Oxidation of anisole over MCM-48 materials modified by incorporation of transition and inner transition metals†
Banani
Kalita
,
Prodeep
Phukan
and
Anup K.
Talukdar
*
Department of Chemistry, Gauhati University, Guwahati-781014, Assam, India. E-mail: anup_t@sify.com; Tel: +91 (0)9864073504
First published on 13th June 2012
Abstract
Aluminosilicates with well-ordered MCM-48 mesostructures were synthesized by one step and two step methods. Single step method was found to be appropriate for metal incorporation. This method was applied to synthesize transition and inner transition metal incorporated MCM-48. These modified MCM-48 materials were characterized by a variety of techniques including XRD, FTIR, DRS-UV-Vis, TGA, N2 adsorption and SEM. Post-synthesis modification of the SiMCM-48 material was also done by impregnating 1%, 3% and 5% vanadium without compromising the integrity of the cubic mesostructure. The transition and inner transition metal incorporated MCM-48 materials were tested for oxidation reaction and were found to exhibit far better activities in anisole oxidation than SiMCM-48. Among the metal introduced MCM-48, vanadium–MCM-48 samples showed highest activities.
1. Introduction
In the early 1990s a class of mesoporous materials, M41S, was first synthesized by Kresge et al. at Mobil Oil Corporation.1 Since then a number of synthesis routes and mechanisms have been proposed for the formation of different mesoporous materials. Synthetic parameters like gel composition, silica source, temperature, aging time, pH etc. assist in manipulating synthesis time for the formation of mesoporous materials. In the past, several research groups have reported different ways to synthesize MCM-48, which is one of the important members of the M41S family.2–6Defect-free mesoporous siliceous molecular sieves with an SiO2 framework are electrically neutral, leading to a lack of strong surface acidity. The M41S materials have high potential for becoming promising catalysts for size and shape selective acid-catalyzed reactions in the petrochemical and fine chemical industries,7 provided the materials are modified to generate oxidation and acid functionality in their structure. Although among the members of the M41S family cubic MCM-48 is preferred over the hexagonal MCM-41 in catalytic applications because of the three-dimensional pore arrangements of the former, one of the reasons for the limited reports on the modification of MCM-48 may be due to the difficulty of its synthesis. There has been a regular approach on improvement in structural and hydrothermal stability of mesostructured MCM-48 materials in the last few years.8–13 In contrast, very few reports are available about the modification of the material for introduction of acid and oxidation functionality.11,12,14–17 Although introducing transition metals into the framework of cubic MCM-48 materials seemed appealing, the attempts so far have been unsystematic in nature. Kao et al.14 reported the preparation of cubic mesoporous aluminosilicate MCM-48 via the assembly of the preformed zeolite precursor and the Gemini surfactant. It required several steps and in total 56 h time duration until final crystallization. The reproducible method, claimed by Campelo et al.,15 used hydroxyl-exchanged cetyl-trimethylammonium bromide (CTAB) to synthesize Al-MCM-48 and NH4F solution (post-synthesis treatment) to improve the quality and acidity of the material is a two step procedure. Other procedures have also been found to be either two step and too lengthy11,17 or post-synthesis grafting.12 Moreover, the procedures were not systematically tested for incorporation of other metals. To modify the composition of the inorganic walls of MCM-48 mesoporous materials, several attempts were made to homogeneous incorporation of transition metal atoms as well as main group elements (Fe, Ti, V, Al, B, Ga etc.) into the framework structure. Metals such as Ti,18–20 Mn,21–24 Cr20,21,25–27 and V20,21,26,28,29 have been reported to have been introduced into M41S mesoporous silica using different synthetic procedures like direct hydrothermal method, wet impregnation, grafting, template ion exchange method etc. Substituting silicon by trivalent cations such as B3+, Ga3+, Al3+ and Fe3+ in the mesoporous silica wall30,31 results a negative framework charge, which can be compensated by protons providing catalytically active acid sites.
In the present work, the synthesis of cubic mesoporous aluminosilicate MCM-48 has been systematically approached through one step and two step procedures and once successful through one step procedure, the same has been applied for incorporation of transition and inner transition metals to MCM-48. The one step procedure is very simple and successful for introduction of acid and oxidation functionality to the material. The catalytic activity of the MCM-48 materials modified by incorporating the transition metals vanadium and zirconium and inner transition metal cerium in anisole oxidation has also been investigated.
2. Experimental
2.1 Synthesis of SiMCM-48 materials
Two synthesis procedures have been tried for the formation of MCM-48 materials in the present study.Step 1: Preparation of surfactant solution. Calculated amount of CTAB was dissolved in 91 mL of deionized water (10 wt%) and to that solution 7.7 g of ethanol was added. The mixture was stirred for 15 min.
Step 2: Preparation of gel. 5 g of fumed silica was added with constant stirring to a NaOH solution. The gel was then stirred for 1 h.
The prepared gel was added slowly with constant stirring to the surfactant solution. Finally, the whole mixture was stirred at pH 9.6 for another 2 h to get a homogeneous gel. The mixture was transferred to an autoclave and heated at 373 K for 3 h and then at 423 K for another 3 h inside an oven. After complete crystallization, the autoclave was quenched and the resultant solid product was separated from mother liquor by centrifugation. The product was first washed with warm water and then repeatedly washed with de-ionized water. The resulting solid product was dried at room temperature overnight and at 383 K for 6 h. For the purpose of removing the surfactant, the sample was calcined in a programmable furnace at 813 K for 6 h with a heating rate of 3 K min−1.
The molar gel composition of the synthesis system was SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Na2O:CTAB:EtOH:H2O = 1:0.21:0.30:4:100
:Na2O:CTAB:EtOH:H2O = 1:0.21:0.30:4:100
After 2 h stirring, the mixture was autoclaved at 373 K for 2 h. The autoclave was quenched and the resultant solid product was separated from mother liquor by centrifugation. Both the products were first washed with warm water and then repeatedly with de-ionized water. The resulting solid products were dried at room temperature overnight and at 383 K for 6 h. For the purpose of removing the surfactant, the samples were calcined in a programmable furnace at 813 K for 6 h with a heating rate of 3 K min−1.
