Asymmetric catalytic hydrogenation for large scale preparation of optically active 2-(N-benzoylamino)cyclohexanecarboxylic acid derivatives†
Takeshi
Kajiwara
*,
Takahiro
Konishi
and
Mitsuhisa
Yamano
Chemical Development Laboratories, Takeda Pharmaceutical Company Limited, 17-85, Jusohonmachi 2-chome, Yodogawa-ku, Osaka, Japan. E-mail: takeshi.kajiwara@takeda.com; Fax: +81-6-6300-6251
First published on 3rd August 2012
Abstract
Asymmetric catalytic hydrogenation methodology was applied to the synthesis of 2-[N-(4-difluoromethoxy)benzoylamino]cyclohexanecarboxylic acid derivatives. During the course of optimization, it was found that chiral Ru(II) dicarboxylate complexes worked efficiently with an acid additive, aqueous HBF4. Regarding chiral ligand, DTBM-BINAP showed the best result with full-conversion and the highest stereoselectivity. Practical conditions for the asymmetric hydrogenation were also established suppressing the amount of the catalyst and multi-kilogram scale synthesis was successfully achieved.
Introduction
β-Amino acid motifs are important chiral building blocks for the synthesis of β-peptides, β-lactams, natural products, and physiologically active substances.1 Among them, chiral cyclic β-amino acids have recently gained much attention as key structural elements of β-peptides.2Several methods have been developed for the preparation of the chiral cyclic β-amino acids and their derivatives. Optical resolution of alicyclic β-amino esters was achieved through direct lipase-catalyzed hydrolysis with high enantiomeric excess.3 Asymmetric synthesis approaches for cis-isomers by stereoselective reduction of olefins of cyclic β-enamino esters to which chiral auxiliaries are attached have been reported.4 However, their cis selectivities were moderate and further purification processes were needed to obtain diastereo- and enantiopure compounds.
Although the asymmetric catalytic hydrogenation approach is often practical, it is generally difficult to develop hydrogenation of tetra-substituted functionalized olefins, which have lower reactivity than tri-substituted functionalized olefins,5 for synthesizing their chiral β-amino acids and derivatives. Zhang et al.,6 Wu and Hoge7 and Beller et al.8 have reported that some optically active (2-amino)cycloalkanecarboxylic acid derivatives have been obtained by applying asymmetric hydrogenation methodology using chiral catalysts, to olefins of cyclic β-(acylamino)acrylates, with high enantiomeric excess. However, the reported substrates of these asymmetric catalytic hydrogenations were only cyclic β-(acetylamino)acrylates and cyclic β-(Boc-amino)acrylates.
Optically active 2-(N-benzoylamino)cycloalkanecarboxylic acid derivatives 1, which are derived from cyclic β-amino acids, are also useful compounds for optical resolution of amines,9 and some are important intermediates of drug substances.10 These compounds were prepared by condensation of chiral cyclic β-amino ester 2 with benzoic acid derivatives 3,10f,11 which were obtained by a multi-step asymmetric synthesis (Fig. 1). An alternative method, involving only optical resolution via diastereomer salt formation of the carboxylic acid derivative 1 (R1, R2 = H), has been reported.12
 | ||
| Fig. 1 Preparation of 1 by condensation of chiral cyclic β-amino ester 2 with benzoic acid derivatives 3. | ||
We attempted to apply the asymmetric catalytic hydrogenation methodology to cyclic (N-benzoylamino)acrylates 6 to develop a more efficient synthetic method of 1 with less steps (Fig. 2). Especially, we had the necessity to produce 1 (R1 = 4-difluoromethoxy) which bears a ‘difluoromethoxy’ moiety (unique biological feature13) at the 4-position of the phenyl ring. Although the asymmetric hydrogenation of the N-benzoyl protected substrate 6 (R1 = H) had lower reactivity than the N-acetyl protected substrate as reported,6 the enantioselectivity of 6 (R1 = H) was relatively high. 6 (R1 = 4-difluoromethoxy) showed better enantioselectivity than non-substituted ones and the reactivity was also improved.
 | ||
| Fig. 2 Strategy for preparing 1 applying the asymmetric catalytic hydrogenation methodology. | ||
Subsequently, we investigated the optimum reaction conditions which were applicable to large-scale synthesis of 6 (R1 = 4-difluoromethoxy). It was found that chiral Ru(II) dicarboxylate complexes worked efficiently for the asymmetric hydrogenation as precatalysts with an acid additive, HBF4. DTBM-BINAP14 as a chiral ligand showed the best results for the reaction from the viewpoint of stereoselectivity and catalyst activity.
We herein describe an efficient, and highly diastereo- and enantioselective synthesis of 1 (R1 = 4-difluoromethoxy)15 based on asymmetric hydrogenation of the corresponding 6 (R1 = 4-difluoromethoxy), by applying [Ru{dtbm-binap}(OAc)2]–HBF4.
