Synthesis, characterization, and catalytic application of networked Au nanostructures fabricated using peptide templates†
Rohit
Bhandari
and
Marc R.
Knecht
*
Department of Chemistry, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA. E-mail: knecht@miami.edu; Fax: (305) 284-4571; Tel: (305) 284-9351
First published on 12th April 2012
Abstract
Bio-inspired-based methods represent new approaches for the fabrication and activation of nanomaterials, all under ambient and energy-neutral conditions. Recent advances have demonstrated the production of non-spherical materials of Pd and Pt; however, the production of similar Au materials remains challenging. Such fabrication routes are highly important as Au-based nanomaterials of selectable morphologies could have immediate applications in catalysis and energy storage. In this contribution, we demonstrate a peptide template-based methodology for the fabrication of Au nanoparticle networks, which are highly branched linear structures that are prepared in water at room temperature. The materials were fully characterized using UV-vis, TEM, XRD, and DLS, from which their catalytic activity was subsequently studied for the reduction of 4-nitrophenol. Using this approach, the materials were shown to be highly reactive as compared to comparable structures, which is likely due to their unique biological template. Together, this research represents a step forward in bio-based methodologies for the fabrication of functional and potentially sustainable materials.
Introduction
The fabrication of catalytic materials of selectable morphologies and arrangements remains a significant challenge. Such structures are critically important for a variety of applications ranging from chemical transformations to energy technologies.1,2 Au materials have become an interesting choice for chemical applications where such particles have demonstrated unique catalytic properties when structured on the nanoscale. For instance, Au nanoparticles possess significant catalytic activity for CO oxidation,3 4-nitrophenol reduction,4 and polymerization of alkylsilanes.5 Furthermore, recent results have demonstrated that this catalytic activity can be tuned and potentially expanded via excitation of the inherent plasmon band of the materials.6 While the correlation between catalytic activity and plasmonic absorption remains unclear, it is evident that Au nanomaterials possess multifunctionality that could be controlled by the particle properties. In general, these properties are embedded into the final system based upon the composite structure of the material. To that end, the size, shape, and ligand passivating layer can be simultaneously tuned to control the optical and catalytic functionality of the nanoparticle. Different shapes of Au nanostructures have been prepared including spherical particles,7,8 wires,9 rods,10 and cubes11 that are passivated by a variety of stabilizing agents. By altering the particle shape, changes to the plasmonic properties will occur.12 Such changes can also introduce additional high energy surface defect atoms that are likely to be highly reactive; however, an optimal Au structure for catalytic-based applications remains unclear.Recently, Au nanoparticle networks (NPNs), which are linear, branching nanomaterials, have become of interest due to their high surface area, unique optical properties, and large frameworks.13–16 Typically, such morphologies are produced when nanoparticles linearly aggregate to form the networked structures; the width of the final materials is determined based upon the size and composition of the individual nanoparticles.16 While the branching framework can extend to distances up to the micron scale, the individual cross section of the materials is typically <20 nm. Furthermore, Au NPNs are typically capped on the surface with a passivating ligand; however, these ligands could be designed to integrate new functionality, cause diminished reactivity, or be removed using standard methods.17,18 Interestingly, the catalytic application of Au NPNs remains under studied where such materials are well situated to make rapid advancements due to their unique structure and extended metallic surface. Unfortunately, efficient methods for their fabrication are limited. Most approaches require the use of non-aqueous solvents and/or complex directed nanoparticle aggregation methods that result in low yields. Recently, Silva et al. have proposed the synthesis of Au NPNs in the presence of deep eutectic solvents (DES) such as ethaline or reline at 40 °C.13 In this study, the linear width of the materials was dependent on the type of solvent and reductant/precursor ratio in the reaction mixture. These materials were found to be catalytically active for the reduction of 4-nitroaniline to diaminophenylene. In similar studies, the inherent instability of Au nanoparticles has been exploited to drive NPN formation using template free approaches.16,19,20 While these materials represent intriguing structures as device components, their catalytic reactivity typically is not studied. To advance such materials toward catalytic applications, new fabrication methods are required that rapidly prepare large quantities of Au NPNs under ambient conditions that limit the synthetic complexity. Furthermore, the passivant could potentially be tuned to engender additional functionality to the system including low temperature reactivity and/or catalytic selectivity.
Biomimetic and peptide-based syntheses have emerged as new methods to fabricate complex and functional inorganic materials for a variety of technological applications.2,21 Recent evidence indicates that such methods are capable of directing the generation of potentially catalytically important, non-spherical Pd and Pt materials including nanocubes,22,23 nanotetrahedra,22 and NPNs24,25 in water at room temperature. This effect would minimize the synthetic environmental impact; however, peptide-based fabrication of non-spherical Au nanomaterials has yet to be demonstrated. Such materials are likely to possess both catalytic and plasmonic capabilities under biological conditions that are presently challenging to achieve, thus raising the interest in such methods. Also, a detailed kinetic analysis of the catalytic activity of such materials may prove useful as the size and shape of the nanoparticles could be correlated to their inherent catalytic capability.