The molar gel composition was TEOS:NH3:EtOH:CTAB:H2O = 1:13:56:0.4:405
2.2 Modification of MCM-48 materials
Synthesis of metal introduced MCM-48. Synthesis of aluminium doped MCM-48 was tried by the above two methods using Al2(SO4)3·18H2O as the aluminium source. After several experiments it was found that synthesis of AlMCM-48 material in pure phase was better by the single step method when Al2(SO4)3·18H2O was added with constant stirring after addition of TEOS. The final mixture was continuously stirred for 2 h and then autoclaved at 373 K for 2 h. The mixture was quenched and the resultant solid product was separated from mother liquor by centrifugation. The products were first washed with warm water and then repeatedly with de-ionized water. The resulting solid products were dried at room temperature overnight and at 383 K for 6 h. For the purpose of removing the surfactant, the samples were calcined in a programmable furnace at 813 K for 6 h with a heating rate of 3 K min−1.
The molar gel composition was TEOS:NH3:EtOH:CTAB:H2O = 1:13:56:0.4:405 and Si/Al molar ratio was 100.
After successful synthesis of AlMCM-48 by the single step method, two samples each of cerium and vanadium doped with Si/metal molar ratios of 100 and 40 and one sample of zirconium incorporated MCM-48 material with Si/Zr molar ratio of 40 was synthesized by this method. The sources of metals were ammonium ceric nitrate, vanadium acetyl acetonate and zirconium nitrate for Ce, V and Zr, respectively. The designation, method details and molar gel composition of in situ and post-synthesis modified MCM-48 samples are given in Table 1.
| Sample | Details | Molar gel composition |
|---|---|---|
| SiMCM-48 (two step) | SiMCM-48 synthesized by two step method | SiO2:Na2O:CTAB:EtOH:H2O = 1:0.21:0.30:4:100 |
| SiMCM-48 | MCM-48 in pure silicon system synthesized by single step method | SiO2:Na2O:CTAB:EtOH:H2O = 1:0.21:0.30:4:100 |
| AlMCM-48 | Al doped MCM-48 with Si/Al molar ratio 100 | TEOS:Al:NH3:EtOH:CTAB:H2O = 1:100:13:56:0.4:405. |
| CeMCM-8(Si/Ce 100) | Ce doped MCM-48 with Si/Ce molar ratio 100 | TEOS:Ce:NH3:EtOH:CTAB:H2O = 1:100:13:56:0.4:405 |
| CeMCM-48(Si/Ce 40) | Ce doped MCM-48 with Si/Ce molar ratio 40 | TEOS:Ce:NH3:EtOH:CTAB:H2O = 1:40:13:56:0.4:405 |
| VMCM-48(Si/V 100) | V doped MCM-48 with Si/V molar ratio 100 | TEOS:V:NH3:EtOH:CTAB:H2O = 1:100:13:56:0.4:405 |
| VMCM-48 (Si/V 40) | V doped MCM-48 with Si/V molar ratio 40 | TEOS:V:NH3:EtOH:CTAB:H2O = 1:40:13:56:0.4:405 |
| ZrMCM-48(Si/Zr 40) | Zr doped MCM-48 with Si/Zr molar ratio 40 | TEOS:Zr:NH3:EtOH:CTAB:H2O = 1:40:13:56:0.4:405 |
| 1%VimpSiMCM-48 | SiMCM-48 with 1% vanadium oxide loading by impregnation method | SiO2:Na2O:CTAB:EtOH:H2O = 1:0.21:0.30:4:100 (1% vanadium) |
| 3%VimpSiMCM-48 | SiMCM-48 with 3% vanadium oxide loading by impregnation method | SiO2:Na2O:CTAB:EtOH:H2O = 1:0.21:0.30:4:100 (3% vanadium) |
| 5%VimpSiMCM-48 | SiMCM-48 with 5% vanadium oxide loading by impregnation method | SiO2:Na2O:CTAB:EtOH:H2O = 1:0.21:0.30:4:100 (5% vanadium) |
Vanadium oxide impregnated MCM-48 materials. In a typical wet impregnation procedure, 1 g of calcined dry SiMCM-48 was added to a solution of 0.0383 g, 0.1149 g or 0.1915 g vanadium acetyl acetonate in 10 mL ethanol to obtain 1%, 3%, 5% vanadium oxide impregnated MCM-48 samples, respectively. The mixture was stirred for 30 min and kept at room temperature until it became dry. The impregnated samples were again dried at 393 K and calcined at 673 K in a programmable furnace with a heating rate of 3 K min−1.
2.3 Characterization of the materials
Low angle X-ray powder diffraction (XRD) patterns were obtained for the identification of mesophase of modified MCM-48 samples using a X'PERT PRO (Philips) diffractometer with Cu-Kα radiation of wavelength 1.54056 Å operated at voltage 40 kV and current 30 mA. The XRD data were collected in the 2θ range of 1.5–10°. A Perkin Elmer RX1 FT-IR spectrophotometer was used for recording the FT-IR spectra of the MCM-48 samples in the form of KBr pellets in mid-IR region of 4000–450 cm−1 (resolution 8 cm−1). Scanning electron micrographs (SEM) of the MCM-48 samples were taken for investigation of the particle morphology using a Jeol JSM-6360 scanning electron microscope. The N2 adsorption–desorption isotherms of the samples were obtained using nitrogen as adsorbate at 77.15 K on a Micromeritics Tristar 3000 analyzer. For this purpose, previously calcined samples were outgassed at 383 K for 3 h and gradually heated to 623 K in a continuous flow of N2. The system was maintained at that temperature for 12 h. The samples were then slowly cooled down to 383 K under N2 atmosphere and the anhydrous weight of the sample was taken prior to N2 adsorption–desorption measurement. Finally, the samples were chilled to 77.15 K using liquid nitrogen and the adsorption of nitrogen was carried out at different equilibrium pressures. Thermal gravimetric analysis (TGA) and differential thermal gravimetry (DTG) of parent and modified uncalcined MCM-48 samples were done on Mettler Toledo TGA/DSC 1, STARe System analyzer in the temperature range 313–1023 K with heating rate of 7 K min−1 in N2 gas atmosphere. The diffuse reflectance UV-Vis spectra (DRS) were recorded in the range 200–800 nm with a Hitachi 4100 spectrometer equipped with a diffuse reflectance attachment using solid sample holder. The base line correction was made using barium sulfate as the reference standard.2.4 Catalytic reaction