Results and discussions
The 2-(N-benzoylamino)cyclohexenecarboxylic acid derivative 6a16 was selected as the first substrate for asymmetric catalytic hydrogenation of β-benzoyl derivatives. While the in situ-generated chiral Ru precatalysts prepared by protonation of a mixture of [Ru(cod)(methallyl)2] and chiral phosphorous ligand with two equivalents of HBF4·Et2O in CH2Cl2 had been reported,5,6a we utilized [Ru(cod)(methallyl)2] as a precatalyst with a chiral phosphorous ligand and an acid additive, aqueous HBF4, for the asymmetric hydrogenation in MeOH.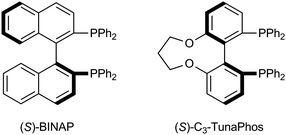
When (S)-BINAP or (S)-C3-TunaPhos6a was used as the chiral phosphorous ligand, the desired product 1a was obtained in 32.2% yield, 84.1% ee (run 1, Table 1) and 14.9% yield, 72.7% ee (run 3, Table 1), respectively. However, the selectivity was relatively high despite the reactivity being significantly reduced, compared with the 2-(N-acetamido)cyclohexanecarboxylic acid derivatives as substrates. We found that the reactions were almost complete at higher temperature (50 °C) (runs 2 and 4, Table 1), but the enantioselectivity was found to be somewhat lower than at 25 °C. Next, we attempted reaction of another substrate 6b, which bears a difluoromethoxy moiety at the para-position of the benzoyl substituent. 6b was synthesized by the reaction of 3a with SOCl2 followed by amidation with 5a, which was synthesized from 4a in high yield as shown in Scheme 1.

| Run | Temp./°C | Ligand | Conv.a (%) | eeb (%) | Configuration |
|---|---|---|---|---|---|
a Conversions were determined by HPLC. Conditions: GL Science Inertsil ODS-3 (15 cm × 4.6 mm i.d.), 50 mM KH2PO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : MeCN = 3:7, 1.0 mL min−1, UV 254 nm, 25 °C: tR 4.0 min for 1a, and 15.1 min for 6a.
b ee's were determined by HPLC. Conditions: DAICEL CHIRALPAK AD-RH (15 cm × 4.6 mm i.d.), 50 mM KH2PO4:MeCN = 6:4, 0.5 mL min−1, UV 254 nm, 25 °C: tR 19.7 min for (1S, 2R) and 21.4 min for (1R, 2S). : MeCN = 3:7, 1.0 mL min−1, UV 254 nm, 25 °C: tR 4.0 min for 1a, and 15.1 min for 6a.
b ee's were determined by HPLC. Conditions: DAICEL CHIRALPAK AD-RH (15 cm × 4.6 mm i.d.), 50 mM KH2PO4:MeCN = 6:4, 0.5 mL min−1, UV 254 nm, 25 °C: tR 19.7 min for (1S, 2R) and 21.4 min for (1R, 2S).
|
|||||
| 1 | 25 | (S)-BINAP | 32.2 | 84.1 | (1S,2R) |
| 2 | 50 | (S)-BINAP | 96.9 | 71.0 | (1S,2R) |
| 3 | 25 | (S)-C3-TunaPhos | 14.9 | 72.7 | (1S,2R) |
| 4 | 50 | (S)-C3-TunaPhos | 94.3 | 70.3 | (1S,2R) |
 | ||
| Scheme 1 Synthesis of cyclic (N-benzoylamino)acrylate 6b. | ||
The hydrogenation of 6b using both [Ru(cod)(methallyl)2] − (S)-BINAP and [Ru(cod)(methallyl)2] − (S)-C3-TunaPhos6a (s/c = 20) was carried out at 25 °C at 5 MPa of H2 atmosphere according to our procedure. It was found that the hydrogenation proceeded to give (1S, 2R)-1b including a small amount of diastereoisomers with 95.3% conversion, 89.5% ee, and 97.1% de when using (S)-C3-TunaPhos ligand (run 1, Table 2). (S)-BINAP ligand showed better reactivity and diastereoselectivity with full-conversion, 86.6% ee, and ∼100% de (run 2, Table 2). These results imply that the improvement of reactivity and the enantioselectivity would be caused by the electron-withdrawing substituents, such as 4-(difluoromethoxy).

| Run | Ru catalyst | s/c | H2/MPa | Temp./°C | Time/h | Conv.a (%) | eeb (%) | deb (%) |
|---|---|---|---|---|---|---|---|---|
|
a Conversions were determined by HPLC. Conditions: GL Science Inertsil ODS-3 (15 cm × 4.6 mm i.d.), 50 mM KH2PO4: MeCN = 3:7, 1.0 mL min−1, UV 220 nm, 25 °C: tR 4.2 min for 1b, 14.1 min for 6b.
b Diastereomer excess and enantiomer excess were determined by chiral HPLC. Conditions: DAICEL CHIRALPAK AD-RH (15 cm × 4.6 mm i.d.), 50 mM KH2PO4:MeCN = 6:4, 0.5 mL min−1, UV 254 nm, 40 °C: tR 16.0 min for (1R,2R), 18.3 min for (1S,2S), 25.8 min for (1S,2R), and 29.4 min for (1R,2S).