Herein, we report a peptide template-based approach for the generation of Au NPNs using the self-assembling R5 peptide (SSKKSGSYSGSKGSKRRIL) under ambient conditions (Scheme 1). The complex networked Au structures were fully analyzed using UV-vis spectroscopy, transmission electron microscopy (TEM), powder X-ray diffraction (XRD), and dynamic light scattering (DLS). The Au NPNs were found to be catalytically active for the reduction of 4-nitrophenol to generate 4-aminophenol in water where the peptide template and the structure of the materials could potentially enhance the reaction efficiency. Such results are important for the development of new template-based catalytic materials syntheses, in understanding peptide-based materials methods, and could be used to elucidate key structure/function relationships of catalytic inorganic nanostructures.
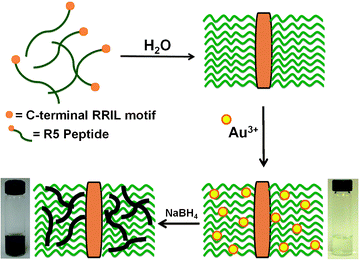 | ||
| Scheme 1 Proposed mechanism for the formation of Au NPNs using the self-assembling R5 peptide as a template. | ||
Experimental section
Chemicals
HAuCl4 (99.9+%) was purchased from Sigma-Aldrich, while NaBH4 (98+%) and 4-nitrophenol (99%) were purchased from Acros Organics. FMOC protected amino acids and WANG resins for the synthesis of the R5 peptide were acquired from Advanced Chemtech (Louisville, KY). Solvents including acetonitrile, methanol, and dimethylformamide (DMF) for peptide synthesis and purification were purchased from Pharmaco-Aaper. All chemicals and solvents were used as received. 18 MΩ cm deionized water (Millipore; Bedford, MA) was used to run all aqueous experiments.Characterization
UV-vis analyses for the synthesis of nanomaterials were conducted using an Agilent 8453 UV-vis spectrophotometer by employing a 2.00 mm quartz cuvette (Starna), while the catalytic reactions were performed using a 1.00 cm cuvette. DLS analyses were carried out using a Malvern Zetasizer Nano ZS instrument with He-Ne laser (633 nm) as the light source with a measurement range of 0.3 nm–10.0 μm. TEM and Energy Dispersive Spectroscopy (EDS) analyses were completed using a JEOL 2010F electron microscope with a resolution of 0.19 nm operating at 200 kV. Additional TEM studies of the control analysis were conducted using a JEOL 1400 TEM with an accelerating voltage of 80 kV. The samples for TEM were prepared by pipetting 5.00 μL of the reaction solution onto 400-mesh carbon coated Cu grids (EM Sciences) and then allowed to dry in a desiccator. Powder XRD studies were performed using Bruker Axis D8 discovery X-ray diffractometer operating with a Cu-Kα radiation source of 1.5418 Å. The diffraction patterns were obtained by running the samples from a 2θ range of 20.0° to 90.0°. Inductively coupled plasma-mass spectrometry (ICP-MS) analyses of the Au materials were conducted using a multi collector-ICP-MS instrument with a detection limit of 100.0 parts per trillion (ppt) using a self aspirating PFA nebulizer paired with a Stable Sample Introduction spray chamber. The samples were diluted and dissolved in 0.45 M HNO3 prior to the analysis and the measurements were made using the secondary electron multiplier as the instrument detector.Quartz Crystal Microbalance (QCM) studies of peptide binding were performed employing a Q-Sense E4 instrument with a frequency range of 1–70 MHz and a maximum mass sensitivity of 0.5 ng cm−2 in liquids. For this, the Au sensor was cleaned using UV/ozone treatment, followed by heating in a mixture of ammonia (25%) and H2O2 (30%) in water at 75 °C. The sensor was then dried under N2, retreated with UV/ozone, and placed in the E4 module. Three different concentrations of the R5 peptide, 2.48 μM, 4.97 μM and 7.45 μM, were flushed through the module to interact with the Au surface from which ka values were calculated based upon the changes in frequency.