The reactions were carried out in glass batch reactor under atmospheric pressure. The temperature was controlled by a temperature controller and stirring was done by a magnetic stirrer. In a typical reaction, X mole of the substrate and Y mole of H2O2 (30% w/v) were reacted over the W g of the catalyst in the presence of solvent acetonitrile under continuous stirring. The products along with the unreacted reactants, if any, of the reaction were collected at various time intervals and analyzed by gas chromatography (Perkin Elmer, Clarus 500, Elite 501 column) to calculate the percentage of conversion and selectivity of the products.3. Results and discussion
3.1 X-Ray diffraction
The XRD patterns of SiMCM-48 samples synthesized by both one step and two step methods (Fig. A in the ESI†) are in good agreement with those reported for the cubic MCM-48 materials and the d spacings are compatible with the cubic Ia3d space group.1,32 The most distinguishable peak along (2 1 1) plane appears at 2θ = ∼2.7° and a distinct shoulder peak appear at 2θ = ∼3° which is attributed to (2 2 0) reflection. The other low intense diffraction peaks appear between 4° and 6°. The value of the ratio d220/d211 is found to be approximately 0.86 (Table 2), which is another characteristic of cubic symmetry.1,33| Sample | d spacing (Å) | d(220)/d(211) | a o (Å) |
|---|---|---|---|
| SiMCM-48 | 32.35 | 0.866 | 79.22 |
| AlMCM-48 | 33.04 | 0.857 | 80.93 |
| CeMCM-48(Si/Ce 100) | 34.19 | 0.866 | 83.83 |
| CeMCM-48(Si/Ce 40) | 36.47 | 0.862 | 89.32 |
| VMCM-48(Si/V 100) | 33.34 | 0.861 | 81.65 |
| VMCM-48(Si/V 40) | 34.44 | 0.854 | 84.34 |
| ZrMCM-48(Si/Zr 40) | 35.33 | 0.825 | 86.52 |
The cubic unit cell parameter ao is calculated by the formula ao = d(h2 + k2 + l2)1/2 (d = interplanar spacing) as calculated by Bragg's law. The d spacings corresponding to (2 1 1) reflection and the values of cubic unit cell constant (ao) of the samples [SiMCM-48, CeMCM-48(Si/Ce 40), CeMCM-48(Si/Ce 100), VMCM-48(Si/V 40), VMCM-48(Si/V 100), ZrMCM-48(Si/Zr 40)] are given in Table 2. The XRD patterns of Al incorporated MCM-48 samples synthesized by both two step and one step methods are shown in Fig. 1. The structural quality of the aluminosilicate MCM-48 samples as evaluated by powder XRD was found to be excellent, showing integrity of the cubic mesostructure when single step was employed for synthesis. The samples herein exhibited a more intense (2 2 0) diffraction peak than those obtained by Kao et al.,14 Li et al.34 and Shih et al.35 who have reported preparation of stable cubic mesostructure from zeolite and claimed to obtain better results than Al–MCM-48 samples prepared by direct Al incorporation. Although, in the last case at high Si/Al ratios the materials exhibited excellent XRD ordering but the (2 2 0) diffraction peak was absent for the samples with lower Si/Al ratio. The unit cell parameter (ao) is found to slightly increase in AlMCM-48 as compared to SiMCM-48 (Table 2). The expansion in the unit cell with incorporation of aluminum is caused by the Al–O bond being longer (1.75 Å) than the Si–O bond (1.60 Å).
 | ||
| Fig. 1 XRD pattern of (a) AlMCM-48 (two step), (b) AlMCM-48 (one step). | ||
The powder XRD patterns of Ce incorporated MCM-48 samples with Si/Ce molar ratio 100 and 40 have shown four main Bragg peaks corresponding to hkl = (2 1 1), (2 2 0), (4 2 0), (3 3 2) which are the characteristic peaks of cubic MCM-48 phase (Fig. B in the ESI†). The XRD results of CeMCM-48(Si/Ce 100) and CeMCM-48(Si/Ce 40) have shown that the d values and cubic unit cell parameters (ao) gradually increase with an increase of Ce content in the framework structure (Table 2). These results are in accordance with the earlier reports for the heteroatom substituted silicate framework.36–38 As the radii of Ce4+ cation (87 pm, 6-coordinate) is larger than that of Si4+ (26 pm, 4-coordinate; 40 pm, 6-coordinate), unit cell parameters are expected to increase when Ce cations are incorporated into the framework structure of MCM-48 materials. Furthermore, in case of Ce incorporated MCM-48 samples, the main diffraction peaks corresponding to (2 1 1) and (2 2 0) reflections shifted to lower 2θ value as compared to SiMCM-48 sample.
The XRD pattern of vanadium incorporated MCM-48 samples with Si/V molar ratio 100 and 40 consist of characteristic Bragg peaks which verify the presence of cubic phase (Ia3d) (Fig. C in the ESI†). The crystallographic XRD data analysis of V incorporated MCM-48 samples show similar changes in the d values and the unit cell parameters with the increase of V content as explained above in case of Ce incorporated MCM-48 samples. The radii of V4+ cation (59 pm) is also larger than that of Si4+ (40 pm). In case of V incorporated MCM-48 samples, the main diffraction peaks corresponding to (2 1 1) and (2 2 0) reflections are also shifted to lower 2θ value as compared to the SiMCM-48 sample. A similar trend in the XRD results is observed in case of Zr doped MCM-48 samples. The XRD pattern of Zr doped MCM-48 sample with Si/Zr molar ratio 40 is shown in Fig. D of the ESI.† A significant change in the values of unit cell constant is also observed for Ce, V and Zr doped MCM-48 samples. The unit cell parameter increases in the order VMCM-48(Si/V 40) < ZrMCM-48(Si/Zr 40) < CeMCM-48(Si/Ce 40). This may be because of the fact that the values of cationic radii also increase in the order V4+ (59 pm) < Zr4+ (72 pm) < Ce4+ (87 pm). Consequently, the crystallographic data analysis from XRD results confirms that Ce, V and Zr atoms have been successfully incorporated into the framework structure of MCM-48 materials by the single step method.
