A highly active Zn(salphen) catalyst for production of organic carbonates in a green CO2 medium†
Masoumeh
Taherimehr
a,
Antonello
Decortes
b,
Syed M.
Al-Amsyar
a,
Warunee
Lueangchaichaweng
a,
Christopher J.
Whiteoak
b,
Eduardo C.
Escudero-Adán
b,
Arjan W.
Kleij
*bc and
Paolo P.
Pescarmona
*a
aCentre for Surface Chemistry and Catalysis, KU Leuven, Kasteelpark Arenberg 23, 3001 Heverlee, Belgium. E-mail: paolo.pescarmona@biw.kuleuven.be; Fax: +32 16321998
bInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007, Tarragona, Spain. E-mail: akleij@iciq.es
cCatalan Institute of Research and Advanced Studies (ICREA), Pg. Lluís Companys 23, 08010, Barcelona, Spain
First published on 20th April 2012
Abstract
Zn(salphen), in combination with Bu4NI, was studied as a binary catalyst system for CO2-fixation in the context of organic carbonate formation. The catalytic potential of this binary catalyst system was considerably improved by working in a solvent-free, CO2-rich environment, thereby increasing the overall contact between the reagents and catalyst. Under these green conditions, excellent conversion and selectivity towards the cyclic carbonate product were obtained with epoxides that are generally less prone to undergo cycloaddition with carbon dioxide. The effect of the reaction conditions and the type of co-catalyst employed together with Zn(salphen) were systematically investigated and optimised.
Introduction
In recent years, the fixation of the greenhouse gas CO2 to yield valuable chemical products has been drawing increasing interest.1 Being the main product of fuel combustion, CO2 is an inexpensive, readily available and renewable C1 raw material. However, the high thermodynamic stability of carbon dioxide represents a challenge for its conversion. This obstacle can be overcome by reacting CO2 with compounds with a relatively high free energy, amongst which are epoxides.2 The addition of carbon dioxide to epoxides is an atom-efficient reaction that can generate either cyclic carbonates or polycarbonates. Cyclic carbonates, which are the thermodynamically favoured products of the reaction, are an attractive family of compounds with broad application potential as polar aprotic solvents with low odour and toxicity, as intermediates in the synthesis of fine chemicals, as starting materials for the production of polymers and as electrolytes for lithium-ion batteries.3 Various homogeneous and heterogeneous catalysts based on free or supported metal complexes3–9 and ionic liquids10–13 have been reported to promote the synthesis of cyclic carbonates by cycloaddition of CO2 to epoxides. Excellent results have been achieved for the conversion of terminal epoxides under mild conditions, while internal epoxides require harsher reaction conditions to produce the respective carbonates. Recently, Zn(salphen) complexes [salphen = N,N′-phenylene-1,2-bis-salicylideneimine] were reported to be very promising homogeneous catalysts for this kind of reaction using a tetrabutylammonium halide [Bu4NX; X = Br, I] as co-catalyst (Scheme 1).14,15 The high activity of the Zn(salphen) complex was ascribed to its constrained geometry imposed by the ligand scaffold, which imparts increased Lewis-acid character to the Zn ion. The proposed mechanism with the Zn(salphen)/Bu4NI catalyst system involves four steps: the activation of the epoxide by the Lewis acid, the opening of the epoxide ring by the halide anion, the insertion of carbon dioxide and the ring-closure yielding the cyclic carbonate.14,16 Although Zn(salphen)s proved to be versatile catalysts for the synthesis of cyclic carbonates from many terminal epoxides, they were rather ineffective for the conversion of internal epoxides.15 Here, we report that the activity of Zn(salphen) in the conversion of sterically more congested epoxides can be remarkably improved by working under solvent-free conditions and by fine-tuning the reaction conditions for each substrate, including the use of carbon dioxide in the supercritical state (sc-CO2: 31.1 °C, 73.8 bar) with the double role of reagent and solvent. Supercritical carbon dioxide represents an attractive and environmentally benign alternative to conventional solvents since it is non-toxic, can be easily separated from reagents and products, has notable dissolving power and displays high molecular diffusivity, low viscosity and low surface tension.17,18 A large variety of synthetically useful reactions can be performed in sc-CO2 with competitive results compared to those obtained in conventional organic solvents.19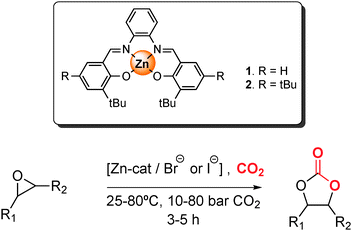 | ||