Synthesis of Au NPNs
The R5 peptide was synthesized using standard FMOC protocols26 on a TETRAS peptide synthesizer (CreoSalus; Louisville, KY), followed by purification via HPLC. The purified peptide was confirmed using MALDI-TOF analysis. For the fabrication of the Au NPNs, 4.93 μL of a 4.97 mM R5 peptide stock solution was dissolved in 3.00 mL of water. To separate reactions, 7.32 μL or 14.6 μL of a 0.1 M HAuCl4 stock solution was added to result in a 30 or 60 fold excess, respectively, of Au3+ to the peptide. After stirring the reactions for 15.0 min, 75.0 μL of a freshly prepared 0.1 M aqueous NaBH4 solution was added to each vial and the reaction was allowed to stir for 1.00 h. After that, the materials were dialyzed against deionized water using cellulose dialysis tubing (MW 14 kDa) for 24.0 h.Catalytic 4-nitrophenol reduction
The procedure employed for the catalytic reduction of 4–nitrophenol was based on previous studies.24,25 For the Au30 catalyzed reaction, 1.00 mL of the NPN sample was added to 0.50 mL of NaBH4 (63.0 mM) in a 1.00 cm quartz cuvette. The reaction was subsequently left undisturbed for 15.0 min. After that, 1.50 mL of a 112.0 μM 4-nitrophenol solution was added to the reaction mixture. Time dependent UV-vis spectra of the reaction were recorded at 30.0 s intervals for 10.0 min at different temperatures ranging from 10.0 °C to 60.0 °C. Identical analyses were conducted for the Au60 reactions where 0.50 mL of the Au60 sample was added to 0.50 mL of freshly prepared NaBH4 (63.0 mM), followed by the addition of 2.00 mL of an 85.0 μM 4-nitrophenol solution. The concentration of the Au catalyst was kept constant (79.0 μM Au) across both reaction systems. Also, different 4-nitrophenol solutions were employed for the Au30- and Au60-based systems to ensure identical reagent concentrations (56.0 μM) in the catalytic reactions. Triplicate analyses were conducted for both sets of materials at all reaction temperatures to achieve a complete statistical analysis.Results and discussion
The fabrication of zerovalent Au NPNs is driven by the self-assembling R5 peptide that serves as a template to direct materials fabrication (Scheme 1).24,25 The sequence assembles via the C-terminal RRIL motif to form peptide aggregates where the functional groups of the amino acids are presented at the interior of the framework.27 Here, the peptide was dissolved in water to a concentration of 8.15 μM and sufficient HAuCl4 was added to vary the Au3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :peptide ratio from 30 to 120. Once added, the Au3+ ions can coordinate to the functional groups of the peptide, thus sequestering the metal ions from solution within the preformed scaffold. The metal ion/peptide complexes were then reduced using excess NaBH4 to generate the zerovalent materials, which was visually confirmed by a solution color change from faint yellow to deep blue. For reactions with Au3+:peptide ratios ≤60, stable colloids were observed with increasing color intensity proportional to the Au3+:peptide ratio (ESI,† Fig. S1); however, at higher ratios, a blue-black precipitate was observed rapidly after reduction. This suggests that the peptide scaffold is saturated with Au3+, thus uncoordinated metal ions remain in solution, resulting in bulk precipitation. Using this fabrication scheme, two Au3+:peptide ratios, 30 and 60, termed Au30 and Au60, respectively, formed stable materials that were studied further.
:peptide ratio from 30 to 120. Once added, the Au3+ ions can coordinate to the functional groups of the peptide, thus sequestering the metal ions from solution within the preformed scaffold. The metal ion/peptide complexes were then reduced using excess NaBH4 to generate the zerovalent materials, which was visually confirmed by a solution color change from faint yellow to deep blue. For reactions with Au3+:peptide ratios ≤60, stable colloids were observed with increasing color intensity proportional to the Au3+:peptide ratio (ESI,† Fig. S1); however, at higher ratios, a blue-black precipitate was observed rapidly after reduction. This suggests that the peptide scaffold is saturated with Au3+, thus uncoordinated metal ions remain in solution, resulting in bulk precipitation. Using this fabrication scheme, two Au3+:peptide ratios, 30 and 60, termed Au30 and Au60, respectively, formed stable materials that were studied further.
Fig. 1a presents the UV-vis analysis of the Au3+/peptide-template complexes prior to reduction. In this study, absorbance peaks are noted at 220 nm and 300 nm, corresponding to the Au3+ ions in the reaction mixture. Nearly identical spectra were observed for both the Au30 and Au60 pre-reduced samples; however, a stronger absorption intensity was noted for the higher ratio, as expected. Fig. 1b displays the post reduction spectra for both samples. Here, no peaks corresponding to the Au3+ ions are present in either the Au30 or Au60 spectra, suggesting full reduction of the materials; however, a very broad absorbance at wavelengths >580 nm is present for both samples. Again, a greater absorption intensity was observed for the Au60 materials, which likely arises from the higher Au concentration in the sample. The broad absorbance and deep blue solution color suggest the formation of larger Au nanostructures.