3.2 FT-IR spectroscopy
The FT-IR spectra of SiMCM-48, CeMCM-48(Si/Ce 40), VMCM-48(Si/V 40) and ZrMCM-48(Si/Zr 40) (Fig. E in the ESI†) shows that a broad band appears at ca. 3468 cm−1 which is ascribed to water. The weak band at 1630 cm−1 corresponds to the bending mode of water.39 The broad band at ca. 1084 cm−1 with a shoulder at 1234 cm−1, and the band at 810 cm−1 are attributed to Si–O–Si asymmetric stretching and Si–O–Si bending vibration respectively.37,40,41All IR spectra exhibit one common feature, i.e. a band at ∼960 cm−1. Some authors have considered this as a proof for the incorporation of the heteroatom into the framework.42,43 In our studies, the intensity of this band marginally increases with increase in the amount of metal ions. Camblor et al.42 have proposed that the band at 960 cm−1 is due to the Si–O stretching vibrations of Si–OH groups present at defect sites. This vibration has also been detected in Ti and V containing silica molecular sieves.44
The FT-IR spectra of vanadium oxide loaded MCM-48 samples (1%VimpSiMCM-48, 3%VimpSiMCM-48, 5%VimpSiMCM-48) are shown in Fig. F in the ESI.†
3.3 Thermogravimetric analysis
The TGA plots of MCM-48 samples are given in Fig. 2. The TGA curves of samples SiMCM-48 (one step), SiMCM-48 (two steps), CeMCM-48(Si/Ce 40), VMCM-48(Si/V 100) and CeMCM-48(Si/Ce 100) showed four steps of weight loss in the temperature ranges 313–413 K, 413–533 K, 533–653 K and 653–1023 K for the first four samples and for CeMCM-48(Si/Ce 100) sample in the temperature ranges 313–413 K, 413–533 K, 533–603 K and 603–1023 K. Samples AlMCM-48, VMCM-48(Si/V 40) and ZrMCM-48(Si/Zr 40) showed five steps of weight loss. The temperature ranges are 313–413 K, 413–483 K, 483–603 K, 603–653 K and 653–1023 K for AlMCM-48, 313–413 K, 413–533 K, 533–653 K, 653–753 K and 753–1023 K for VMCM-48 (Si/V 40) and 313–413 K, 413–533 K, 533–653 K, 653–773 K and 773–1023 K for ZrMCM-48(Si/Zr 40). It is clear from the TGA analysis results reported in Table 3 that ZrMCM-48(Si/Zr 40) has the highest total weight loss (64.8%), whereas AlMCM-48 has the lowest total weight loss (25.4%) when heated up to 1023 K. | ||
| Fig. 2 TGA/DTG plot of MCM-48 samples. | ||
| Samples | Mass percentage loss | ||||
|---|---|---|---|---|---|
| 313–413 K | 413–533 K | 533–653 K | 653–1023 K | Total | |
| SiMCM-48 (two steps) | 3.2 | 37.2 | 14.1 | 8.3 | 62.8 |
| SiMCM-48 | 6.0 | 29.2 | 16.5 | 5.2 | 56.9 |
| AlMCM-48 | 2.1 | 5.0 (413–483 K) | 10.5 (483–603 K) | 6.0 | 25.4 |
| 1.8 (603–653 K) | |||||
| CeMCM-48 (Si/Ce 100) | 3.2 | 28.2 | 5.0 (533–603 K) | 10.8 (603–1023 K) | 47.2 |
| CeMCM-48 (Si/Ce 40) | 3.2 | 27.2 | 11.3 | 7.3 | 49.0 |
| VMCM-48 (Si/V 100) | 4.2 | 26.1 | 16.2 | 8.2 | 54.7 |
| VMCM-48 (Si/V 40) | 5.5 | 28.1 | 16.2 | 3.4 (753–1023 K) | 56.9 |
| 3.7 (653–753 K) | |||||
| ZrMCM-48 (Si/Zr 40) | 4.2 | 30.4 | 16.1 | 8.1 (773–1023 K) | 64.8 |
| 6.0 (653–773 K) | |||||
The initial small weight loss in the range of 313–413 K is due to desorption of physically adsorbed water. The removal of organic surfactants is completed in two steps. This occurs in SiMCM-48 (both samples), CeMCM-48(Si/Ce 40) and VMCM-48(Si/V 100) in the ranges 413–533 K and 533–653 K and in CeMCM-48(Si/Ce 100) in the ranges 413–533 K and 533–603 K. However, this process completes in three steps for samples AlMCM-48, VMCM-48(Si/V 40) and ZrMCM-48(Si/Zr 40) e.g. in the temperature ranges of 413–483 K, 483–603 K and 603–653 K for AlMCM-48, and 413–533 K, 533–653 and 6533–753 K for VMCM-48(Si/V 40) and ZrMCM-48(Si/Zr 40).
The low-temperature weight loss in SiMCM-48 (both samples), CeMCM-48(Si/Ce 40), VMCM-48(Si/V 100) and CeMCM-48(Si/Ce 100) is assigned to the decomposition of CTAB surfactant occluded inside the channels. In addition to this low-temperature weight loss, the high-temperature weight loss observed in TGA of AlMCM-48 is assigned to the decomposition of protonated amines balancing the framework negative charge resulting from the incorporation the trivalent metal into MCM-48. From the TGA analysis, it is also observed that with an increase in level of metal content in the MCM-48 structure, the total weight loss also increases for the samples CeMCM-48(Si/Ce 40) and VMCM-48(Si/V 40).