|
||||||||
| 1 | [Ru(cod)(methallyl)2] + (S)-C3-TunaPhos | 20 | 5 | 25 | 18 | 95.3 | 89.5 | 97.1 |
| 2 | [Ru(cod)(methallyl)2] + (S)-BINAP | 20 | 5 | 25 | 18 | 100.0 | 86.6 | ∼100 |
| 3 | [Ru(cod)(methallyl)2] + (S)-BINAP | 20 | 1 | 25 | 89 | 100.0 | 85.8 | 92.4 |
| 4 | [Ru {(S)-binap]}(OAc)2] | 20 | 1 | 25 | 18 | 79.2 | 86.2 | 87.8 |
| 5 | [Ru{(S)-binap}Cl2] | 20 | 1 | 25 | 18 | 0.5 | — | — |
| 6 | [Ru{(S)-xylyl-binap}(OAc)2] | 20 | 1 | 25 | 18 | 100.0 | 89.3 | 88.0 |
| 7 | [Ru{(S)-dadmp-binap}(OAc)2] | 20 | 1 | 25 | 18 | 100.0 | 92.0 | 91.9 |
| 8 | [Ru{(S)-dtbm-binap}(OAc)2] | 20 | 1 | 25 | 18 | 100.0 | 95.0 | 98.7 |
| 9 | [Ru{(S)-dtbm-binap}(OAc)2] | 40 | 1 | 25 | 48 | 90.6 | 88.5 | 98.3 |
| 10 | [Ru{(S)-dtbm-binap}(OAc)2] | 20 | 1 | 45 | 18 | 100.0 | 90.0 | 95.8 |
| 11 | [Ru{(S)-dtbm-binap}(OAc)2] | 80 | 1 | 45 | 40 | 100.0 | 89.4 | 94.5 |
| 12 | [Ru{(S)-dtbm-binap}(OAc)2] | 160 | 1 | 45 | 40 | 100.0 | 90.6 | 95.7 |
Also, we investigated the application of lower H2 pressure for scale-up synthesis using (S)-BINAP ligand, and found that the selectivity was preserved even when the H2 pressure was lowered to 1 MPa (run 3, Table 2) although the reactivity and diastereoselectivity were slightly decreased.

Using (S)-BINAP as the ligand, several Ru complexes for this reaction were examined to reduce the reaction time under a pressure of 1 MPa H2 (run 4, Table 2). It was revealed that chiral Ru(II) dicarboxylate complexes were more reactive than [Ru(cod)(methallyl)2]–(S) − BINAP, however, the diastereoselectivity was slightly lower and the conversion was almost 80% (run 3, Table 2). [Ru{(S)-binap}Cl2] showed almost no reaction (run 5, Table 2). Our focus moved to a modified BINAP ligand comprised of a chiral Ru(II) dicarboxylate complex to improve reactivity, diastereo- and enantioselectivity, because the reactivity and stereoselectivity are highly dependent upon the substituents of the phenyl groups on the phosphorous atom of the axially chiral diphosphine ligand.17 When (S)-xylyl-BINAP18 was used in this reaction system, the conversion was 100% even when the reaction time was 18 h while preserving the diastereo- and enantioselectivity (run 6, Table 2). When using (S)-DADMP-BINAP,14b,19 which bears more electron-donating substituents, higher enantioselectivity was obtained compared to that with the xylyl group (run 7, Table 2). (S)-DTBM-BINAP,14 with a substituted 4-methoxy group and a t-butyl group at the 3 and 5-position, showed the best result with full-conversion and the highest stereoselectivity (95.0% ee, 98.7% de) (run 8, Table 2).
For the large scale asymmetric hydrogenation, suppressing the amount of the expensive catalyst would be the critical issue on the aspect of the cost effective production. Therefore, we investigated the reaction conditions in order to use less amount of catalyst. When the s/c was raised to 40 at 25 °C, the conversion was decreased to 90.6% even if the reaction time was prolonged to 48 h (run 9, Table 2). In order to enhance the reaction rate by elevation of reaction temperature, we conducted this reaction at 45 °C with s/c = 20, and found that a slight decrease of selectivity was observed (run 10, Table 2). The decrease of selectivity in this reaction was found to be acceptable because it would be possible to obtain diastereo- and enantiopure 1c, a hydrolyzed product of 1b as described below. Therefore, we conducted this reaction using less amount of catalyst at 45 °C, and found that the reaction was completed using s/c = 80 or s/c = 160 condition (runs 11 and 12, Table 2) although the reaction time was increased (40 h). These results revealed that the reactivity was improved by conducting the reaction at 45 °C while retaining the selectivity within acceptable levels.
The examination of solvent (s/s = 35; solvent/substrate ratio) was conducted seeking higher solubility, because 6b has moderate solubility in alcohol solvent. The results are summarized in Table 3 with the condition of 18 h reaction time at 25 °C using [Ru{(S)-dtbm-binap}(OAc)2] (s/c = 20) and aqueous HBF4 (2 equiv.) under 1 MPa of H2 atmosphere. When the reaction was conducted in toluene, DMF, and THF, no conversion was observed (runs 4 to 6, Table 3). EtOH was less effective than MeOH for enantioselectivity, and isopropyl alcohol was less effective for both reactivity and enantioselectivity (runs 2 and 3, Table 3). From these results, MeOH was identified as the most appropriate solvent for this hydrogenation reaction.
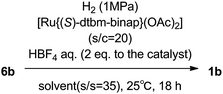
We carried out the screening of acid additives with the conditions of [Ru{(S)-dtbm-binap}(OAc)2] (s/c = 160) at 45 °C as shown in Table 4. The conversion was only 21.4% when this reaction was conducted without an acid additive (run 1, Table 4), which revealed that the addition of acid was necessary for enhancing the reactivity of catalysts as reported by Zhang et al.5,6a HBF4-etherate gave almost the same effect as aqueous HBF4 (run 2 vs. 3, Table 4). MeSO3H, (−)-10-CSA, and CF3SO3H were found to be almost as effective as HBF4. p-TsOH·H2O and 7-amino-1,3,6-naphthalenetrisulfonic acid were less effective in spite of bearing sulfonic acid structures (runs 4 to 8, Table 4). NH4Cl (run 9, Table 4) and sulfanilic acid (run 10, Table 4) were also less effective, and these results revealed that acids bearing amino moieties were not suitable for this reaction system (runs 8 to 10, Table 4). Other mineral acids, such as HCl, were also investigated, however, the conversion was limited to 94.0% even when the reaction time was 90 h (run 11, Table 4). Reducing the amount of acid additive from 2.0 equiv. to 1.0 equiv. showed almost no effect on the reactivity (run 2 vs. 12, Table 4). When a greater equivalency (10 equiv.) of HBF4 was used, a decrease in selectivity was observed (run 13, Table 4).