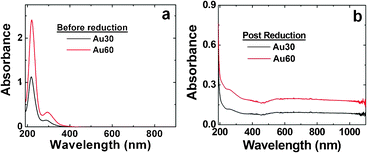 | ||
| Fig. 1 UV-vis analysis of the synthesized Au materials. Part (a) presents the UV-vis spectra of the Au3+:peptide template complexes before reduction, while part (b) displays the spectra of the materials after reduction with excess NaBH4. | ||
After reduction, XRD analysis was conducted to fully characterize the materials. Fig. 2a and b present XRD plots for the Au30 and Au60 structures, respectively. Both zerovalent samples demonstrated identical reflections associated with the face centered cubic (fcc) structure of Au. Additional reflections from two different crystal forms of NaCl are present in the samples, which arise from salts formed during the drying process. These salts are generated from the counter ions of the starting reagents employed during materials synthesis. Scherrer's analysis using the Au (111) reflection for the Au30 and Au60 samples indicated particle crystallite sizes of 6.2 nm and 6.4 nm, respectively. This suggests that the materials are indeed composed of nanoscale-based structures, which is evident by the width of the reflections in the plot; however, imaging of the materials using electron microscopy is required to confirm this structural detail.
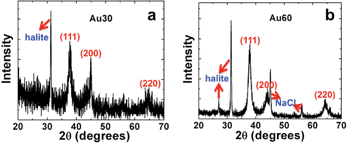 | ||
| Fig. 2 XRD analysis of the synthesized Au materials. Part (a) presents the diffraction pattern for the Au30 NPNs, while part (b) displays the diffraction pattern for the Au60 materials. The peaks are labelled with their appropriate reflections. | ||
TEM analysis of the materials is shown in Fig. 3. Specifically, Fig. 3a presents a low magnification TEM image for the Au30 sample, while the analysis of the Au60 sample is displayed in Fig. 3b. For both materials, networked, linear-like NPNs were observed. The Au30 structures possessed an average width of 6.7 ± 1.1 nm, while the Au60 materials demonstrated a width of 7.1 ± 1.3 nm. Such values correspond well with the results determined using the Scherrer's analysis of the XRD patterns for both samples. High-resolution TEM (HR-TEM) analyses for both materials are presented in Fig. 3c and d. For the Au30 materials (Fig. 3c), a polycrystalline structure was observed that demonstrated lattice fringes with a d spacing of 2.4 Å. This value is consistent with the known d spacing for the {111} fringe of fcc Au.24Fig. 3d displays the HR-TEM analysis of the Au60 NPNs where similar polycrystalline Au materials are observed. Again, Au {111} lattice fringes are noted with a d spacing of 2.4 Å. EDS analysis of the Au30 materials is presented in the ESI,† Fig. S2, which confirms the Au composition of the materials. Further ICP-MS studies of both the Au30 and Au60 materials demonstrated that the structures were fully comprised of Au; no trace contamination by Pd or Pt was observed as both metals were below the ppt limit of detection. Together, this suggests that the fabrication method employing the peptide template forms highly networked inorganic materials with a polycrystalline structure composed of fcc Au.
 | ||
| Fig. 3 TEM analysis of the Au materials. Part (a) presents a low magnification image of the Au NPNs produced in the Au30 sample, while part (c) presents a high-resolution lattice-based image of the material. Parts (b and d) present the corresponding analysis for the Au60-based structures. | ||
Based upon the results above, it is likely that the self-assembling R5 peptide acts as a template to form the Au NPNs. It is known that the R5 sequence is able to self assemble to form large peptide aggregates via the C-terminal RRIL motif.27 To that end, biological scaffolds are present that are able to sequester metal ions within the framework via the peptide functional groups.24 This event is similar to the mechanism by which poly(amidoamine) dendrimers can be used to template the formation of dendrimer-encapsulated nanoparticles.28 As shown in Scheme 1, when the peptide is dissolved in water, the scaffold is generated where the metal ion binding functional groups are displayed at the framework interior. Upon addition of Au3+, binding to these groups occurs to sequester the ions within the scaffold, which are subsequently reduced to form the Au NPNs. Initially, nanoparticles are likely generated that then linearly aggregate to form linear, branched metallic morphologies. The peptide branches of the framework then act as a materials-directing template to generate the inorganic structures. This aggregation-based method is supported by the HR-TEM images where clear bulging points are observed in the NPNs that arise from the initial spherical nanoparticles. This is similar to results observed for the fabrication of comparable Pd NPNs.24,25 Interestingly, we were unable to observe spherical materials within the scaffold, even at the lowest Au:peptide ratio. This is likely due to the very strong metal bonding of Au,29,30 which drives the nanoparticle aggregation process to form the NPNs. Based upon this motif, the colloidal stability of the materials is directly owed to the composite structure; by being contained within the peptide scaffold, no exposed Au is present to stabilize the system. The materials remain suspended in water for at least one month on the benchtop without observable precipitation.