| Scheme 1 Cycloaddition of CO2 to epoxides under various reaction conditions using Zn(salphen) complex 1 as catalyst. | ||
Experimental
1. Materials
Zn(salphen) complexes 1 and 2 were prepared according to a literature procedure,7,15 using a one-pot condensation–metalation that involves ortho-phenylenediamine, 3-tert-butylsalicylaldehyde or 3,5-di-tert-butylsalicylaldehyde and Zn(OAc)2·2H2O in MeOH. All the epoxides and co-catalysts were purchased from Aldrich, Fluka or Acros and used without any further purification (purity ≥95%). The solvents were purchased from Merck (purity ≥98%).2. Crystallographic studies
For the crystallographic studies, the use of both Zn(salphen) complexes 1 and 2 proved to be useful depending on the respective ligand used; the only difference between 1 and 2 is the presence of two additional distant tBu groups that do not interfere with the coordination potential of the studied epoxides. The measured crystals for adducts 2·(2,3-dimethyloxirane) and 1·[(3-methyloxetan-3-yl)methanol] were stable under atmospheric conditions; nevertheless they were treated under inert conditions immersed in perfluoropolyether as protecting oil for manipulation. Data collection: measurements were made on a Bruker-Nonius diffractometer equipped with an APPEX 2 4K CCD area detector, a FR591 rotating anode with MoKα radiation, Montel mirrors and a Kryoflex low temperature device (T = −173 °C). Full-sphere data collection was used with ω and φ scans. Programs used: data collection Apex2 V2011.3 (Bruker-Nonius 2008), data reduction Saint + Version 7.60A (Bruker AXS 2008) and absorption correction SADABS V. 2008-1 (2008). Structure solution: SHELXTL Version 6.10 (Sheldrick, 2000)20 was used. Structure refinement: SHELXTL-97-UNIX Version.
Crystallographic summary for
2
·(2,3-dimethyloxirane): formula: C40H54N2O3Zn, Mr = 676.22, triclinic, P-1, a = 10.9698(5) Å, b = 12.9718(6) Å, c = 13.3427(7) Å, α = 91.834(3)°, β = 108.509(6)°, γ = 92.660(3)°, V = 1836.16(15) Å3, Z = 2, ρ = 1.223 mg m−3, μ = 0.707 mm−1, λ = 0.71073 Å, T = 100(2) K, F(000) = 724, crystal size = 0.40 × 0.40 × 0.40 mm, θ(min) = 1.92°, θ(max) = 37.15°, 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 091 reflections collected, 13539 reflections unique (Rint = 0.0579), GoF = 1.043, R1 = 0.0431 and wR2 = 0.1182 [I > 2σ(I)], R1 = 0.0597 and wR2 = 0.1279 (all indices), min/max residual density = −0.653/1.746 [e Å−3]. The structure has been deposited at the CCDC with reference number 855909.
091 reflections collected, 13539 reflections unique (Rint = 0.0579), GoF = 1.043, R1 = 0.0431 and wR2 = 0.1182 [I > 2σ(I)], R1 = 0.0597 and wR2 = 0.1279 (all indices), min/max residual density = −0.653/1.746 [e Å−3]. The structure has been deposited at the CCDC with reference number 855909.
Crystallographic summary for
1
·[(3-methyloxetan-3-yl)methanol]: formula: C68.5H85N4O9Zn2, Mr = 1239.15, monoclinic, C2/c, a = 42.190 Å, b = 9.719 Å, c = 31.932 Å, β = 110.08°, V = 12297.7 Å3, Z = 8, ρ = 1.339 mg m−3, μ = 0.841 mm−1, λ = 0.71073 Å, T = 100(2) K, F(000) = 5248, crystal size = 0.30 × 0.10 × 0.10 mm, θ(min) = 1.03°, θ(max) = 35.06°, 26810 reflections collected, 19976 reflections unique (Rint = 0.0437), GoF = 1.024, R1 = 0.0393 and wR2 = 0.0984 [I > 2σ(I)], R1 = 0.0630 and wR2 = 0.1120 (all indices), min/max residual density = −0.714/0.833 [e Å−3]. The structure has been deposited at the CCDC with reference number 867372.