| Run | Additivesa | Time/h | Conv. (%) | ee (%) | de (%) |
|---|---|---|---|---|---|
| a The number in the parentheses is the equivalency to the ruthenium complex. b Generation of methyl ester (32.7%) was observed. | |||||
| 1 | None | 40 | 21.4 | 89.1 | 97.0 |
| 2 | aqueous HBF4 (2.0) | 40 | 100.0 | 90.1 | 95.8 |
| 3 | HBF4·Et2O (2.0) | 40 | 100.0 | 90.7 | 95.6 |
| 4 | (−)-10-CSA (2.0) | 100 | 100.0 | 89.7 | 94.7 |
| 5 | MeSO3H (2.0) | 20 | 99.6 | 90.1 | 94.8 |
| 6 | CF3SO3H (2.0) | 40 | 100.0 | 90.3 | 95.0 |
| 7 | TsOH·H2O (2.0) | 100 | 95.8 | 88.9 | 91.2 |
| 8 | 7-Amino-1,3,6-naphthalene trisulfonic acid (1.0) | 20 | ∼0 | — | — |
| 9 | NH4Cl (1.0) | 23 | 46.6 | 89.4 | 96.3 |
| 10 | Sulfanilic acid (1.0) | 90 | 78.3 | 91.1 | 96.6 |
| 11 | HCl–AcOEt (1.0) | 90 | 94.0 | 89.8 | 95.8 |
| 12 | aqueous HBF4 (1.0) | 40 | 100.0 | 90.6 | 95.5 |
| 13 | aqueous HBF4 (10)b | 90 | 100.0 | 89.2 | 91.6 |
In order to obtain diastereo- and enantiopure 1c from crude 1b produced by asymmetric hydrogenation of 6b, which was expected to be used as an intermediate of drug substance or as an optical resolution agent, an effective purification method of 1b or 1c was required. We investigated the purification method which was applicable to large scale synthesis for crude 1b or crude 1c derived from 1b by hydrolysis. As a result, it was difficult to obtain diastereo- and enantiopure 1b by recrystallization of crude 1b, however, we found that the diastereo- and enantiopure 1c was easily obtained by recrystallization using a MeCN–H2O solvent mixture from crude 1c (Scheme 2).
 | ||
| Scheme 2 Preparation of diastereo- and enantiopure 1c. | ||
Finally, the preparation of 1c on a multi-kilogram scale was conducted with the conditions (run 2, Table 4) as shown in Scheme 3. Regarding the cost effective production for asymmetric catalytic hydrogenation, the reaction conditions of [Ru{(S)-dtbm-binap}(OAc)2] (s/c = 160) and HBF4 (2.0 equiv.) at 45 °C were chosen. Also, 0.9 MPa of H2 was applied for the scale-up. When 18 kg of 6b was hydrogenated using the selected condition in two batches, the reactions proceeded smoothly to give 1b in full-conversion, >96% ee, and >91% de, respectively. After concentration of the reaction mixture without purification, 1b was hydrolysed using an aqueous NaOH–EtOH mixture without epimerization, followed by recrystallization with MeCN–H2O to afford 1c in 74.4% yield (12.36 kg) and 69.1% yield (11.49 kg) from 6b with >99.9% ee and >99.9% de respectively.
 | ||
| Scheme 3 The result of multi-kilogram scale synthesis of 1c. | ||
Conclusion
In conclusion, we found that the asymmetric hydrogenation using chiral Ru(II) complexes with an acid additive, aqueous HBF4, was applicable to cyclic (N-benzoylamino)acrylates 6, but they were less reactive than those of 2-(N-acetamido)-derivatives. During the course of optimization for asymmetric hydrogenation of 6b, chiral Ru(II) dicarboxylate complexes were more reactive than [Ru(cod)(methallyl)2] − (S)-BINAP under the pressure of 1 MPa H2. Regarding chiral ligand, (S)-DTBM-BINAP showed the best result with full-conversion and highest stereoselectivity (95.0% ee, 98.7% de) for the asymmetric hydrogenation of 6b. Furthermore, we established a practical asymmetric hydrogenation condition for 6b suppressing the amount of the expensive catalyst for cost effective production and also, diastereo- and enantiopure 2-[N-(substituted)benzoylamino]cycloalkanecarboxylic acid 1c derived from crude 1b obtained by asymmetric hydrogenation of 6b was easily purified by recrystallization using a MeCN–H2O solvent mixture. Finally, we succeeded in multi-kilogram scale synthesis of 1c. This methodology would be applied to 2-[N-(substituted)benzoylamino]cycloalkanecarboxylic acid derivatives as intermediates of drug substances, novel optical resolution agents or versatile building blocks for a range of chiral compounds.Experimental section
Melting points were measured by a SIBATA B-545 apparatus. 1H NMR and 13C NMR spectra were measured in CDCl3 solutions at 500 and 125 MHz, respectively, using a Bruker Avance-III spectrometer. IR spectra were measured using a SHIMADZU IR Prestige-21. HRMS spectra were measured by SHIMADZU LCMS-IT-TOF. Diastereomer excess and enantiomer excess were determined by chiral HPLC.Ethyl 2-aminocyclohexene-1-carboxylate (5a)
To a 1 L three-necked flask was added 4a (50 g, 0.293 mol) and EtOH (250 mL) at room temperature. 25% ammonia aq. (125 mL) was added, and the reaction mixture was stirred at room temperature for 17 h. Crystallization was gradually performed by adding H2O (125 mL) and seed crystals for 10 min, then additional H2O (500 mL) was added for 15 min at room temperature. The reaction mixture was stirred for 15 min at room temperature, and then for 30 min at 5 °C. The resultant crystals were collected by filtration, washed with H2O (500 mL), and dried under reduced pressure (50 °C) to afford the titled compound 5a (42.6 g, 85.9% yield) as white crystals.Mp: 66–67 °C.