3.4 UV-Vis DRS spectroscopy
The UV-Vis DRS spectra of CeMCM-48 samples (Fig. 3) show a single broad absorption band at ca. 300 nm. The band intensity increases with increase in Ce content for the CeMCM-48 materials. The absorption band position of ligand to metal charge transfer (O2− → Ce4+) depends on the ligand field symmetry surrounding the Ce center and the individual transitions are affected by different ligands, temperature, medium and so on.22,45 The electronic transitions from oxygen to cerium require higher energy for tetra-coordinated Ce4+ than for hexa-coordinated one. Therefore, the absorption band at near 310 nm for CeMCM-48 is due to the presence of the Ce4+ cations with tetra-coordination in the framework and the adsorption band at higher wavelength (405 nm) for CeO2 may be assigned to hexa-coordinated Ce4+ species. From the UV-Vis results of Ce doped MCM-48 samples, it may be inferred that the presence of strong absorption at 300 nm without the absorption bands at 405 nm indicates the presence of large amounts of Ce in the framework structure of MCM-48 materials (presumably in tetra-coordination).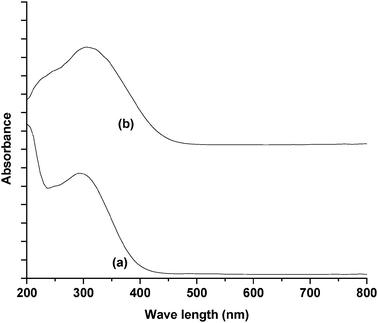
The UV-Vis DRS spectrum of the VMCM-48(Si/V 40) sample shows two bands in the region 250–390 nm (Fig. 4). According to Sen et al. the first band in the region 250–300 nm is characteristic of V4+ charge transfer band of VO2+ species and the band in the region 330–390 nm is due to charge transfer resulting from V5+ in Td environment.45

3.5 Adsorption measurement
Low-temperature N2 adsorption isotherms give the most reliable information about the mesoporous structure of solids. Fig. 5(a), (b), (c) and (d) show the amount of nitrogen physisorbed at 77 K versus the relative pressure for the samples SiMCM-48, CeMCM-48(Si/Ce 40), VMCM-48(Si/V 40), ZrMCM-48(Si/Zr 40) respectively. The isotherms are of type IV, typical of mesoporous solids.46 A well-defined step occurs between p/p0 = 0.2 and 0.3, which is indicative of the filling of the mesopores. The p/p0 coordinate of the inflection point depends on the pore size. The sharpness in this step suggests a uniform size pore system. The MCM-48 materials result from an excellent organization of the wall structure right across the particles and the cubic mesophase must be the predominant phase in the system, which can be seen in the nitrogen adsorption–desorption isotherm and Barrett–Joyner–Halenda (BJH) pore size distribution (calculated from the adsorption isotherm) for this samples. The extremely narrow pore size distribution also clearly indicates that the materials have a high order pore system. The absence of any detectable micropore filling at low p/p0 is confirmed by the fact that the initial region can be extrapolated back to the origin. | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms of (a) SiMCM-48, (b) CeMCM-48(Si/Ce 40), (c) VMCM-48(Si/V 40), (d) ZrMCM-48(Si/Zr 40). | ||
Pore size distribution (PSD) was obtained from the adsorption data by means of the BJH formula.47 The mesopore diameter (DBJH), cumulative pore volume (VBJH), and Brunauer–Emmett–Teller (BET) surface area (ABET) are listed in Table 4. The cumulative BJH desorption pore volumes are found in the range of 0.21–0.73 cm3 g−1. The BJH adsorption average pore diameters (Table 4) for the physisorption of N2 on MCM-48 samples are found to be 2.4, 2.7, 3.0 and 2.5 nm for SiMCM-48, CeMCM-48(Si/Ce 40), VMCM-48(Si/V 40), and ZrMCM-48(Si/Zr 40), respectively. The BET surface area of metal incorporated MCM-48 increases with ZrMCM-48 (Si/Zr 40) having highest value (1133.96 m2 g−1). The surface area values increase in the order of SiMCM-48 < VMCM-48 (Si/V 40) < CeMCM-48 (Si/Ce 40) < ZrMCM-48 (Si/Zr 40).
| Physical properties | Sample name | |||
|---|---|---|---|---|
| SiMCM-48 | CeMCM-48 (Si/Ce 40) | VMCM-48 (Si/V 40) | ZrMCM-48 (Si/Zr 40) | |
| BET surface area (m2 g−1) | 683.81 | 1105.31 | 715.21 | 1133.96 |
| BJH adsorption average pore diameter (nm) | 2.4 | 2.7 | 3.0 | 2.5 |
| BJH desorption cumulative pore volume (cm3 g−1) | 0.21 | 0.73 | 0.31 | 0.37 |
3.6 Scanning electron microscopy
The SEM images of MCM-48 samples are reported in Fig. 6. From the micrographs, it is clearly observed that all the particles are spherical in shape with size 0.3–0.6 μm. | ||
| Fig. 6 Scanning-electron micrographs of (a) SiMCM-48, (b) CeMCM-48(Si/Ce 40) (c) VMCM-48(Si/V 40), (d) ZrMCM-48(Si/Zr 40). | ||
3.7 Anisole oxidation reactions
 | ||
| Fig. 7 Effect of reaction time on conversion and selectivity in the anisole oxidation reaction using VMCM-48(Si/V 40); anisole:H2O2:acetonitrile:1:5:5 (molar ratio), temperature: 323 K, catalyst amount: 20 (w/w)% with respect to anisole. | ||
:5. The concentration of the catalyst in the reaction system was 20 (w/w)% with respect to anisole. No detectable activity was observed in the GC analysis for SiMCM-48 sample even for duration up to 6 h. The reaction was investigated under similar conditions using transition metals incorporated and impregnated catalysts [VMCM-48(Si/V 40), VMCM-48(Si/V 100), CeMCM-48(Si/Ce 40), CeMCM-48(Si/Ce 100), ZrMCM-48(Si/Zr 40), 5%Vimp.SiMCM-48, 3%VimpSiMCM-48, 1%VimpSiMCM-48]. When the reaction was carried out using transition metals incorporated or impregnated MCM-48, the two products o-hydroxyanisole (o-HA) and p-hydroxyanisole (p-HA) were observed straight away after one hour, although in low concentration. The conversions and selectivities to the products of anisole oxidation reaction after 6 h of reaction time for different catalysts under similar reaction conditions are compared graphically in Fig. 8(a). When Si/Me (Me = Ce, V) ratio was decreased from 100 to 40 (metal content increased) the conversion was increased by 63.5% for vanadium incorporated and 71.7% for cerium incorporated MCM-48 samples. It may be inferred that presence of highly dispersed tetra co-ordinated M4+ (M = metal) species into the silica network in MCM-48 materials activated anisole oxidation reaction. The ratio of o- to p-hydroxyanisole is small when metal incorporation is more, indicating the tendency of formation of more para isomer when the metal content in the MCM-48 materials is more.
 | ||
| Fig. 8 Comparison of (a) metal incorporated MCM-48 materials on anisole oxidation reaction; (b) V incorporated and V-impregnated MCM-48 materials on anisole oxidation reaction; anisole:H2O2:acetonitrile: 1:5:5 (molar ratio), temperature: 323 K, catalyst amount: 20 wt% with respect to anisole. | ||
Comparison of percentage of conversion and product distribution in anisole oxidation for different metals incorporated MCM-48 with same Si/M molar ratio of 40 under the similar reaction conditions reveals that vanadium incorporated MCM-48 shows highest conversion (15.7%) among the three catalysts (Fig. 8(a)). This is probably due to the fact that V can act as a better electron pair acceptor and able to expand its coordination number easily. The order of activity is found to be VMCM-48 >ZrMCM-48 > CeMCM-48. The product distribution is more or less same over VMCM-48 and ZrMCM-48, with CeMCM-48 showing slight preference for formation of the para-isomer.