3. Catalytic tests
The catalytic tests were performed in a batch reactor with borosilicate glass windows enabling the direct visualisation of the phase behaviour of the system under study, or in a high-throughput array of 24 parallel batch reactors allowing the rapid and reliable comparison of the performance of the catalyst under different reaction conditions (Fig. 1).21,22 The visualisation reactor and the 24-HT reactor block are part of a high-throughput unit for the study of catalytic systems under (supercritical) CO2, manufactured by Integrated Lab Solutions (ILS) and Premex and controlled with a software by ProControl. Two ISCO pumps are employed to achieve the controlled pressurisation of the reactors with CO2. The batch reactors in the 24-HT reactor block are pressurised simultaneously while check valves prevent backflow and cross-contamination among them. The temperature in these reactors is regulated through electric heating elements and a Huber thermostat. The heating of the visualisation reactor is controlled through a thermocouple, which is immersed in the reaction mixture. Both the visualisation reactor and the 24 parallel batch reactors are operated with individual magnetic stirring. The entrance and exit lines of the reactors are controlled by valves, which can be opened and closed using the software of the unit. An automated and stepwise depressurisation protocol is employed for depressurising the reactors. Protection against overpressure during the catalytic tests is ensured by automated depressurisation protocols and by the presence of rupture discs. | ||
| Fig. 1 (a) Visualisation reactor: (1) pressurisation line; (2) depressurisation line; (3) rupture disc; (4) thermocouple; (5) viewing window; (6) heating block; (7) magnetic stirrer. (b) 24-HT reactor block: (1) pressurisation line; (2) depressurisation line; (3) check valves. | ||
In a typical catalytic test, the catalyst, Zn(salphen) 1 (0.025 g, 5.0 × 10−5 mol), the co-catalyst (5 × 10−5 mol) and the internal standard, mesitylene (0.27 mL, 2.0 × 10−3 mol), were weighed in a glass vial. To this mixture, the epoxide (2.0 × 10−3 mol) was added and the solution was stirred until complete dissolution occurred. The vial containing this solution and a magnetic stirring bar was transferred into the selected stainless-steel reactor of the high-throughput unit. Air was removed from the lines and from the reactor by purging with N2 for 5–10 min. Next, the system was purged with CO2 at 15 bar for 15 min before increasing the CO2 pressure to the desired value. In a typical test carried out at a CO2 pressure of 80 bar, first the unit was pressurised to 60 bar (∼1 h), while keeping the block at room temperature. Next, the temperature was increased to the desired value (e.g. 45 °C) and the pressure was adjusted to the final value of 80 bar. As soon as the desired conditions were reached, the entrance valve of the reactor was closed and stirring at 900 rpm was started. The sample was allowed to react for the required time (3 or 5 h). Then, the stirring was stopped and the reactors were cooled down. The depressurisation was started when the reactor had cooled down to room temperature (to prevent loss of reagents and products). These conditions were reached in 1.5–2 h when the 24-HT reactor block was used and in about 1 h when the visualisation reactor was used. The depressurisation was allowed to continue until the pressure inside the reactor(s) decreased to 2 bar (these conditions were reached in about 2 h).21 Then, the reactor block was opened and the glass vials were removed from the block. The reaction samples were analysed by gas chromatography (GC) and 1H NMR, using mesitylene as internal standard. Gas chromatography (GC) was performed on a Trace GC Ultra from Interscience (RTX-5 column, 5 m, 0.1 mm), equipped with an ultrafast module allowing the rapid analysis of the reaction solutions (2 min per sample). The assignment of peaks related to unknown products was performed by gas chromatography–mass spectrometry (GC-MS) on an Agilent 6890N Gas Chromatograph (WCOT fused silica column, 30 m, 0.25 mm) coupled to an Agilent 5973 MSD Mass Spectrometer. 1H NMR spectra were measured with a 300 MHz spectrometer (7.0 T). 32 scans were accumulated with a recycle delay of 1 s. The pulse length was 8.0 μs and the power level was 0.0 dB. The samples for analysis by 1H NMR were prepared by adding ca. 1 mL of a deuterated solvent (CDCl3 or CD2Cl2) to an aliquot of the reaction solution at the end of the catalytic test.
After each reaction, the reactor was cleaned in order to remove reaction residues. Approximately one third of the reactor volume was filled with ethanol. The closed reactor was heated up to 90 °C for 20 min. Then, the lines and the reactor were purged with 65 bar CO2 for 5 min. Once the reactor cooled down to room temperature, the lines and the reactor were purged with N2 for about 5 min. The reactor was opened and the ethanol solution was analysed by GC. The cleaning procedure was repeated until no reaction residues were detected by GC analysis. Finally, the empty reactor was purged with N2 for 20 min at 150 °C to remove residual ethanol.
Results and discussion
The catalytic cycloaddition of CO2 to epoxides is a complex system: a good interaction between epoxide, CO2, catalyst and co-catalyst is extremely important for achieving the best possible kinetics, which would lead to high carbonate yields. On the basis of these considerations, we initially aimed at improving the performance of the binary Zn(salphen)/Bu4NI system in catalysing the synthesis of cyclic carbonates derived from the more challenging substrates styrene oxide and cyclohexene oxide; the latter is one of the least reactive substrates for this reaction.3,14,15 Our strategy involves a reaction with CO2 in the supercritical state,19,23,24 thus acting both as reagent and solvent. In this way we expected to be able to optimise the contact among all reaction components and the catalyst system, so as to improve the yield of the carbonate synthesis while meeting the requirements for a green, solvent-free process.In previous work, the Zn(salphen) complex 1 (Scheme 1) was employed using a solvent that aided the dissolution of the catalyst and co-catalyst in the reaction mixture, which is crucial for achieving good activity.9,14,15 Under these conditions we found that styrene carbonate can be produced in 13% yield after 3 h of reaction at 45°C and 10 bar of CO2 with dichloromethane (DCM) as solvent (Table 1, entry 1).