1H NMR (CDCl3, 500 MHz, TMS): δ 1.27 (3H, t, J = 7.3 Hz), 1.51–1.69 (4H, m), 2.10–2.38 (4H, m), 4.14 (2H, q, J = 7.3 Hz), 6.00 (2H, br s) ppm.
13C NMR (CDCl3, 125 MHz, TMS): δ 14.6, 22.2, 23.2, 23.4, 30.6, 58.8, 92.2, 156.5, 170.5 ppm.
IR (ATR, cm−1): 3425, 3321, 2976, 2930, 2901, 2839, 1649, 1607, 1530, 1418, 1302, 1273, 1246, 1215, 1173, 1082, 1069, 1041, 1018, 906, 879, 855, 826, 777.
Anal. calcd for C9H15NO2: C, 63.88; H, 8.93; N, 8.28%; found: C, 63.81; H, 9.19; N, 8.16%.
HRMS: calcd for C9H15NO2 (M + H)+ = 170.1176, found: 170.1196.
Ethyl 2-[N-{4-(difluoromethoxy)benzoyl}]aminocyclohexene-1-carboxylate (6b)
To a 100 mL three-necked flask was added 3a (5.0 g, 26.6 mmol), toluene (25 mL) and DMF (0.025 mL) at room temperature, then thionyl chloride (3.48 g, 29.2 mmol) was added at once. The reaction mixture was heated at 60 °C for 2.5 h. The reaction mixture was concentrated and MeCN (10 mL) was added to prepare the acid chloride solution. To another 200 mL three-necked flask was added 5a (4.94 g, 29.2 mmol), pyridine (2.3 g, 29.2 mmol) and MeCN (15 mL), before it was heated at 40 °C. The acid chloride solution, prepared above, was then added dropwise over 10 min, and the mixture was stirred at 60 °C for 1.5 h. After cooling the reaction mixture, H2O (50 mL) was added slowly at 25–30 °C, and stirring continued for 30 min. The resultant solid was collected by filtration, washed with H2O (50 mL), and dried under reduced pressure at 60 °C to afford the titled compound 6b (7.98 g, 88.4% yield) as a white solid.Mp: 88–90 °C.
1H NMR (CDCl3, 500 MHz, TMS): δ 1.32 (3H, t, J = 7.3 Hz), 1.60–1.72 (4H, m), 2.37–2.41 (2H, m), 3.11–3.16 (2H, m), 4.22 (2H, q, J = 7.3 Hz), 6.58 (1H, t, J = 73.5 Hz), 7.20 (2H, d, J = 8.8 Hz), 7.99 (2H, d, J = 8.8 Hz), 12.58 (1H, br s) ppm.
13C NMR (CDCl3, 125 MHz, TMS): δ 14.2, 21.7, 21.9, 24.4, 28.7, 60.4, 105.6, 115.5 (t, J = 259 Hz), 119.0, 129.6, 131.9, 152.4, 153.9, 164.1, 170.4 ppm.
IR (ATR, cm−1): 3196, 2943, 1676, 1651, 1618, 1585, 1476, 1458, 1246, 1221, 1200, 1179, 1126, 1109, 1047, 1015, 988, 945, 851, 818, 775, 754, 705, 681, 664.
Anal. calcd for C17H19NO4F2: C, 60.17; H, 5.64; N, 4.13; F, 11.20%; found: C, 60.14; H, 5.79; N, 4.01; F, 11.08%.
HRMS: calcd for C17H19NO4F2 (M + H)+ = 340.1355, found: 340.1345.
(1S,2R)-Ethyl 2-[N-{4-(difluoromethoxy)benzoyl}]aminocyclo-hexane-1-carboxylate (1b)
(All operations were carried out under an argon atmosphere)To a 120 mL pressure-resistant vessel were added 6b (5.00 g, 14.7 mmol), [Ru{(S)-dtbm-binap}(OAc)2] (0.125 g, 0.0886 mmol, s/c = 160), and 42% aqueous HBF4 (14.3 mL, 0.1772 mmol) in MeOH (25 mL). The mixture was purged with H2 (1 MPa), then stirred for 40 h at 45 °C. After the H2 absorption was stopped, the H2 pressure was released. The reaction mixture was concentrated to afford the diastereomer–enantiomer mixture of the crude titled compound 1b (5.00 g, 100%) as a pale-brown solid–oil.
Conversion: 100%, 95.5% de, 90.6% ee.
Pure 1b (4.25 g, 85.0%) was obtained by silica-gel chromatography (Hexane–AcOEt = 2:1, Rf 0.66) as a white solid.
Mp: 92–94 °C.