Comparison of percentage of conversion and product distribution in anisole oxidation for different percentage of vanadium impregnated SiMCM-48 materials (vanadium as 1%, 3% and 5% vanadium oxide) are given in Fig. 8(b). Reactions were carried out under the same experimental conditions as reported above. Both the activity and selectivity for vanadium impregnated MCM-48 samples increase with increase of vanadium oxide impregnation amount. Vanadium oxide impregnated samples gave almost equal selectivity towards ortho and para products when impregnation was 1 and 3%. However, at higher loading of vanadium (5%) the ortho to para ratio increased up to 1:3. Comparing the results of V-impregnated MCM-48 materials, it is observed that conversion increases with increase of vanadium oxide loading. From this result it may be inferred that there is a critical concentration of V4+ species in the channels of MCM-48 above which the formation and diffusion of ortho-product are prevented due to steric factors. The higher activity of V impregnated MCM-48 with 5% vanadium oxide for oxidation of anisole than the corresponding V incorporated-MCM-48 samples may be attributed to the presence of highly dispersed vanadium oxides inside the channels. Under these conditions, oxidation of anisole gave preferentially the para-isomer.
Vanadium complexes and oxides due to their high reactivity and remarkable stability have emerged as efficient catalysts for a variety of oxidation reactions using hydrogen peroxide as oxidant,48–51 but there are few literature reports on the hydroxylation of aromatics.52–54 A comparison of the catalytic efficiency of various heterogeneous vanadium-based catalysts55,56 in anisole hydroxylation is presented in Table 5. Under almost similar reaction conditions our catalysts are found to be more efficient (entry 3 and 4, Table 5) for conversion in hydroxylation of anisole with hydrogen peroxide.
| Entry | Ref. | Catalyst | Conditions | Conv. (mol%) | Selectivity of products (%) | ||
|---|---|---|---|---|---|---|---|
| PHA | OHA | Others | |||||
| T = temperature, R = ratio of substrate to H2O2, t = time of reaction. | |||||||
| 1 | 51 | TS-1(Si/Ti = 100) | T = 353 K, R = 2, t = 3 h | 1.4 | 58 | 42 | — |
| 2 | 52 | Vanadyltetraphenoxyphthalocyanine | T = 298 K, R = 5, t = 8 h | 11.0 | 48 | — | 52 |
| T = 353 K, R = 5, t = 8 h | 22.0 | 39 | — | 61 | |||
| 3 | This work | VMCM-48(S/V = 40) | T = 323 K, R = 5, t = 8 h | 27.7 | 66 | 34 | — |
| 4 | This work | 5% VimpMCM-48 | T = 323 K, R = 5, t = 8 h | 35.5 | 68 | 32 | — |
It is believed that the new bridges of M–O–Si were formed (M = V, Zr, Ce) in the in situ metal incorporated MCM-48 structure as shown in Scheme 1(a),57 while isolated monomeric tetrahedral vanadium species may be present on the vanadium impregnated MCM-48 surface (Scheme 1(b)). We envisioned that the M–O–Si bonds in incorporated MMCM-48 structure and V species in the impregnated VMCM-48 may be the active centre for the hydroxylation of aniosole.58
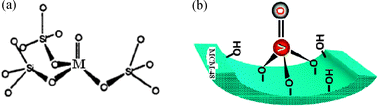 | ||
| Scheme 1 (a) Formation of M–O–Si in the structure (for Zr and Ce, M = O will be M–O), (b) isolated monomeric tetrahedral vanadium species on the vanadium impregnated MCM-48 surface. | ||
Based on the study of Nizova et.al.59 oxidation by H2O2 in acetonitrile for vanadium we can propose the following mechanism for the formation of o-hydroxyanisole (OHAP) and p-hydroxyanisole (PHAP):
| V5+ + H2O2 ⇌ Complex |
| Complex → V4+ + H+ + HO2˙ |
| V5+ + HO2˙ → V4+ + H+ + O2˙ |
| V4+ + H2O2 → V5+ + HO− + HO˙ |
| 2 Anisole + 2HO˙ → PHAP + OHAP + H |
| 2 |
| O |
The excess formation of para isomers in hydroxylation of anisole over MMCM-48 (M = Zr, Ce, V) requires the assumption of some effects favouring formation of these isomer molecules. It might be explained by the different adsorption ability of the para and ortho products on the catalysts or by the different rate of polymerization of both products.60 In the present case the stronger adsorption of the ortho molecule from the hydroxyl and methoxy groups, ortho to each other, enabling a two fold attachment to an active centre, leading to lower diffusion, may be the cause lower selectivity to o-isomer (Scheme 2). Obviously, the kinetics is influenced by the rate law comprising a term which considers the different strengths of adsorption of both products.
 | ||
| Scheme 2 Two fold adsorption of OHA. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :H2O2 (molar ratio).
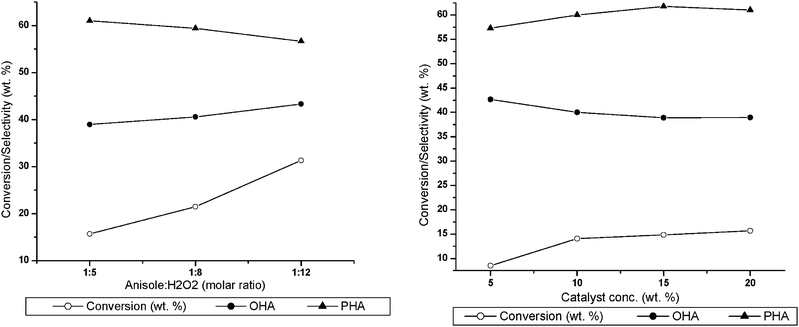
Fig. 9(a) depicts the effect of anisole:H2O2 molar ratio on conversion and product distribution in anisole oxidation after 6 h at 323 K with VMCM-48(Si/V 40) catalyst amount 20 (w/w)% with respect to anisole. It is observed that with a decrease in the anisole:H2O2 ratio there is a gradual increase in conversion. Another prominent feature is that the selectivity for the para-isomer increases with increase in the anisole:H2O2 molar ratio. With the presence of a relatively larger amount of H2O2 (low anisole:H2O2 molar ratio), the surface active species and the active species inside the pore are both indistinguishable.