| Entry | T/°C | p CO2/bar | Solvent | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: styrene oxide (2.0 × 10−3 mol), Zn(salphen) (5 × 10−5 mol), NBu4I (5 × 10−5 mol), mesitylene (0.27 mL, 2.0 × 10−3 mol), 5 mL of solvent (if present), 3 h. Complete selectivity was observed in all cases. b The yields were calculated by 1H NMR (CDCl3) and confirmed by GC. c Taken from ref. 15. d The reaction was performed with 2.0 × 10−4 mol of mesitylene. | ||||
| 1 | 45 | 10 | DCM | 13 |
| 2 | 45 | 10 | DCM | 66 (18 h)c |
| 3 | 45 | 80 | DCM | 15 |
| 4 | 45 | 80 | — | 96 |
| 5 | 25 | 80 | — | 52 |
| 6 | 45 | 40 | — | 86 |
| 7 | 45 | 10 | — | 90 |
| 8 | 45 | 10 | — | 98d |
This result is consistent with the previously reported 66% yield under similar conditions but with a much longer reaction time (18 h, see entry 2).15 Increasing the CO2 pressure beyond the supercritical point using DCM as a co-solvent led only to a minor improvement in styrene carbonate yield (15% after 3 h, entry 3). To our delight we found that by working under solvent-free conditions with CO2 in the supercritical state, a large increase in the carbonate yield was observed with nearly complete conversion after only 3 h at 45 °C (entry 4). We ascribe this significant improvement in the reaction kinetics to a much better contact between all the involved reagents and catalysts.
When performing the reaction at the same CO2 pressure (80 bar) but at much lower temperature (25 °C) having CO2 in the liquid state, a lower but appreciable yield of styrene carbonate was obtained (Table 1, entry 5). Performing the cycloaddition of CO2 to styrene oxide at 45 °C and at lower CO2 pressures (entries 6 and 7) did not significantly affect the styrene carbonate yield compared with the reaction operated under sc-CO2 conditions (entry 4). In all experiments in which CO2 was in the supercritical or liquid state (entries 3–5), we did not observe the formation of a single phase between CO2 and the solution containing the catalysts and the epoxide, but the volume of the latter increased sensibly after reaching the reaction conditions indicating a relevant dissolution of CO2 (see Fig. 2a). On the other hand, in the tests in which CO2 was in the gaseous state (entries 2 and 6–8), no evident change in the volume of the liquid phase could be observed. On the basis of these observations and of the catalytic results in Table 1, we can infer that the main factor hindering the contact between CO2, styrene oxide and the catalyst system is the presence of the solvent rather than the degree of dissolution of CO2 in the reaction mixture.
 | ||
| Fig. 2 Visualisation of the reaction of CO2 with: (a) styrene oxide at 45 °C and 80 bar CO2 (entry 4 in Table 1) – the dotted line indicates the approximate liquid level before adding CO2 to the reactor; (b) cyclohexene oxide at 45 °C and 80 bar CO2 (entry 1 in Table 2); (c) cyclohexene oxide at 80 °C and 80 bar CO2 (entry 3 in Table 2). | ||
The reactions discussed above were performed using a relatively large amount of mesitylene (i.e., the internal standard for NMR and GC analyses). To test whether mesitylene plays a relevant role as a co-solvent, we carried out a reaction with styrene oxide using a much lower amount of mesitylene (Table 1, entry 8). In this case, a slight increase in the carbonate yield was observed, indicating that mesitylene does not play a positive role as a co-solvent in the reaction. This result supports our conclusion that a mixture of carbon dioxide and epoxide alone represents a suitable reaction medium for this cycloaddition reaction: the two reagents – and the cyclic carbonate product – are suitable solvents for the catalyst and co-catalyst, and the absence of an additional solvent guarantees an improved contact between all components.