1H NMR (CDCl3, 500 MHz, TMS): δ 1.22–1.35 (4H, m), 1.40–1.85 (6H, m), 2.15–2.25 (1H, m), 2.80–2.95 (1H, m), 4.10–4.40 (3H, m), 6.56 (1H, t, J = 73.4 Hz), 7.15 (2H, d, J = 8.8 Hz), 7.32 (1H, d, J = 8.5 Hz), 7.79 (2H, d, J = 8.8 Hz) ppm.
13C NMR (CDCl3, 125 MHz, TMS): δ 14.2, 22.4, 24.3, 27.5, 44.3, 48.5, 60.7, 115.5 (t, J = 259 Hz), 119.1, 128.8, 131.9, 153.4, 165.2, 174.5 ppm.
IR (ATR, cm−1): 3372, 2945, 2859, 1715, 1628, 1611, 1537, 1503, 1454, 1406, 1335, 1317, 1240, 1223, 1198, 1180, 1119, 1047, 1020, 845, 829, 766, 685.
Anal. calcd for C17H21NO4F2: C, 59.82; H, 6.20; N, 4.10; F, 11.13%; found: C, 59.95; H, 6.30; N, 3.99; F, 11.12%.
HRMS: calcd for C17H21NO4F2 (M + H)+ = 342.1511, found: 342.1491.
(1S,2R)-2-[N-{4-(Difluoromethoxy)benzoyl}]aminocyclo-hexane-1-carboxylic acid (1c)
To a 100 mL three-necked flask was added crude 1b (14 g, 41.0 mmol, 94.8% de, 88.7% ee) and EtOH (42 mL). The mixture was heated at 40 °C. 5 M NaOH aq. (9 mL, 45 mmol) was added, and the reaction mixture was stirred at 40 °C for 2.5 h. After cooling to 25 °C, H2O (89 mL), toluene (28 mL) and n-heptane (28 mL) were added. The reaction mixture was stirred for 10 min. The water layer was collected and washed with a mixture of toluene (28 mL) and n-heptane (28 mL). To the water layer was added MeCN (14 mL), 6 M HCl aq. and H2O (47 mL) for crystallization. The mixture was stirred at 25 °C for 1 h. The resultant crystals were collected by filtration, washed with H2O (70 mL), and dried under reduced pressure at 60 °C to afford the crude titled compound 1c (10.8 g, 82.5%) as a light-purple solid.The crude compound obtained above (10 g, 31.9 mmol) was recrystallized with MeCN (80 mL)–H2O (120 mL) to afford the titled compound 1c (8.3 g, 83.0%) as a light-grey solid.
Mp: 157–160 °C.
>99.9% de, >99.9% ee.
1H NMR (CDCl3, 500 MHz, TMS): δ 1.30–1.42 (1H, m), 1.43–1.54 (1H, m), 1.55–1.64 (1H, m), 1.67–1.85 (4H, m), 2.17–2.27 (1H, m), 2.96–3.02 (1H, m), 4.30–4.40 (1H, m), 6.55 (1H, t, J = 73.5 Hz), 7.14 (2H, d, J = 8.6 Hz), 7.22 (1H, d, J = 9.2 Hz), 7.77 (2H, d, J = 8.5 Hz) ppm.
13C NMR (CDCl3, 125 MHz, TMS): δ 22.3, 24.4, 27.5, 29.3, 44.1, 48.4, 115.4 (t, J = 260 Hz), 119.1, 128.9, 131.5, 153.5, 165.6, 178.6 ppm.
IR (ATR, cm−1): 3360, 3300–2800, 2938, 2868, 1701, 1608, 1580, 1528, 1501, 1391, 1229, 1179, 1107, 1057, 1015, 885, 839, 768, 714, 683, 664.
Anal. calcd for C15H17NO4F2: C, 57.50; H, 5.47; N, 4.47; F, 12.13%; found: C, 57.52; H, 5.56; N, 4.41; F, 12.13%.
HRMS: calcd for C15H17NO4F2 (M + H)+ = 314.1198, found: 314.1214.
(1S,2R)-2-[N-{4-(Difluoromethoxy)benzoyl}]aminocyclo-hexane-1-carboxylic acid (1c) (multi-kilogram scale)
To a 500 L pressure-resistant reactor were added 6b (18.0 kg, 53.06 mol), [Ru{(S)-dtbm-binap}(OAc)2] (467.8 g, 0.3316 mol), dehydrated MeOH (118 L), and 42% aqueous HBF4 (138.7 g, 0.6632 mol). H2 (0.85 MPa) was purged into the reactor, and stirred for 42 h 45 min by keeping the H2 pressure (0.85–0.90 MPa) at 45 °C. After H2 absorption was stopped, the H2 pressure was released. The HPLC analysis of the reaction mixture revealed that the conversion, de, and ee were 100%, 96.2% de, and 91.7% ee, respectively. The reaction mixture was transferred to a 300 L reactor and concentrated to afford the crude hydrogenated ester. To the mixture was added EtOH (54 L) and 5 M NaOH aq. (11.7 L, 58.5 mol). The reaction mixture was stirred for 2 h at 35–40 °C. After cooling to 25 °C, toluene (36 L), n-heptane (36 L) and H2O (114 L) were added, and the mixture was stirred for 10 min. The water layer was collected, and washed with a mixture of toluene (36 L) and n-heptane (36 L). To the water layer was added MeCN (18 L), 6 M HCl aq. (10.2 L, 61.2 mol) and H2O (61 L) for crystallization at 10 °C. The mixture was stirred overnight at 10 °C. The resultant crystals were centrifuged, and washed with H2O (80 L) to afford the crude, wet titled compound 1c (16.90 kg) as a light-purple solid.To the 300 L reactor was added the wet crude solid obtained above (16.75 kg), MeCN (86 L) and H2O (14 L). The mixture was heated to 48 °C to dissolve the crude solid. Active charcoal (1.8 kg) was added, and then the mixture was stirred for 10 min. The insoluble fraction was filtered off, and washed with MeCN–H2O (2:1, 20 L). Both filtrates were combined, and heated to 35–40 °C. H2O (72 L) and seed crystal (3.6 g) were added, then the mixture was stirred for 30 min (crystallization occurred). H2O (72 L) was added, and the slurry was stirred for 30 min at 35–40 °C, overnight at 25 °C, then 1 h at 10–20 °C. The slurry was centrifuged, washed with H2O (67 L), and dried under reduced pressure at 60 °C to afford the titled compound 1c (12.36 kg, 74.4% (2 steps)) as a light-pink solid.