:H2O2 (molar ratio).
Fig. 9(a) depicts the effect of anisole:H2O2 molar ratio on conversion and product distribution in anisole oxidation after 6 h at 323 K with VMCM-48(Si/V 40) catalyst amount 20 (w/w)% with respect to anisole. It is observed that with a decrease in the anisole:H2O2 ratio there is a gradual increase in conversion. Another prominent feature is that the selectivity for the para-isomer increases with increase in the anisole:H2O2 molar ratio. With the presence of a relatively larger amount of H2O2 (low anisole:H2O2 molar ratio), the surface active species and the active species inside the pore are both indistinguishable.
 | ||
| Fig. 9 Effect of (a) H2O2 using VMCM-48(Si/V 40); (b) catalyst concentration VMCM-48(Si/V 40). | ||
:5. The results are shown Fig. 9(b). It is observed that maximum conversion is obtained when the catalyst concentration is 20 (w/w)% with respect to the substrate. This may be due to the fact that with an increase in the concentration of the catalyst, the number of active metal species involved in the reaction increases and hence the reaction rate. The para selectivity of the product also increases on increasing the catalyst concentration from 5 (w/w)% to 20 (w/w)%. This can also be explained from the fact that at higher concentration, the number of active vanadium species is more and as the amount of H2O2 remains the same, the probability of utilization of H2O2 for the formation of product is also quite high at 20% catalyst and low at 5% catalyst.
:5 and catalyst concentration of 20%. With an increase of temperature from 313 to 353 K the conversion increased from 19.1 to 43.1%. As the activation energy for a reaction was increasingly available at higher temperature, so the reaction became faster at 353 K compared to 313 and 343 K. At higher conversion p-hydroxyanisole selectivity was higher, against lower selectivity towards the ortho-isomer.
 | ||
| Fig. 10 Effect of temperature on conversion and selectivity in the anisole oxidation reaction using VMCM-48(Si/V 40); anisole:H2O2:acetonitrile: 1:5:5 (molar ratio), reaction time: 10 h catalyst amount: 20 (w/w)% with respect to anisole. | ||
4. Conclusion
SiMCM-48 was successfully synthesized by both single step and two step methods. The procedure for formation of metal incorporated MCM-48 appeared to be more successful by single step method as evidenced from the intense reflections in XRD patterns. V, Ce and Zr were, therefore, incorporated into the framework structure of MCM-48 materials with the help of his method. The XRD results of V and Ce incorporated MCM-48 samples with Si/Me (Me = V, Ce) 40 and 100 showed that the d values and cubic unit cell parameters (ao) gradually increased with an increase of metal content in the framework structure. FT-IR spectroscopy, UV-Vis DRS technique and TGA technique also proved successful incorporation of metal cations into the framework position.Transition and inner transition metal incorporated MCM-48 samples were found to be much more active for oxidation of anisole than SiMCM-48. Among all the metal incorporated samples VMCM-48 was the better catalyst for oxidation of anisole by H2O2.
Acknowledgements
The authors are grateful to DST, New Delhi for financial assistance. They also acknowledge the help of IIT, Guwahati and OIL, Duliajan for characterization of the samples.References
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- A. Sayari, J. Am. Chem. Soc., 2000, 122, 6504–6505 CrossRef CAS.
- K. Schumacher, M. Grün and K. K. Unger, Micropor. Mesopor. Mater., 1999, 27, 201–206 CrossRef CAS.
- Q. Huo, D. I. Margolese, U. Ciesla, D. G. Demuth, P. Feng, T. E. Gier, P. Sieger, A. Firouzi, B. F. Chmelka, F. Schüth and G. D. Stucky, Chem. Mater., 1994, 6, 1176–1191 CrossRef CAS.
- S. Zhang, Y. Fujii, H. Yamashita, K. Koyano, T. Tatsumi and M. Anpo, Chem. Lett., 1997, 7, 659–660 CrossRef.
- J. Xu, Z. Luan, H. He, W. Zhou and L. Kevan, Chem. Mater., 1998, 10, 3690–3698 CrossRef CAS.
- O. J. Sjöblom and M. Stöcker, Adv. Colloid Interface Sci., 2001, 89–90, 439–466 Search PubMed.
- W. Zhao and Q. Li, Chem. Mater., 2003, 15, 4160–4162 CrossRef CAS.
- Y. Xia and R. Mokaya, J. Phys. Chem. B., 2003, 107, 6954–6960 CrossRef CAS.
- H. Kosslick, G. Lischke, H. Landmesser, B. Parlitz, W. Storek and R. Fricke, J. Catal., 1998, 176, 102–114 CrossRef CAS.
- S. E. Dapurkar and P. Selvam, Appl. Catal., A, 2003, 254, 239–249 CrossRef CAS.
- H. W. Lee, J. Ki Jeon, S. Hoon Park, K. E. Jeong, H. J. Chae and Y. K Park, Nanoscale Res. Lett., 2011, 6(500), 1–7 Search PubMed.
- H. Liu, M. Wang, H. Hu, Y. Liang, Y. Wang, W. Cao and X. Wang, J. Solid State Chem., 2011, 184(3), 509–515 CrossRef CAS.
- H. M. Kao, H. M. Wu, Y. W. Liao and S. T. Anthony, Micropor. Mesopor. Mater., 2005, 86, 256–267 CrossRef CAS.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas, A. A. Romero, J. J. Calvino and M. P. Rodríguez-Luque, J. Catal., 2005, 230, 327–338 CrossRef CAS.
- P. Selvam and S. E. Dapurkar, Catal. Today, 2004, 96, 135–141 CrossRef CAS.
- J. K. Shon, J. Y. Sim, S. Singh Thakur, E. M. Ko, S. S. Kong, J. Y. Choi, M. Kang, B. K. Senapati, D. S. Choi, D. H. Ryu and J. M. Kim, Bull. Korean Chem. Soc., 2008, 29(10), 1993–1997 CrossRef CAS.
- A. Corma, M. T. Navarro and J. Pérez-Pariente, J. Chem. Soc., Chem. Commun., 1994, 147–148 RSC.