Inspired by the results obtained for styrene oxide, we then examined cyclohexene oxide as a substrate (Table 2). The reaction conditions that gave the best results in the conversion of styrene oxide (3 h, 45 °C, pCO2 = 80 bar) led to a very low yield of cyclohexene carbonate (entry 1, 3%). This low yield cannot only be ascribed to the low reactivity of the cyclohexene oxide but also to the observed low solubility of complex 1 and of Bu4NI in this substrate (see Fig. 2b). In an attempt to improve this aspect, we added a minimal amount of toluene as a co-solvent that should help to dissolve the catalyst and co-catalyst and be miscible with the sc-CO2 phase. However, we observed virtually no formation of the cyclic carbonate product (entry 2), underlining again the negative effect of dilution of the reaction mixture with co-solvents. On the other hand, increasing the reaction temperature to 80 °C (at 80 bar of CO2, entry 3) had the double role of achieving full dissolution of complex 1 and of Bu4NI in the reaction mixture and enhancing the reaction rate giving an appreciable yield of cyclohexene carbonate (29%). This is a significant improvement compared to the previously reported binary Zn(salphen)/Bu4NI catalyst system operated in DCM that proved to be completely ineffective at a temperature as high as 105 °C (entry 4).15 This result is also promising compared to that of catalysts described in the literature, which generally require higher temperatures and longer reaction times to afford respectable yields of cyclohexene carbonate.3,25–27

| Entry | T/°C | p CO2/bar | Solvent | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: cyclohexene oxide (2.0 × 10−3 mol), Zn(salphen) (5 × 10−5 mol), NBu4I (5 × 10−5 mol), mesitylene (0.27 mL, 2.0 × 10−3 mol), 5 mL of solvent (if present), 3 h. b The yields were calculated by 1H NMR (CDCl3) and confirmed by GC. c Toluene: 0.63 mL (0.55 g) was used. d Taken from ref. 15. e Conditions as in (a) but with 2.0 × 10−4 mol of mesitylene. f Conditions as in (a) but using 1 × 10−5 mol of Zn(salphen). g Conditions as in (a) but with 1 × 10−5 mol of Zn(salphen) and 1 × 10−5 mol of NBu4I. h Conditions as in (a) but without Zn(salphen). | ||||
| 1 | 45 | 80 | — | 3 |
| 2 | 45 | 80 | Toluenec | 0 |
| 3 | 80 | 80 | — | 29 |
| 4 | 105 | 10 | DCM | 0 (18 h)d |
| 5 | 80 | 20 | — | 4 |
| 6 | 80 | 20 | DCM | 2 |
| 7e | 80 | 80 | — | 37 |
| 8f | 80 | 80 | — | 25 |
| 9g | 80 | 80 | — | 3 |
| 10h | 80 | 80 | — | 0 |
Cyclohexene carbonate was the only product obtained apart from a minor amount (<5%) of epoxide ring opening products due to the presence of residual ethanol, which is the solvent used for cleaning the reactor (see Experimental section). During the reaction of cyclohexene oxide at 80 °C and 80 bar, two phases were observed (Fig. 2c): a supercritical CO2 phase (top) and a liquid cyclohexene oxide solution phase (bottom). When a lower CO2 pressure was employed (20 bar, Table 2, entry 5), cyclohexene oxide tended to shift to the gas phase, thus causing precipitation of complex 1 and of Bu4NI. Consequently, the interactions of catalyst and co-catalyst with epoxide and CO2 were limited, and the conversion dropped significantly compared to the reaction performed in sc-CO2 (entry 3). A similar phase separation behaviour was observed when performing the reaction in DCM (entry 6), thus rationalising the previously reported poor performance of the Zn(salphen)/Bu4NI catalyst system in the conversion of cyclohexene at higher temperatures (entry 4).15
By comparing the results obtained with cyclohexene oxide (bp = 129–130 °C) and styrene oxide (bp = 194 °C), we can conclude that for epoxides such as cyclohexene oxide a higher reaction temperature (80 °C) is required to give substantial conversion. Furthermore, for substrates having a lower boiling point, sc-CO2 plays an essential role in maintaining a good interaction among all components involved in the reaction and thus achieving a good level of conversion. On the other hand, for epoxides such as styrene oxide that readily dissolves the catalyst and co-catalyst and also allows good conversion levels at relatively low temperature (45 °C), a high CO2 pressure is not needed and the advantage of working with sc-CO2 is less relevant. Hence, our results show that the use of a sc-CO2 medium for the synthesis of cyclohexene carbonate may also be beneficial for other types of catalysts, although with cyclohexene oxide the focus is usually on the formation of polycarbonates, which is beyond the current scope.
The yield of cyclohexene oxide could be improved up to 37% by lowering the amount of internal standard (Table 2, entry 7), in line with what was observed for the reaction of styrene oxide (vide supra). Lowering the amount of Zn(salphen) catalyst only slightly affected the carbonate yield (entry 8), while decreasing the amount of both catalyst and co-catalyst led to much lower carbonate yield (entry 9). Finally, a control reaction (Table 2, entry 10) was carried out using only the co-catalyst (Bu4NI): virtually no conversion of cyclohexene oxide was observed after 3 h (80 °C, pCO2 = 80 bar) emphasising the crucial role of the Zn(salphen) complex 1 in the activation of the epoxide substrate.