HPLC (GL Science Inertsil ODS-3, 220 nm, 25 °C, 1.0 mL min−1, 50 mM KH2PO4:MeCN = 6:4).
t R: 7.3 min (99.9 area%).
HPLC (DAICEL CHIRALPAK AD-RH, 254 nm, 40 °C, 0.2 mL min−1, 50 mM KH2PO4:MeCN = 7:3).
t R: 27.4 min (99.9 area%) >99.9% de, >99.9% ee.
Anal. calcd for C15H17NO4F2: C, 57.50; H, 5.47; N, 4.47; F, 12.13%; found: C, 57.52; H, 5.45; N, 4.45; F, 12.12%.
References
- (a) T. Kanamaru, S. Shinagawa, M. Asai, H. Okazaki, Y. Sugiyama, T. Fujita, H. Iwatsuka and M. Yoneda, Life Sci., 1985, 37, 217 CrossRef CAS; (b) P. Crews, L. V. Manes and M. Boehler, Tetrahedron Lett., 1986, 27, 2797 CrossRef CAS; (c) S. Shinagawa, T. Kanamaru, S. Harada, M. Asai and H. Okazaki, J. Med. Chem., 1987, 30, 1458 CrossRef CAS; (d) M. Namikoshi, K. L. Rinehart, A. M. Dahlem, V. R. Beasley and W. W. Carmichael, Tetrahedron Lett., 1989, 30, 4349 CrossRef CAS; (e) D. C. Cole, Tetrahedron, 1994, 50, 9517 CrossRef CAS; (f) C. Shih, L. S. Gossett, J. M. Gruber, C. S. Grossman, S. L. Andis, R. M. Schultz, J. F. Worzalla, T. H. Corbett and J. T. Metz, Bioorg. Med. Chem. Lett., 1999, 9, 69 CrossRef CAS; (g) R. Roers and G. L. Verdine, Tetrahedron Lett., 2001, 42, 3563 CrossRef CAS; (h) J. H. Cohen, A. F. Abdel-Magid, H. R. Almond Jr and C. A. Maryanoff, Tetrahedron Lett., 2002, 43, 1977 CrossRef CAS; (i) M. Liu and M. P. Sibi, Tetrahedron, 2002, 58, 7991 CrossRef CAS; (j) A. K. Awasthi, M. L. Boys, K. J. Cain-Janicki, P. J. Colson, W. W. Doubleday, J. E. Duran and P. N. Farid, J. Org. Chem., 2005, 70, 5387 CrossRef CAS.
- (a) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015 RSC; (b) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS; (c) D. Seebach, S. Abele, K. Gademann, G. Guichard, T. Hintermann, B. Jaun, J. L. Matthews and J. V. Schreiber, Helv. Chim. Acta, 1998, 81, 932 CrossRef CAS.
- (a) E. Forró and F. Fülöp, Szegedi Tudomanyegyetem, PCT Pat. Appl., PCT/HU, 000006, 2007 Search PubMed; (b) E. Forró and F. Fülöp, Chem.–Eur. J., 2007, 13, 6397 CrossRef.
- (a) C. Cimarelli and G. Palmieri, J. Org. Chem., 1996, 61, 5557 CrossRef CAS; (b) C. Cimarelli, G. Palmieri and E. Volpini, Synth. Commun., 2001, 31, 2943 CrossRef CAS; (c) J. Matsuo, M. Okano, K. Takeuchi, H. Tanaka and H. Ishibashi, Tetrahedron: Asymmetry, 2007, 18, 1906 CrossRef CAS.
- (a) D. A. Dobbs, K. P. M. Vanhessche, E. Brazi, V. Rautenstrauch, J. Lenoir, J. Genêt, J. Wiles and S. H. Bergens, Angew. Chem., Int. Ed., 2000, 39, 1992 CrossRef CAS; (b) P. Dupau, C. Bruneau and P. H. Dixneuf, Adv. Synth. Catal., 2001, 343, 331 CrossRef CAS.
- (a) W. Tang, S. Wu and X. Zhang, J. Am. Chem. Soc., 2003, 125, 9570 CrossRef CAS; (b) W. Tang and X. Zhang, The Penn State Research Foundation, US Pat., 7,214,813, 2004 Search PubMed.
- H. Wu and G. Hoge, Org. Lett., 2004, 6, 3645 CrossRef CAS.
- S. Enthaler, G. Erre, K. Junge, J. Holz, A. Börner, E. Alberico, I. Nieddu, S. Gladiali and M. Beller, Org. Process Res. Dev., 2007, 11, 568 CrossRef CAS.