To confirm the peptide-template synthetic mechanism, DLS analyses were conducted of the materials before and after reduction, as presented in Table 1. For the R5 peptide dissolved in water, aggregates of 960 ± 43 nm were observed, demonstrating the formation of the template. Upon introduction of Au3+ ions to the peptide scaffold, aggregates of 1534 ± 27 nm and 2554 ± 23 nm were determined at Au3+:peptide ratios of 30 and 60, respectively. This change in aggregate size likely arises from peptide/metal ion crosslinking in solution. In this event, Au3+ ions near the template surface could be bound by multiple peptides from different templates, which would increase the average aggregate size by coupling multiple scaffolds. This is supported by the larger aggregates observed at higher Au3+:peptide ratios where more metal ions would be available. When the Au3+/peptide complexes were reduced using NaBH4, dispersed aggregates of 115 ± 0.1 nm and 176 ± 2 nm were observed for the Au30 and Au60 samples, respectively. This change in size likely arises due to the formation of the zerovalent structures; the metallic Au surface could change both peptide-metal and peptide-peptide interactions as compared to the Au3+/peptide complex. These changes occur to facilitate the encapsulated nanoparticle networks. Indeed, QCM analysis of the binding of the R5 peptide to a flat Au surface (Fig. S3 of the ESI†) demonstrated a strong ka value of 3.2 × 105 M−1. Furthermore, only minor peptide desorption was noted when the peptide-bound system was washed with water, demonstrating the strong interaction between the peptide and metallic surface. In this sense, once the NPNs are formed, changes to the peptide scaffold may be possible, which give rise to the observed size changes after reduction. Nevertheless, the DLS results are consistent with the proposed peptide-templation synthesis for the fabrication of the Au NPNs.
By comparing the TEM, XRD, and DLS results for the characterization of the two different Au NPNs (Au30 and Au60), an interesting observation concerning the global structure of the materials arises. For instance, as noted by the XRD and TEM results, materials of roughly the same inorganic morphology are observed with similar dimensions, within the limits of the error; however, DLS demonstrates that larger composite structures are observed from the Au60 NPNs as compared to the Au30 materials. This suggests that while the inorganic components of the materials are roughly the same, a higher degree of template crosslinking is observed in the Au60 structures, leading to larger 3D composite materials. Such differences could affect the properties of the materials, especially for catalysis and energy applications.
The overall composite structure of the materials, linear Au components encapsulated by a peptide framework, is well situated for catalysis due to the large inorganic/catalytic motif and the permeability of the open biological scaffold.31–33 To test the reactivity of these materials, their capabilities for the reduction of 4-nitrophenol to 4-aminophenol was tested.25 For this reaction, the substrate is catalytically reduced by the Au NPNs in the presence of NaBH4. In this system, the solution is saturated with borohydride to the point that its concentration remains essentially constant during the reaction; therefore, pseudo first order reaction kinetics with respect to 4-nitrophenol can be assumed.34,35 Furthermore, the reagent exhibits a strong absorbance at 400 nm under basic conditions due to the formation of the 4-nitrophenolate ion, thus the reaction can readily be monitored using UV-vis.25 As shown in Fig. 4a, upon introduction of the Au30 NPN catalyst at a reaction concentration of 79.0 μM Au and a temperature of 20.0 °C, the 400 nm reagent peak begins to diminish concomitant with the generation of a new peak at 310 nm. This new peak corresponds to the formation of the 4-aminophenol product. As a control experiment in the absence of the noble metal catalyst, no change in the intensity of the 400 nm peak was observed, indicating that NaBH4 alone could not reduce 4-nitrophenol. Unfortunately, consistent with previous reports, no isosbestic point was observed for this reaction;25,34 H2 gas bubbles are developed during the process that cause scattering to prevent observation of the anticipated point. As such, pseudo first order reaction rate constants (k) can be isolated by monitoring the decomposition of the 400 nm reagent absorbance as a function of time.34,36,37
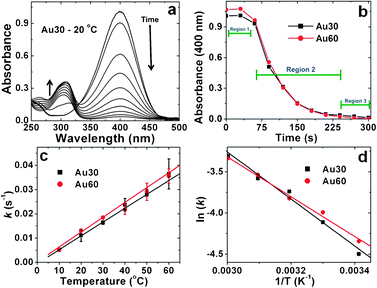 | ||
| Fig. 4 Kinetic analysis for the 4-nitrophenol reduction reaction catalyzed by the Au NPNs. Part (a) demonstrates the time-resolved UV-vis plot for the reduction reaction driven by the Au30 materials at 20.0 °C, while part (b) presents an analysis of the decreasing 400 nm reagent absorbance as a function of time for both the Au30 and Au60 materials. Part (c) displays the rate constants for both systems as a function of temperature, while part (d) presents the Arrhenius plots for the kinetic analysis of the Au NPNs where the slope of the trend line is used to determine the Ea values. | ||