Next, the substrate scope for Zn(salphen) 1 was further broadened by studying and comparing the carbonate yields with increasing sterically demanding/challenging substrates (A–G, Table 3) using various co-catalyst structures (3–5). These tests were performed in parallel in a high-throughput reactor, using the conditions optimised for the reaction with cyclohexene oxide (vide supra) and with a reaction time of 3–5 h. The highest yield of carbonate with each epoxide substrate was obtained with iodide as a nucleophile (3, entries 1, 6–8), in line with previous reports.15 Lower, but in most cases still relatively good yields were achieved with the two co-catalysts containing bromide (4 and 5). Notably, when Bu4NI was used as a co-catalyst, good carbonate yields were obtained with both terminal, disubstituted epoxides (C: 2-methyl-2-vinyloxirane, entry 3, 52%) as well as internal epoxides (D, cyclohexene oxide, entry 4, 38%). As expected, upon increasing the steric impediment (cf. substrates B, C and D, entries 6–8), lower yields of the carbonate product were obtained, which reflects the higher complexity of the ring opening step of the epoxide by the nucleophile. Complete selectivity towards the cyclic carbonate was achieved with substrates A–C. For vinyl oxirane (A), full conversion to the cyclic carbonate could be achieved also with a shorter reaction time (entries 2 and 3), and very high carbonate yields were obtained even with lower amounts of catalyst (entries 4 and 5). Despite the excellent results obtained with various challenging epoxides, we found that substrates E–G could not be converted smoothly to their cyclic organic carbonates: very low carbonate yields were observed with oxetane F,28 while no conversion was found with the more sterically hindered oxetane G and with the internal epoxide E (entries 9–11). We ascribe the low activity of Zn(salphen)/NBu4I in the conversion of oxetanes to the intrinsic lower tendency of these substrates to undergo ring-opening reaction compared to the more strained epoxide rings. For substrates E and F we also observed a poor mass balance indicating that under these experimental conditions the conversion to carbonate is hindered by the vaporisation of the substrates due to their low boiling points, resulting in poor mixing at high temperature with the other components of the reaction mixture. The low activity of the binary Zn(salphen)/NBu4I system in the conversion of substrates E–G is thus attributed to the lower reactivity of these compounds and to their volatility (for E and F), and not to a decreased ability of the substrate to coordinate to the Zn(salphen) complex. Furthermore, for the sterically more hindered substrates C–E the ring-opening of the coordinated epoxide by the nucleophile should be more difficult compared to terminal epoxides A and B, increasing the challenge in this type of conversion.

| Entry | Substrate | Reaction time/h | Yieldb (%) | ||
|---|---|---|---|---|---|
| 3 | 4 | 5 | |||
| a Reaction conditions: epoxide (2.0 × 10−3 mol), Zn(salphen) (5 × 10−5 mol), co-catalyst (5 × 10−5 mol), mesitylene (0.27 mL, 2.0 × 10−3 mol), no solvent, 80 °C, 80 bar. b The yields were calculated by 1H NMR and confirmed by GC. c Conditions as in (a) but using 2.0 × 10−4 mol of mesitylene. d Conditions as in (a) but using 1 × 10−5 mol catalyst and 5 × 10−5 co-catalyst. e Conditions as in (a) but using 1 × 10−5 mol catalyst/co-catalyst. f Poly(TMC) [2%] was present in addition to 1% of TMC, TMC = trimethylene carbonate. | |||||
| 1 | A | 5 | >99 | 85 | 81 |
| 2 | A | 3 | >99 | — | — |
| 3c | A | 3 | >99 | — | — |
| 4d | A | 3 | 93 | — | — |
| 5e | A | 3 | 86 | — | — |
| 6 | B | 5 | 97 | — | 90 |
| 7 | C | 5 | 52 | — | 30 |
| 8 | D | 5 | 38 | 17 | 7 |
| 9 | E | 3 | 0 | — | — |
| 10 | F | 3 | 3f | — | — |
| 11 | G | 3 | 0 | — | — |
This hypothesis is further supported by analysis of the X-ray structures of the Zn(salphen) in the presence of 2,3-dimethyloxirane (E, Fig. 3), (3-methyloxetan-3-yl)methanol (G, Fig. 4) and by comparison with the previously reported structure for 2·[cyclohexene oxide] depicted in Fig. 5 (substrate D).15