- (a) Jpn. Kokai Tokkyo Koho, JPS58-24545; (b) Jpn. Kokai Tokkyo Koho, JPS63-54341; (c) Jpn. Kokai Tokkyo Koho, JPS63-54342; (d) H. Nohira, M. Nohira, S. Yoshida, A. Osada and D. Terunuma, Bull. Chem. Soc. Jpn., 1988, 61, 1395 CrossRef CAS.
- (a) J. Duan, G. Ott, L. Chen, Z. Lu, T. P. Maduskuie Jr, M. E. Voss and C. -B. Xue, Du Pont Pharmaceuticals Company, PCT Pat. Appl., PCT/US, 08334, 2001 Search PubMed; (b) J. X. Qiao, Bristol-Myers Squibb Company, PCT Pat. Appl., PCT/US, 032990, 2004 Search PubMed; (c) J. J.-W. Duan, L. Chen, Z. Lu, C.-B. Xue, R.-Q. Liu, M. B. Covington, M. Qian, Z. R. Wasserman, K. Vaddi, D. D. Christ, J. M. Trzaskos, R. C. Newton and C. P. Decicco, Bioorg. Med. Chem. Lett., 2008, 18, 241 CrossRef CAS; (d) G. R. Ott, N. Asakawa, Z. Lu, R.-Q. Liu, M. B. Covington, K. Vaddi, M. Qian, R. C. Newton, D. D. Christ, J. M. Traskos, C. P. Decicco and J. J.-W. Duan, Bioorg. Med. Chem. Lett., 2008, 18, 694 CrossRef CAS; (e) G. R. Ott, N. Asakawa, Z. Lu, R. Anand, R.-Q. Liu, M. B. Covington, K. Vaddi, M. Qian, R. C. Newton, D. D. Christ, J. M. Trzaskos and J. J.-W. Duan, Bioorg. Med. Chem. Lett., 2008, 18, 1577 CrossRef CAS; (f) K. Miura and Y. Nishikimi, Takeda Pharmaceutical Company limited, PCT Pat. Appl., PCT/JP, 060616, 2008 Search PubMed; (g) K. Miura and Y. Nishikimi, Takeda Pharmaceutical Company limited, PCT Pat. Appl., PCT/JP, 069799, 2010 Search PubMed.
- (a) F. Folöp, G. Bemáth, G. Argay, A. Kálmán and P. Sohár, Tetrahedron, 1984, 40, 2053 CrossRef; (b) J. B. Santella, J. Hynes and D. S. Gardner, Bristol-Myers Squibb Company , PCT Pat. Appl., PCT/US, 070804, 2008 Search PubMed.
- W. L. F. Armarego and T. Kobayashi, J. Chem. Soc. C, 1970, 1597 RSC.
- (a) S. Krusekopf, I. Roots, A. G. Hildebrandt and U. Kleeberg, Xenobiotica, 2003, 33, 107 CrossRef CAS; (b) D. B. Horne, M. D. Bartberger, M. R. Kaller, H. Monenschein, W. Zhong and S. A. Hitchcock, Tetrahedron Lett., 2009, 50, 5452 CrossRef CAS; (c) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (d) B.-M. Swahn, J. Holenz, J. Kihlström, K. Kolmodin, J. Lindström, N. Plobeck, D. Rotticci, F. Sehgelmeble, M. Sundström, S. von Berg, J. Fälting, B. Georgievska, S. Gustavsson, J. Neelissen, M. Ek, L. Olsson and S. Berg, Bioorg. Med. Chem. Lett., 2012, 22, 1854 CrossRef CAS.
- (a) M. Goto, M. Yamano and S. Kawaguchi, Takeda Pharmaceutical Company limited, PCT Pat. Appl., PCT/JP, 015536, 2003 Search PubMed; (b) M. Goto, T. Konishi, S. Kawaguchi, M. Yamada, T. Nagata and M. Yamano, Org. Process Res. Dev., 2011, 15, 1178 CAS.
- Y. Kondo and Y. Nishikimi, Takeda Pharmaceutical Company limited, PCT Pat. Appl., PCT/JP, 005005, 2009 Search PubMed.
- (a) W. L. F. Armarego and T. Kobayashi, J. Chem. Soc. C, 1971, 3222 RSC; (b) R. T. Buckler, H. E. Hartzler and B. M. Phillips, J. Med. Chem., 1975, 18, 509 CrossRef CAS.
- (a) K. Mashima, K. Kusano, N. Sato, Y. Matsumura, K. Nozaki, H. Kumobayashi, N. Sayo, Y. Hori, T. Ishizaki, S. Akutagawa and H. Takaya, J. Org. Chem., 1994, 59, 3064 CrossRef CAS; (b) T. V. RajanBabu, T. A. Ayers and A. L. Casalnuovo, J. Am. Chem. Soc., 1994, 116, 4101 CrossRef CAS; (c) G. Trabesinger, A. Albinati, N. Feiken, R. W. Kunz, P. S. Pregosin and M. Tschoerner, J. Am. Chem. Soc., 1997, 119, 6315 CrossRef CAS; (d) P. Dotta, P. G. Anil Kumar and P. S. Pregosin, Organometallics, 2004, 23, 2295 CrossRef CAS.
- Y. Okeda, T. Hashimoto, Y. Hori and T. Hagiwara, Tagasago International Corporation, Eur. Pat. Appl., EP, 0955303, 1999 Search PubMed.
- M. Yamano, M. Goto, S. Kawaguchi, M. Yamada and J. Kawakami, Takeda Pharmaceutical Company limited, PCT Pat. Appl., PCT/JP, 319095, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20307c |
| This journal is © The Royal Society of Chemistry 2012 |