- B. J. Aronson, C. F. Blanford and A. Stein, Chem. Mater., 1997, 9, 2842–2851 CrossRef CAS.
- Z. Y. Yuan, W. Zhou, Z. L. Zhang, J. Q. Liu, J. Z. Wang, H. X. Li and L. M. Peng, Surf. Interface Anal., 2001, 32, 193–197 CrossRef CAS.
- Q. Zhang, Y. Wang, S. Itsuki, T. Shishido and K. Takehira, J. Mol. Catal. A, 2002, 88, 189–200 CrossRef.
- J. Xu, Z. Luan, M. Hartman and L. Kevan, Chem. Mater., 1999, 11, 2928–2936 CrossRef CAS.
- B. J. Aronson, C. F. Blanford and A. Stein, J. Phys. Chem. B, 2000, 104, 449–459 CrossRef CAS.
- V. Caps and S. C. Tsang, Catal. Today, 2000, 61, 19–27 CrossRef CAS.
- W. Zhang and T. J. Pinnavaia, Catal. Lett., 1996, 38, 261–265 CrossRef CAS.
- E. P. Reddy, L. Davydov and P. G. Smirniotis, J. Phys. Chem. B, 2002, 106, 3394–3401 CrossRef CAS.
- C. Pak and G. L. Haller, Micropor. Mesopor. Mater., 2001, 48, 165–170 CrossRef CAS.
- N. Lang, P. Delichere and A. Tuel, Micropor. Mesopor. Mater., 2002, 56, 203–217 CrossRef CAS.
- K. Schumacher, C. Du Fresne von Hohenesche, K. K. Unger, R. Ulrich, A. Du Chesne, U. Wiesner and H. W. Spiess, Adv. Mater., 1999, 11, 1194–1198 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- S. Wang and J. A. Guin, Stud. Surf. Sci. Catal., 2004, 147, 439–444 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- A. Monnier, F. Schuth, Q. Huo, D. Kumar, D. Margolese, R. S. Maxwell, G. D. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi, M. Janicke and B. F. Chmelka, Science, 1993, 261, 1299–1303 CAS.
- Y. Li, J. Shi, H. Chen, Z. Hua, L. Zhang, M. Ruan, J. Yan and D. Yan, Micropor. Mesopor. Mater., 2003, 60, 51 CrossRef CAS.
- P. C. Shih, H. P. Lin and C. Y. Mou, in Nanotechnology in Mesostructured Materials, Studies in Surface Science and Catalysis, ed. S. E. Park, R. Ryoo, W. S. Ahn, C. W. Lee and J. S. Chang, Elsevier, Amsterdam, 2003, vol. 146, p. 557 Search PubMed.
- S. C. Laha, P. Mukherjee, S. R. Sainkar and R. Kumar, J. Catal., 2002, 207, 213–223 CrossRef CAS.
- Z. Wangcheng, L. Guanzhong, G. Yanglong, G. Yun, W. Yanqin, W. Yunsong, Z. Zhigang and L. Xiaohui, J. Rare Earths, 2008, 26, 515–522 CrossRef.
- X. Ge, L. Shi, J. Wei, S. Huang and G. Ma, J. Rare Earths, 2005, 23, 521–525 Search PubMed.
- L. H. Little, Infrared Spectra of Adsorbed Species, Academic Press, London, 1966 Search PubMed.
- A. A. Romero, M. D. Alba and J. Klinowski, J. Phys. Chem. B, 1998, 102, 123–128 CrossRef CAS.
- F. L. Galeener, A. J. Leadbetter and M. W. Stringfellow, Phys. Rev. B, 1983, 27, 1052–1078 CrossRef CAS.
- M. A. Camblor, A. Corma and J. Perez-Pariente, J. Chem. Soc., Chem. Commun., 1993, 557–559 RSC.
- D. Kumar, K. T Pillai, V. Sudersanan, G. K. Dey and N. M. Gupta, Chem. Mater., 2003, 15, 3859–3865 CrossRef CAS.
- A. Thangaraj, R. Kumar, M. P. Mirajkar and P. Ratnasamy, J. Catal., 1991, 130, 1–8 CrossRef CAS.
- T. Sen, P. R. Rajamohanan, S. Ganapathy and S. Sivasankar, J. Catal., 1996, 163, 354–364 CrossRef CAS.
- S. Brunauer, L. S. Deming, W. E. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723–1732 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- A. G. J. Ligtenbarg, R. Hage and B. L. Feringa, Coord. Chem. Rev., 2003, 237, 89–101 CrossRef CAS.
- C. Bolm, Coord. Chem. Rev., 2003, 237, 245–256 CrossRef CAS.
- V. Conte, F. DiFuria and G. Licini, Appl. Catal., A, 1997, 157, 335–361 CrossRef CAS.
- M. J. Clague, N. N. Keder and A. Butler, Inorg. Chem., 1993, 32, 4754–4761 CrossRef CAS.
- J. Zhang, Y. Tang, G. Li and C. Hu, Appl. Catal., A, 2005, 278, 251–261 CrossRef CAS.
- K. Lemke, H. Ehrich, U. Lohse, H. Berndt and K. Jähnisch, Appl. Catal., A, 2003, 243, 41–51 CrossRef CAS.
- M. Iwamoto, J. I. Hirata, K. Matsukami and S. Kagawa, J. Phys. Chem., 1983, 87, 903–905 CrossRef CAS.
- N. Georgi, Vayssilov, P. Zdravka and T. Alain, Chem. Eng. Technol., 1997, 20, 333–337 CrossRef.
- J. K. Joseph, S. Singhal, S. L. Jain, R. Sivakumaran, B. Kumar and B. Sain, Catal. Today, 2009, 141, 211–214 CrossRef CAS.
- P. Selvam and S. E. Dapurkar, J. Catal., 2005, 229, 64–71 CrossRef CAS.
- J. Gholamia, A. R. Badieia, G. Mohammadi Ziarani and A. R. Abbasi, Journal of Nanostructures, 2012, 1, 69–75 Search PubMed.
- G. V. Nizova, Y u. N. Kozlov and G. B. Shuĺpin, Russ. Chem. Bull., 2004, 53(10), 2330–2333 CrossRef CAS.
- K. Kulawik, G. S. Ekloff, J. Bathonsky, A. Zukal and Z. Had, Chem. Commun., 1995, 60, 951–956 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20198d |
| This journal is © The Royal Society of Chemistry 2012 |