![X-ray molecular structure for 2·[trans-2,3-dimethyl-oxirane]. H-atoms, co-crystallised solvent molecules and rotational disorder in the coordinated epoxide are omitted for clarity, and only a partial numbering scheme is provided. Selected bond lengths (Å) and angles (°): Zn(1)–O(1) = 1.9438(9), Zn(1)–O(2) = 1.9399(9), Zn(1)–O(3) = 2.2086(11), Zn(1)–N(1) = 2.0638(9), Zn(1)–N(2) = 2.0577(10); O(1)–Zn(1)–O(2) = 95.27(4), N(1)–Zn(1)–N(2) = 79.95(4), N(1)–Zn(1)–O(1) = 90.41(4), N(2)–Zn(1)–O(2) = 90.60(4), O(1)–Zn(1)–O(3) = 96.70(4), N(1)–Zn(1)–O(3) = 90.25(4).](/image/article/2012/CY/c2cy20171b/c2cy20171b-f3.gif) | ||
| Fig. 3 X-ray molecular structure for 2·[trans-2,3-dimethyl-oxirane]. H-atoms, co-crystallised solvent molecules and rotational disorder in the coordinated epoxide are omitted for clarity, and only a partial numbering scheme is provided. Selected bond lengths (Å) and angles (°): Zn(1)–O(1) = 1.9438(9), Zn(1)–O(2) = 1.9399(9), Zn(1)–O(3) = 2.2086(11), Zn(1)–N(1) = 2.0638(9), Zn(1)–N(2) = 2.0577(10); O(1)–Zn(1)–O(2) = 95.27(4), N(1)–Zn(1)–N(2) = 79.95(4), N(1)–Zn(1)–O(1) = 90.41(4), N(2)–Zn(1)–O(2) = 90.60(4), O(1)–Zn(1)–O(3) = 96.70(4), N(1)–Zn(1)–O(3) = 90.25(4). | ||
![X-ray molecular structure for 1·[(3-methyloxetan-3-yl)methanol]. H-atoms, co-crystallised solvent molecules and rotational disorder in the coordinated oxetane are omitted for clarity. Selected bond lengths (Å) and angles (°): Zn(1B)–O(1B) = 1.9352(7), Zn(1B)–O(2B) = 1.9622(6), Zn(1B)–O(3B) = 2.1434(7); O(1B)–Zn(1B)–O(2B) = 96.96(3), O(2B)–Zn(1B)–O(3B) = 99.24(3), O(1B)–Zn(1B)–O(3B) = 94.14(3), N(1B)–Zn(1B)–N(2B) = 79.84(3), N(1B)–Zn(1B)–O(3B) = 101.56(3), N(1B)–Zn(1B)–O(2B) = 157.09(3), N(1B)–Zn(1B)–O(1B) = 91.04(3).](/image/article/2012/CY/c2cy20171b/c2cy20171b-f4.gif) | ||
| Fig. 4 X-ray molecular structure for 1·[(3-methyloxetan-3-yl)methanol]. H-atoms, co-crystallised solvent molecules and rotational disorder in the coordinated oxetane are omitted for clarity. Selected bond lengths (Å) and angles (°): Zn(1B)–O(1B) = 1.9352(7), Zn(1B)–O(2B) = 1.9622(6), Zn(1B)–O(3B) = 2.1434(7); O(1B)–Zn(1B)–O(2B) = 96.96(3), O(2B)–Zn(1B)–O(3B) = 99.24(3), O(1B)–Zn(1B)–O(3B) = 94.14(3), N(1B)–Zn(1B)–N(2B) = 79.84(3), N(1B)–Zn(1B)–O(3B) = 101.56(3), N(1B)–Zn(1B)–O(2B) = 157.09(3), N(1B)–Zn(1B)–O(1B) = 91.04(3). | ||
![X-ray molecular structure for 2·[cyclohexene oxide] (taken from ref. 15). Note the steric impediment around both C-atoms of the oxirane unit. The tBu groups in the 5-positions show rotational disorder.](/image/article/2012/CY/c2cy20171b/c2cy20171b-f5.gif) | ||
| Fig. 5 X-ray molecular structure for 2·[cyclohexene oxide] (taken from ref. 15). Note the steric impediment around both C-atoms of the oxirane unit. The tBu groups in the 5-positions show rotational disorder. | ||
These crystal structures demonstrate that all these substrates are involved in coordination to the Zn(II) centre through the O-atoms of the epoxide/oxetane unit. However, our catalytic results indicate that this interaction is a necessity but not a sufficient condition alone to activate the substrate towards the ring-opening by the nucleophile.
All these results, combined with previous findings of some of us,14,15 demonstrate the wide scope of the Zn(salphen)/Bu4NI system in the preparation of cyclic carbonates, also in the case of substrates that are generally more difficult to convert.3,4,15,29,30
Conclusions
We have demonstrated that excellent yields in the synthesis of cyclic carbonates by cycloaddition of CO2 to epoxides can be achieved with the binary Zn(salphen)/Bu4NI catalyst system in short time frames. We obtained nearly complete conversion of styrene oxide to its carbonate, promisingly good yields of cyclohexene carbonate and excellent yields with other epoxides. This significant increase in the catalytic performance was achieved by working in a solvent-free, CO2-rich environment.31 This means that we were able to improve the performance of the Zn(salphen)/Bu4NI catalyst system while working in greener, solvent-free conditions. An efficient interaction between CO2, the epoxide, the Zn(salphen) catalyst and the Bu4NI co-catalyst has proven to be a key factor for obtaining high cyclic carbonate yields. Combined with the other advantageous properties of the Zn(salphen) complex (low cost/toxicity, good stability), this catalyst system thus shows high potential in the context of organic carbonate production in a green CO2 medium.Acknowledgements
We thank the programs START1, CREA and Methusalem (KU Leuven, Flemish Government), and ICIQ, ICREA, the Spanish Ministerio de Economía y Competitividad (MINECO) through project CTQ2011-27385 for funding.Notes and references
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS.
