edn, 2001, p. 443 Search PubMed.
- C. Blecker, C. Fougnies, J. C. Van Herck, J. P. Chevalier and M. Paquot, J. Agric. Food Chem., 2002, 50(6), 1602 CrossRef CAS.
- M. Hatanaka, E. Yokoyama, M. Sano, S. Kumazawa and T. Takagi, UK Patent, GB 2168980 A, 1985 Search PubMed.
| This journal is © The Royal Society of Chemistry 2013 |