Fig. 4b presents the decrease in intensity of the 400 nm 4-nitrophenol absorbance during the reactions catalyzed by the Au30 and Au60 NPNs at 20.0 °C. As is evident, three distinct regions are present in the graph: an induction period (region 1: t < 60.0 s), a reaction period (region 2: t = 60.0 s–240 s), and a completion period (region 3: t > 240 s). The induction period (t0) observed here can be attributed to the adsorption of the two reagents, 4-nitrophenol and BH4−, onto the metallic surface. Following the Langmuir-Hinshelwood mechanism for the reaction,37,38 both reagents must be fully adsorbed onto the particle surface for the reaction to proceed. In this sense, the BH4− will react with the catalytic particle to result in the formation of surface-bound H-species that are required for the reduction. In our system, the Au NPNs are premixed with NaBH4 prior to addition of the substrate to fully saturate the metallic surface with hydrogen. In the second step, 4-nitrophenol must adsorb onto the unoccupied area of the metallic surface, from which the reduction reaction can occur. The final product, 4-aminophenol, subsequently desorbs from the catalytic materials, releasing a new reaction site.37 In the present system, t0 is thus attributed to the 4-nitrophenol adsorption process, which is known to be rapid as compared to the rate limiting reduction step.37,38 As a result, pseudo-first order kinetics are studied during the reaction step of the process, region 2, consistent with previous studies employing Au nanocatalysts.36,39
To fully study the kinetics of the Au NPN catalyzed reaction, a graph of ln(Ct/C0), where Ct represents the reagent concentration at time t and C0 represents the starting reagent concentration, as a function of time was plotted. The analysis over region 2 was used to calculate the pseudo first order rate constant k using the equation below.34,35
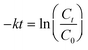
Using this approach, the k values were determined for both the Au30 and Au60 systems over a temperature range of 10.0 °C to 60.0 °C where the rate constants are listed in Table 2 and shown in Fig. 4c. From this analysis, it is evident that the rate constants for both biocatalytic systems increase proportional to the reaction temperature. For instance, when the reaction was catalyzed using the Au30 NPNs, a k value of 0.005 ± 0.0005 s−1 was observed at 10.0 °C that increased to 0.036 ± 0.006 s−1 at 60.0 °C. For the Au60 materials, the pseudo first order rate constants ranged from 0.004 ± 0.0005 s−1 at 10.0 °C to 0.035 ± 0.003 s−1 at 60.0 °C. When these rate constants are plotted in Fig. 4c as a function of temperature, it is evident that the values for the two different systems are statistically equivalent at all temperatures for both the Au30 and Au60 compositions (within the error limit). This suggests that both systems are equivalent catalytic structures to drive the 4-nitrophenol reduction reaction.
| T/°C | R5-Au material [10−2k (s−1)] | |
|---|---|---|
| Au30 | Au60 | |
| 10 °C | 0.5 ± 0.05 | 0.4 ± 0.05 |
| 20 °C | 1.1 ± 0.2 | 1.3 ± 0.05 |
| 30 °C | 1.6 ± 0.07 | 1.8 ± 0.1 |
| 40 °C | 2.4 ± 0.2 | 2.1 ± 0.2 |
| 50 °C | 2.8 ± 0.2 | 2.9 ± 0.3 |
| 60 °C | 3.6 ± 0.6 | 3.5 ± 0.3 |
| E a (kJ mol−1) | 29.0 ± 1.4 | 27.7 ± 1.6 |
Further analyses were conducted to derive a relationship between the rate constants and activation energy (Ea) associated with both sets of NPNs. For that, an Arrhenius plot was prepared, presented in Fig. 4d, from which the slope of the trend line was used to calculate the activation energies for both catalytic samples. Using this approach, an Ea value of 29.0 ± 1.4 kJ mol−1 and 27.7 ± 1.6 kJ mol−1 was determined for the Au30 and Au60 systems, respectively. It is not surprising that nearly identical Ea values were determined for both materials as they are composed of the same noble metal nanocatalyst (Au) with very similar overall structural motifs (NPNs). Interestingly, these activation energies are greater than those observed for Pd NPNs generated with the R5 peptide (∼7.5 kJ mol−1),25 where similar structural motifs are present for both the Au and Pd materials. This suggests that the Pd structures possess a higher degree of catalytic activity for this reaction as compared to the Au materials, which is likely due to the different inorganic compositions. When comparing the present materials to other comparable Au nanostructures, the biomimetic Au NPNs possessed significantly faster rate constants. For instance, Au NPNs formed using DESs demonstrated rate constants of 3.0 × 10−2 min−1 at 40.0 °C for the reduction of 4-aminophenol,13 which is 40 times lower than the values observed using the peptide-based Au NPNs (2.1–2.4 × 10−2 s−1). This suggests that while the inorganic composition strongly affects the reactivity rates, the organic/biological template structure of the materials is also critically important to the functionality. To that end, by possessing higher rate constants, the peptide-based framework of the materials is potentially optimized for faster reactivity for the reduction reaction, which likely arises from more efficient exposure of the inorganic catalyst. This could be achieved via the open/porous framework of the peptide scaffold that is permeable by the reagents in solution. Fully elucidating this structure/function relationship is highly important to realize next generation catalytic materials optimized for reactivity under ambient conditions.