- D. Bai, Q. Wang, Y. Song, B. Li and H. Jing, Catal. Commun., 2011, 12, 684 CrossRef CAS.
- M. Fondo, A. M. García-Deibe, M. R. Bermejo, J. Sanmartín and A. L. Llamas-Saiz, J. Chem. Soc., Dalton Trans., 2002, 4746 RSC.
- A. W. Kleij, M. Kuil, D. M. Tooke, M. Lutz, A. L. Spek and J. N. H. Reek, Chem.–Eur. J., 2005, 11, 4743 CrossRef CAS.
- M. Alvaro, C. Baleizao, D. Das, E. Carbonell and H. Garcia, J. Catal., 2004, 228, 254 CrossRef CAS.
- J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323 CrossRef.
- P. Ramidi, S. Z. Sullivan, Y. Gartia, P. Munshi, W. O. Griffin, J. Darsey, A. Biswas, A. Shaikh and A. Ghosh, Ind. Eng. Chem. Res., 2011, 50, 7800 CrossRef CAS.
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639 RSC.
- J. Sun, S.-i. Fujita, F. Zhao and M. Arai, Green Chem., 2004, 6, 613 RSC.
- J. Wang, X. Yue, F. Cai and L. He, Catal. Commun., 2007, 8, 167 CrossRef CAS.
- A. Decortes and A. W. Kleij, ChemCatChem, 2011, 3, 831 CrossRef CAS.
- A. Decortes, M. Martínez Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580 RSC.
- Y. Ren, C.-H. Guo, J.-F. Jia and H.-S. Wu, J. Phys. Chem. A, 2011, 115, 2258 CrossRef CAS.
- A. Baiker, Chem. Rev., 1999, 99, 453 CrossRef CAS.
- R. Sheldon, Green Chem., 2005, 7, 267 RSC.
- W. Leitner, Acc. Chem. Res., 2002, 35, 746 CrossRef CAS.
- G. M. Sheldrick, SHELXTL Crystallographic System Version 6.10, Bruker AXS, Inc., Madison (Wisconsin), 2000 Search PubMed.
- C. Aprile, F. Giacalone, L. Liotta, J. A. Martens, P. P. Pescarmona and M. Gruttadauria, ChemSusChem, 2011, 4, 1830 CrossRef CAS.
- J. Jammaer, C. Aprile, S. W. Verbruggen, S. Lenaerts, P. P. Pescarmona and J. A. Martens, ChemSusChem, 2011, 4, 1457 CrossRef CAS.
- S. Campestrini and U. Tonellato, Curr. Org. Chem., 2005, 9, 31 CrossRef CAS.
- B. Subramaniam and D. H. Busch, CO2 Conversion and Utilization, American Chemical Society, 2002, p. 364 Search PubMed.
- H. Jing and S. R. Nguyen, J. Mol. Catal. A: Chem., 2007, 261, 12 CrossRef CAS.
- A. Ion, V. Parvulescu, P. Jacobs and D. de Vos, Appl. Catal., A, 2009, 363, 40 CrossRef CAS.
- H. Xie, S. Li and S. Zhang, J. Mol. Catal. A: Chem., 2006, 250, 30 CrossRef CAS.
- See for a successful example of oxetane conversion using Cr(salen) complexes: D. J. Darensbourg, A. I. Moncada, W. Choi and J. H. Reibenspies, J. Am. Chem. Soc., 2008, 130, 6523 CrossRef CAS.
- B. Barkakaty, K. Morino, A. Sudo and T. Endo, Green Chem., 2010, 12, 42 RSC.
- L. Han, H.-J. Choi, S.-J. Choi, B. Liu and D.-W. Park, Green Chem., 2011, 13, 1023 RSC.
- For a previous work on DMC (dimethylcarbonate) synthesis in sc-CO2 see: Y. Chang, T. Jiang, B. Han, Z. Liu, W. Wu, L. Gao, J. Li, H. Gao, G. Zhao and J. Huang, Appl. Catal., A, 2004, 263, 179 CrossRef CAS.
Footnote |
| † CCDC 855909 and 867372. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cy20171b |
| This journal is © The Royal Society of Chemistry 2012 |