It is interesting to note that the two different Au NPNs demonstrated statistically equivalent catalytic activity, regardless of the reaction temperature. While the inorganic component is roughly the same, different degrees of crosslinking are observed where larger peptide frameworks are present for the Au60 NPNs as compared to the Au30 materials. This suggests that the biological scaffold is potentially quite flexible and does not overly inhibit the diffusion of the reagents to access the catalytic surface. This likely arises from the inorganic morphology, which may possess direct influence over the peptide-assembled structure; however, additional studies are required to confirm this hypothesis.
As a control, the Au30 and Au60 materials were fabricated under identical conditions; however, no peptide was added to the reaction. Surprisingly, after reduction with NaBH4, stable nanomaterials were observed in solution (ESI,† Fig. S4 and S5). For the peptide-free Au30 system, a mixture of spherical Au nanoparticles with a diameter of 7.7 ± 2.3 nm and short linear structures with widths of 10.3 ± 1.9 nm were observed. For the Au60 system in the absence of the R5 peptide, a variety of shapes were developed including spherical nanoparticles (18.3 ± 4.2 nm diameter), large nanocubes (177 ± 57 nm edge length), linear materials (15.0 ± 2.5 nm width), and fern leaf shaped structures. The relative distribution of the different shaped materials is presented in the ESI,†Fig. S5. This polydispersity in shape arises from the lack of the peptide framework in the reaction that directs the growth of only NPNs in the template-based samples. Furthermore, the widths of the R5-encapsulated materials were smaller as compared to the linear structures in the peptide-free systems. This is an important point as the smaller material widths enhance the surface-to-volume ratio to optimize catalytic activity. Unfortunately, catalytic analysis of the peptide-free materials was unable to completed as the structures rapidly aggregated and precipitated at temperatures >25 °C (room temperature), where such conditions are required for the analysis of the reduction reaction.
Conclusions
In summary, we have demonstrated an ambient, bio-template-based synthetic route for the fabrication of non-spherical Au NPNs with a significant degree of catalytic reactivity. The materials were fabricated using a simple two-step, template-based approach that resulted in highly crystalline materials. NPN-based structures were observed at all metal loadings within the peptide scaffold, due to the strong metal-metal interactions of Au. The linear-based character of the materials is likely due to particle aggregation that is influenced by the peptide strands of the template. While the inorganic components were quite similar, different degrees of peptide scaffold crosslinking were observed as a function of the metal concentration that likely altered the composite 3D structure. The materials were found to be highly catalytically active for the 4-nitrophenol reduction reaction with increased efficiency as compared to other networked Au nanostructures, which is an effect of the bio-organic surface layer. Furthermore, the reactivity of the materials was shown to be insensitive to the degree of peptide crosslinking, suggesting that reagent diffusion through the framework does not inhibit the reactivity. Taken together, these materials represent a new class of Au-based shaped nanostructures that could potentially serve as candidates for multiple applications ranging from catalysis and energy production to biosensing.Acknowledgements
This study is based upon work supported by the National Science Foundation under Grant DMR-1145175. Further partial support from the University of Miami and the ACS Petroleum Research Fund is acknowledged. The authors wish to thank Dr. Ali Pourmand for assistance in acquiring the ICP-MS data. We would also like to acknowledge the Dauer EM lab (UM Biology) for assistance with TEM analysis.Notes and references
- N. J. Halas, Nano Lett., 2010, 10, 3816–3822 CrossRef CAS.
- Y. J. Lee, Y. Lee, D. Oh, T. Chen, G. Ceder and A. M. Belcher, Nano Lett., 2010, 10, 2433–2440 CrossRef CAS.
- A. Horváth, A. Beck, G. Stefler, T. Benkó, G. Sáfrán, Z. Varga, J. Gubicza and L. Guczi, J. Phys. Chem. C, 2011, 115, 20388–20398 Search PubMed.
- S. Praharaj, S. Nath, S. K. Ghosh, S. Kundu and T. Pal, Langmuir, 2004, 20, 9889–9892 CrossRef CAS.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen, V. Zaikovski and K. J. Klabunde, J. Am. Chem. Soc., 2003, 125, 10488–10489 CrossRef CAS.
- Z. Liu, W. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Nano Lett., 2011, 11, 1111–1116 CrossRef CAS.
- A. M. Alkilany and C. J. Murphy, Langmuir, 2009, 25, 13874–13879 CrossRef CAS.
- J.-J. Yuan, A. Schmid, S. P. Armes and A. L. Lewis, Langmuir, 2006, 22, 11022–11027 CrossRef CAS.
- J. U. Kim, S. H. Cha, K. Shin, J. Y. Jho and J. C. Lee, Adv. Mater., 2004, 16, 459–464 CrossRef CAS.
- Z. Li, J. Tao, X. Lu, Y. Zhu and Y. Xia, Nano Lett., 2008, 8, 3052–3055 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- M. Chirea, A. Freitas, B. S. Vasile, C. Ghitulica, C. M. Pereira and F. Silva, Langmuir, 2011, 27, 3906–3913 CrossRef CAS.
- T. Wang, X. Hu and S. Dong, J. Phys. Chem. B, 2006, 110, 16930–16936 CrossRef CAS.
- L. Pei, K. Mori and M. Adachi, Langmuir, 2004, 20, 7837–7843 CrossRef CAS.
- G. Ramanath, J. D'Arcy-Gall, T. Maddanimath, A. V. Ellis, P. G. Ganesan, R. Goswami, A. Kumar and K. Vijayamohanan, Langmuir, 2004, 20, 5583–5587 CrossRef CAS.
- M. Crespo-Quesada, J.-M. Andanson, A. Yarulin, B. Lim, Y. Xia and L. Kiwi-Minsker, Langmuir, 2011, 27, 7909–7916 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS.
- B.-K. Pong, H. I. Elim, J.-X. Chong, W. Ji, B. L. Trout and J.-Y. Lee, J. Phys. Chem. C, 2007, 111, 6281–6287 CAS.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939–13948 CrossRef CAS.
- M. B. Dickerson, K. H. Sandhage and R. R. Naik, Chem. Rev., 2008, 108, 4935–4978 CrossRef CAS.
- C.-Y. Chiu, Y. Li, L. Ruan, X. Ye, C. B. Murray and Y. Huang, Nat. Chem., 2011, 3, 393–399 CrossRef CAS.
- L. M. Forbes, A. P. Goodwin and J. N. Cha, Chem. Mater., 2010, 22, 6524–6528 CrossRef CAS.
- A. Jakhmola, R. Bhandari, D. B. Pacardo and M. R. Knecht, J. Mater. Chem., 2010, 20, 1522–1531 RSC.
- R. Bhandari and M. R. Knecht, ACS Catal., 2011, 1, 89–98 CrossRef CAS.
- W. C. Chan and P. D. White, Oxford Univ. Press, New York, 2000.
- M. R. Knecht and D. W. Wright, Chem. Commun., 2003, 3038–3039 RSC.
- V. S. Myers, M. G. Weir, E. V. Carino, D. F. Yancey, S. Pande and R. M. Crooks, Chem. Sci., 2011, 2, 1632–1646 RSC.
- J. K. Burdett, O. Eisenstein and W. B. Schweizer, Inorg. Chem., 1994, 33, 3261–3268 CrossRef CAS.
- H. Schmidbaur, Gold Bull., 1990, 23, 11–21 CrossRef CAS.
- M. Haruta, Gold Bull., 2004, 37, 27–36 CrossRef CAS.
- H.-L. Jiang, B. Liu, T. Akita, M. Haruta, H. Sakurai and Q. Xu, J. Am. Chem. Soc., 2009, 131, 11302–11303 CrossRef CAS.
- F. Gao, Y. Wang and D. W. Goodman, J. Am. Chem. Soc., 2009, 131, 5734–5735 CrossRef CAS.
- Z. V. Feng, J. L. Lyon, J. S. Croley, R. M. Crooks, D. A. Vanden Bout and K. J. Stevenson, J. Chem. Educ., 2009, 86, 368 CrossRef CAS.
- S. Behrens, A. Heyman, R. Maul, S. Essig, S. Steigerwald, A. Quintilla, W. Wenzel, J. Bürck, O. Dgany and O. Shoseyov, Adv. Mater., 2009, 21, 3515–3519 CrossRef CAS.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2010, 10, 30–35 CrossRef CAS.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814–8820 CAS.
- M. A. Mahmoud and M. A. El-Sayed, Nano Lett., 2011, 11, 946–953 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: image of AuNPN solutions, EDS analysis, and characterization of the peptide-free control materials. See DOI: 10.1039/c2cy20149f |
| This journal is © The Royal Society of Chemistry 2012 |